

Abstract
Photocatalytic lithography couples light with photoreactive coated mask materials to pattern surface chemistry. We excite porphyrins to create radical species that photocatalytically oxidize, and thereby pattern, chemistries in the local vicinity. The technique advantageously is suited for use with a wide variety of substrates. It is fast and robust, and the wavelength of light does not limit the resolution of patterned features. We have patterned proteins and cells to demonstrate the utility of photocatalytic lithography in life science applications.
1. Introduction
Our research examines innovative photocatalytic techniques as well as the development of novel hybrid materials. We seek to engineer dictated chemical patterning and host response. Our long-term goal is the rapid, reproducible, and inexpensive patterning of surface arrays with nanometer-scale features. Deterministic collection and organization of proteins, DNA, viruses, and cells into ordered arrays holds enormous potential across multiple disciplines, including materials science, synthetic chemistry, biology and synthetic biology (presentation of nanoscale ligands), as well as medicine.
Several techniques for micrometer- or larger-scale patterning of chemistries for biomolecular and other purposes currently exist.1 These include photolithography,2–4 microcontact printing,5–7 etching with incorporation of elastomeric stencils,8 and selective molecular assembly patterning (SMAP9,10).
Photocatalytic patterning using metallic oxide catalysts has also been reported.11–13 These publications utilize TiO2 as a photocatalytic semiconductor activated with UV energy to degrade underlying chemistry. They do not report use of photocatalytic patterning techniques in the study of biophysical processes.
This article presents initial results using porphyrin photosensitizers for photocatalytic patterning. Photocatalytic patterning with photosensitizers represents a versatile new method for patterning surface chemistry with simple, variable wavelength energy sources, such as light-emitting diodes (LED). Advantageously, the technique does not require photoresist; is inexpensive, fast, and robust; primarily operates in the molecular, as opposed to the physical, domain; can accommodate various mask materials, chemistries, and substrates; is not limited by mass transfer of a self-assembled monolayer; and is capable of patterning from the macro- to the nanoscale (in progress).
To demonstrate the technique, we photocatalytically patterned silane using porphyrins and then covalently grafted a nonfouling background to the remaining silane. The patterned substrates were analyzed by atomic force microscopy (AFM) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). Additionally, the patterned surfaces were exposed to protein and cells to confirm the robustness of the patterning technique and the nonfouling background while illustrating the technique’s applicability to life science applications.
Although this publication concentrates on micrometer-scale results, we note that the wavelength of light used to activate the photosensitizer does not determine or limit feature resolution as it does in photolithography. As will be described in more detail in future publications, we therefore can pattern at the nanoscale using inexpensive and off-the-shelf broadband, low-energy light sources.
2. Experimental Section
2.1. Master and PDMS Mask Fabrication
Silicon masters were fabricated by standard photoresist photolithography methods, according to the manufacturer’s data sheets. Briefly, a Si wafer was dehydrated in an oven at 200 °C for 30 min and exposed to hexamethyldisilazane (HMDS; Clariant, Somerville, NJ), which served as an adhesion promoter. Next, AZ1518 photoresist (Hoechst Celanese, Somerville, NJ) was spin-coated onto the wafer to a thickness of 1.8 μm. The wafer was baked on a hotplate at 90 °C for 1 min and exposed to collimated UV light through a chrome/glass mask (Advance Reproduction Corporation, North Andover, MA). The wafer was then developed in AZ 1:1 developer and dried under nitrogen. RIE etching was performed in an etcher (Surface Technology System, Newport, U.K.) for approximately 2 min, or eight cycles. The remaining photoresist was removed in acetone, and the etched wafers were cleaned in a Piranha etch comprised of concentrated H2SO4/30% H2O2 (5:1 vol/vol %) for 20 min. Note that extreme caution should be used when mixing and working with Piranha solutions. After etching, the wafers were rinsed thoroughly in ultrapure water (UPW; 18 MΩ cm) before being blown dry with nitrogen and transferred to Fluoroware wafer holders. All chemicals were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise noted.
Next, the silicon masters were exposed to oxygen plasma (Plasma Prep II, SPI, West Chester, PA) at 50 mA, 300 mTorr vacuum. Heptadecafluoro-1,1,2,2-tetrahydrodecyl-1-trichlorosilane (United Chemicals, Bristol, PA) was prepared in anhydrous toluene (0.05 vol %), within a glove box purged with nitrogen. Immediately after preparation, the silane mixture was transferred to a laminar flow hood, and the substrates were immersed in the mixture for 1 min, which was followed by three toluene rinses of 1 min each. After exposure to the fluorosilane, the masters were baked in an oven for 5 min at 120 °C to accelerate covalent bond formation between the silane and the SiO2 surface. Fluorosilane coating renders the silicon masters hydrophobic and therefore assists in separating PDMS masks from silicon masters.
Poly(dimethylsiloxane) (PDMS) prepolymer (Sylgard 184, Dow Corning, Midland, MI) was prepared in a beaker by adding 1 part curing agent to 10 parts PDMS base. After mixing, the material was degassed until all bubbles were removed. The etched, silane-coated Si masters were placed in polystyrene dishes, and the degassed PDMS prepolymer was poured on top of the Si masters to a thickness of a few millimeters, after which the dishes were placed in a 60 °C oven for at least 1 h to cure the PDMS. After curing, the PDMS was peeled off of the Si masters and cut with a razor blade to appropriate dimensions for use as PDMS masks, selectively patterned as the negatives of the silicon masters (i.e., etched silicon wells became PDMS steps). The PDMS photomasks were sonicated in ethanol for 60 min and then left in fresh ethanol overnight to remove any unreacted monomer. The following day, the cured and sonicated masks were blown with nitrogen and allowed to outgas for another day prior to use.
2.2. Substrate Silanization
Silicon (cut to approximately 1 cm2, <100> from Micralyne, Edmonton, Alberta, CA) and glass coverslip (number 2, VWR, West Chester, PA) substrates were placed in Fluoroware (Chaska, MN) baskets and sonicated first in UPW, then in 2-propanol, and finally in UPW. Each sonication lasted for 10 min. The substrates then were immersed in a Piranha etch bath comprised of concentrated H2SO4/30% H2O2 (5:1 vol/vol %) for 20 min, followed by thorough rinsing in UPW. Substrates were individually blown dry under a filtered nitrogen stream and exposed to oxygen plasma (SPI) at 50 mA, 300 mTorr vacuum. Allyltrichlorosilane (ATC; United Chemicals, Bristol, PA) was prepared in anhydrous toluene (1.25% by volume) in a glove box purged with nitrogen. Immediately after preparation, the silane mixture was transferred to a laminar flow hood, and the substrates were immersed for 5 min, followed by three toluene rinses of 1 min each. The substrates (still contained in Fluoroware) were placed in a 120 °C oven for 5 min to accelerate covalent bond formation between the silane and the SiO2 surface.
2.3. Photocatalytic Patterning
We have employed numerous porphyrin photosensitizers as photocatalyts and have examined their UV absorption characteristics (Table 1). Chlorophyllin copper sodium salt (ethanol, Sigma Aldrich, St. Louis, MO), hematoporphyrin IX dihydrochloride (methanol, Frontier Scientific, Logan, UT), and magnesium phthalocyanine (ethanol or acetone, Frontier Scientific) were added to their respective solvent at concentrations ranging from 1 to 4 mg/mL. A cotton swab dipped in solvated porphyrin was used to coat the previously described PDMS photomasks with the porphyrin photocatalyst. The porphyrin-coated PDMS masks were blown dry with nitrogen and then placed by hand on top of the ATC-coated (section 2.2) Si chips or glass coverslips.
Table 1.
Absorption Peaks for Photosensitizers as Determined by UV/Vis Spectroscopy
| Photosensitizer | solvent | primary Abs peak (nm) | secondary Abs peak (nm) |
|---|---|---|---|
| chlorophyllin copper sodium salt | ethanol | 411 | 633 |
| hematoporphyrin IX dihydrochloride | methanol | 417 | 559 |
| magnesium phthalocyanine | acetone | slowly increases from 350 nm (m = −7.82308 × 10−5) | |
| magnesium phthalocyanine | methanol | slowly decreases from 350 nm (m = 7.38426 × 10−5) | |
Controlled patterning and removal of the ATC was achieved by local oxidation via approximately 10 s activation of the photocatalyst on the topographically patterned PDMS masks with either 480 nm blue or 660 nm red LED light (LUMEX, Glenview, IL; or Superbright LEDs, St. Louis, MO), or with UV light (Greenspot UV Source, Inc., Lebanon, IN). In addition, a common flashlight (Restoration Hardware, San Francisco, CA) with intensity peaks at 455 and 550 nm was successfully used to pattern substrates on the order of seconds. Localized patterning and removal of the ATC occurred at locations in close contact to the excited porphyrin on the PDMS photomasks, that is, elevated areas of the masks that were selectively created from the Si masters. ATC areas positioned under recessed PDMS regions remained intact (Scheme 1). Surfaces then were sonicated in solvent for 1 min and blown dry with nitrogen. Control experiments exposed ATC-coated substrates to PDMS photomasks without porphyrin in the presence of excitation energy (light). Selective patterning was not observed. ATC-coated substrates were chosen for demonstration and derivitization purposes; however, virtually any type of thin chemical layer may be patterned this way. This includes saturated and unsaturated chemistries as well as layers bonded to substrates via silanes, thiols, electrostatic interactions, and so forth. Additionally, the photomasks are reusable and can be cleaned by washing or sonicating in ethanol or another appropriate solvent.
Scheme 1. Schematic of Photocatalytic Patterning Processa.
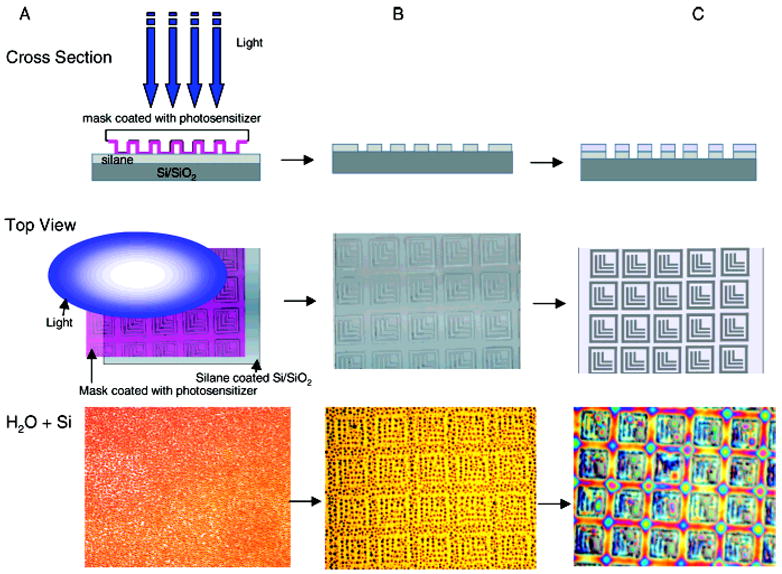
a Cross section (top), top down view (middle), and hydrated result after substrate exposure to water vapor (bottom): (A) patterning process performed through PDMS photomask coated with photosensitizer from volatile solvent onto silane-coated silicon substrate; (B) patterned silane substrate upon selective silane removal from regions subjected to chemical decomposition by reactive oxygen species from excited photosensitizer; and (C) polymer grafting of thin acrylamide hydrogel layer onto remaining silane. Ls remain as Si/SiO2 substrate.
2.4. Polymer Grafting
On the ATC-coated silicon or glass substrates, a nonfouling, interpenetrating network (IPN) chemistry of poly(acrylamide) and poly(ethylene glycol), P(AAm)-co-EG14, was covalently grafted to the SiO2 regions that retained the ATC layer post-patterning (Scheme 1). Briefly, acrylamide (AAm; Polysciences, Warrington, PA) was first photopolymerized onto the unsaturated allyl silane groups using N,N-methylene-bis-acrylamide (BIS) as a cross-linker, camphorquinone (Polysciences) as a surface-based photoinitiator, and acetone as a solvent. Polyethylene glycol methacrylate (PEG; Polysciences) then was introduced into the AAm layer by swelling the layer in methanol and highly cross-linking with BIS. IPN/silicon patterned substrates fabricated for use in protein- or cell-based experiments were exposed to aminopropylsilane (APS; United Chemical) in methanol (~2 vol %) to create adhesive regions on the freshly patterned (bare) regions of silicon or glass. Substrates then were stored in desiccators until use.
2.5. Optical Microscopy
Surface patterning was monitored at each step of the patterning process by exposing the patterned substrates to water vapor15 and acquiring images with a Nikon D100 camera mounted on a reflectance-based Nikon Labophot 2 microscope. A few images were acquired in quick succession after introduction of water vapor to view the differences in surface energy between the patterned and background substrate regions (Scheme 1). The same microscope/camera combination was used to image patterned eukaryotic HeLa cells (see below).
Detection of fluorescein isothiocyanate (FITC)-labeled neutravidin (Molecular Probes, Eugene, OR) on surfaces exposed to fluorescent proteins was performed using a Zeiss Axiovert 200M materials microscope equipped with darkfield, epifluorescence, a FITC filter set, and a Zeiss Axiocam HRM high-resolution digital camera. Images were captured using Zeiss Axiovision software.
2.6. Atomic Force Microscopy
Topographic features on patterned silicon substrates were imaged using a Digital Instruments Dimension 3100 atomic force microscope (Digital Instruments/Veeco Metrology Group, Inc., Santa Barbara, CA) with SiN (DNP-S) probes.
2.7. Time-of-Flight Secondary Ion Mass Spectometry (ToF-SIMS)
ToF-SIMS measurements were conducted on a PHI-TRIFT III instrument (Physical Electronics USA, Chanhassen, MN) equipped with a gallium liquid metal ion gun (Ga LMIG). The ion gun was operated at either 25 kV (unbunched mode) for high image resolution or 15 kV (bunched mode) for high mass resolution. Analyses were done utilizing Ga+ ions at room temperature. ToF-SIMS measurements were conducted over a 100 μm × 100 μm area for 10 min. The positive mass spectra were calibrated using common hydrocarbon fragment peaks at CH3+, C2H3+, and C4H7+, while the negative mass spectra were calibrated using CH−, OH−, C2H−. Spectra for background controls were acquired by analyzing clean silicon areas on the wafers.
2.8. Proteins and Cell Culture
FITC-neutravidin (Molecular Probes, Eugene, OR) was dissolved in phosphate buffered saline (PBS) at a working concentration of 50 μg/mL. Photocatalytically patterned samples exposed to FITC-neutravidin were stored in polystyrene dishes sealed with Parafilm and wrapped in aluminum foil to keep out light. Dishes were placed on a shaker table for 60 min. Substrates were subsequently rinsed three times in PBS, then rinsed in distilled water, and dried before imaging.
HeLa cells (Cambrex, East Rutherford, NJ) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies Inc., Gaithersburg, MD) that was supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin/fungizone (antibiotic, Life Technologies). Cultures were maintained in T25 flasks (Corning, Corning, NY) inside an incubator held at 37 °C and 5% CO2. Cells were removed from the flasks using trypsin (Life Technologies) and were resuspended in supplemented DMEM. Cells were counted with a hemocytometer, diluted if necessary, and plated onto substrates at approximate cell densities of 10 000–100 000 cells/mL.
3. Results
3.1. Surface Modification and Characterization
We demonstrate porphyrin-based photocatalytic lithography using silane patterning. Surfaces patterned according to this technique may serve as a foundation on which a nonfouling polymer network may be grafted to facilitate biomedical research. Use of unsaturated silanes as patterning media provides convenient synthetic routes to covalent modification, such as the free radical polymerization techniques described here. We have also grafted thermoreversible polymers (poly(N-isopropylacrylamide), NiPAAm) to patterned silane substrates, and we have covalently coupled bacteria to patterned aminosilane-coated substrate surfaces. Furthermore, we have photocatalytically patterned poly-(L-lysine)-g-poly(ethylene glycol) (PLL-g-PEG),16,17 thiols,18 and poly(ethylene glycol)-bl-poly(propylenesulfide)-bl-poly(ethylene glycol) (PPS-bl-PEG)19,20 (results in forthcoming publications). We believe that the ability to pattern variously functionalized silanes, thiols, and sulfides illustrates the robust and flexible patterning capacity of the method.
Atomic force microscopy provided topographic and deflection measurements (Figure 1).
Figure 1.

Contact mode AFM image of P(AAm-co-EG)/Si substrate patterned with a LLNL logo photomask. Allyltrichlorosilane (ATC)-coated substrate was patterned photocatalytically with MgPC and a blue LED. A hydrogel layer, P(AAm-co-EG), was then photo-polymerized onto the remaining silane. The substrate was sonicated in water and then blown dry with nitrogen before imaging. The height of the hydrogel is on the order of 20 nm. The line width of the Ls is 4 μm.
Comparing plane data from a region of the unpatterned matrix with the patterned “L” features in the substrate of Figure 1 indicated an IPN thickness of 17 nm, consistent with previous ellipsometric results.14 Figure 1 illustrates the homogeneity of the coating and the patterned elements.
Time-of-flight secondary ion mass spectrometry (Figure 2) confirmed chemical patterning via selected m/z fragment analysis. Note that while PDMS did present surface contamination on a few samples examined by ToF-SIMS, it is a useful material for our process, as it is transparent to radiation at the wavelengths employed, can act as a lens material,21–23 is reusable, and contains large amounts of oxygen to facilitate radical formation. Other potentially suitable mask materials include a polyolefin elastomer from Dow, POP,24 polyimide, and polyethylene.
Figure 2.

ToF-SIMS conveys chemical proof of patterning. Excerpts of ToF-SIMS positive ion imaging data for AAm/Si patterned substrate (top) and P(AAm-co-EG)/aminopropylsilane (IPN/APS, middle). Images include total ion as well as m/z ratios indicative of O, Si, CH, and CN. The bottom spectrum shows high-resolution data from positive ion imaging of P(AAm-co-EG). Counts vs mass/charge (m/z) for the window of 28 to 45 conveys peaks to confirm the presence of Si, hydrocarbons, and PEG fragments. Scale bars are 5 μm.
ToF-SIMS of the patterned ATC layer indicated more hydrocarbon fragments on the retained ATC matrix than on the regions where ATC had been photocatalytically ablated away. Due to the small size of the grafted ATC molecule (SiC3H5), contrast with the silicon substrate was not as high as on acrylamide or IPN grafted layers, so results are not shown. ToF-SIMS indicated that the IPN is characterized by intense hydrocarbon fragments at m/z 13, 15, and 27 (CH, CH3, and C2H3), acrylamide related fragments at 26 and 42 (CN and CNO), and PEG related fragments at 41, 43, and 45 (C3H5, C2H3O, and C2H5O). Peaks due to contamination from sodium (m/z = 23), calcium (39), potassium (40), or PDMS (73) were present on some samples. The oxidatively patterned silicon regions were characterized by intense silicon and oxygen containing fragments, including Si, SiH+, CH3Si+, and SiO2 (m/z = 28, 29, 43, and 60). Figure 2 includes a selection of positive ion imaging peaks for the AAm/Si layer and then the IPN/APS layers. Contrast is similar in most of the images. However, the total ion image on the IPN/APS substrate shows less contrast, as does the CN peak at 27. While it is unclear why the contrast in the total ion image decreases, we believe that the CN imaging presents less contrast in the IPN/APS sample than in the AAm/Si sample because of the nitrogen present on the spots after backfilling with the amino containing silane. The bottom of Figure 2 shows the high-resolution spectral analysis window from m/z 28 to 45 to convey the Si, hydrocarbon, and PEG fragments.
3.2. Biomolecular Adsorption
As shown in Figure 3, the fluorescently tagged protein FITC-neutravidin provided the first biological test for photocatalytically patterned silicon. A large area (over 1 cm2 in total area) was patterned successfully with resolution on the micrometer-scale in just a few seconds. Protein selectively adsorbed onto the amino-terminated APS-coated Si where ATC had been removed, and protein was repelled from matrix regions where the IPN had been built up. This process worked equally well for both silicon and glass substrates.
Figure 3.

A photomask bearing LLNL logos was used to locally oxidize, and therefore pattern, ATC on silicon. A nonfouling polymer layer (IPN) was then synthesized on the patterned ATC on silicon. After back-filling bare silicon regions with aminopropylsilane (APS), the substrate was incubated with a solution of fluorescein-labeled Neutravidin. The fluorescence micrograph shows that protein selectively adsorbs to APS regions and is repelled by the nonfouling polymer (IPN) regions (20× magnification, line width = 4 μm).
Figure 4 shows the result of a HeLa cell plating experiment. Adhesion of the single cells or cell clusters was limited to regions of the adhesive chemistry, as confirmed through optical microscopy. Although the goal of these experiments did not include cell culture longevity, we note that the IPN chemistry (photolithographically patterned) has been proven to maintain cell patterns for up to 60 days.2 We have carried out cell transfection experiments on photocatalytically patterned substrates with the IPN chemistry and demonstrated cell viability and successful transfection over a period of at least 5 days.25 Depending on the goals and targeted substrate materials for experiments, PLL-g-PEG16 or PPS-bl-PEG19 can also be photocatalytically patterned to provide similar protein resistance for biotechnological use.
Figure 4.

Individual and cell cluster patterning results. Images show HeLa cells plated on IPN/APS photocatalytically patterned substrates: (A) 30 μm circles and (B) letters with line width of 200 μm.
4. Discussion
This publication describes initial results of a novel, porphyrin-based technique for patterning surface chemistry, which has applications in a broad variety of settings. Our initial work demonstrates the technique’s usefulness in life science applications. Porphyrins, comprised of four pyrrole residues that are linked by four methine bridging groups formulating an aromatic macrocyclic ring, absorb light energy to carry out chemical reactions.26 They are nature’s most prominent catalysts and carry out a spectrum of bioenergetic reactions, ranging from the photosynthetic energy transduction that converts absorbed light in green chlorophyll pigment to usable energy, to the biochemical transductions responsible for oxygen storage and transport throughout the body in hemoglobin and myoglobin, to the conversion of carbon dioxide into hydrocarbons. Porphyrins (and, more generally, photosensitizers) have a long history of use in the field of photodynamic therapy (PDT), treatments that use light to induce beneficial reactions within patients. For a review, see Moan.27,28
We employ and excite porphyrins to create radical species that photocatalytically oxidize, and thereby pattern, chemistries in the local vicinity by ablation. Photosensitizers have a stable electronic configuration: a singlet state in their ground state energy level. Absorption of a photon of light of specific wavelength results in a porphyrin molecule being promoted to a very short-lived excited singlet state. Wavelengths longer than the specific wavelength do not result in excitation; wavelengths shorter than the specific wavelength typically result in a molecule being promoted to an excited state but do not fundamentally alter this step function-type event. The porphyrin molecule may also convert to the triplet state via intersystem crossing which involves a spin change of an electron. Although the triplet state photosensitizer has lower energy than the singlet state, it has a longer lifetime (typically >500 ns), and this increases the probability of energy transfer to other molecules such as oxygen, resulting in production of radical species. Advantageously, photosensitizer excitation does not necessarily destroy the photosensitizer, which may return to its ground state. Thus, the photosensitizer may repeat radical-inducing energy transfer numerous times, which means that multiple patterned surfaces may be made from a single porphyrin mask. The photosensitizer may also return to the ground state by emitting fluorescence or by dissipating excess energy as heat after an internal energy conversion and loss.
There are two mechanisms by which the triplet state photosensitizer can react with molecules; these are known as the Type I and Type II reactions. Type I involves electron/hydrogen transfer directly from the photosensitizer to produce ions, or electron/hydrogen abstraction from a substrate molecule to form free radicals. These radicals then react rapidly, usually with oxygen, resulting in the production of highly reactive oxygen species (ROS, e.g., the superoxide and the peroxide anions).
Type II reactions produce the electronically excited and highly reactive state of oxygen known as singlet oxygen. Direct interaction of the excited triplet state photosensitizer with molecular oxygen results in the photosensitizer returning to its singlet ground state and the formation of singlet oxygen. During photocatalytic patterning with photosensitizers, there is probably a contribution from both Type I and II processes, suggesting that the mechanism of ablation is dependent on oxygen tension and photosensitizer concentration.
Radical oxygen species diffusion is expected to limit photocatalytic patterning resolution. It is estimated that the distance diffused by singlet oxygen is on the order of 10–20 nm, corresponding to a lifetime of 10–40 ns,29 while the distance diffused by hydroxyl radicals in C–H environments is about 4 nm.30 When exposed to a polymer substrate, the reactive species likely react and oxidize the polymer via a shortest mean free path within a few nanometers of their origin. As such, we believe that species migration (in terms of ROS and mask placement) will be sufficiently constrained to allow feature generation on the order of 50 nm.
The previously discussed articles from Tatsuma et al.11 and Kubo et al.12 report excitation times of 20 min to affect metallic oxide photo-oxidation on substrates. Here, we demonstrate localized oxidation via Type I and Type II photosensitizer reactions on the order of seconds. Electron–hole pairs are generated in photocatalysts, such as TiO2, upon irradiation from UV light, which excites TiO2 above its band gap energy. Electron-hole pairs must migrate out of the particles within which they are generated, and they must reach the external environment to contact oxygen and generate the ROS. Electron–hole pair diffusion constants depend on the medium in which they are generated as well as on incident energy intensity. However, one may generalize and say that electron-hole pairs generated in TiO2 may migrate ~75 nm from their source in a free field region.29 Unlike semiconductors such as TiO2, excited porphyrins generate ROS directly, thus eliminating concerns about electron-hole pair diffusion.
Our process has similarities to the TiO2-based lithography reported first by Tatsuma et al.11 in that reactive oxygen species are created upon light excitation of the catalyst and these reactive oxygen species can decompose underlying chemistries. Tatsuma reported the placement of a polyimide spacer between a quartz mask coated with anatase TiO2 and an oxidizable surface (silane on Si); light shown through a photomask on top of the quartz plate was used to pattern surface substrate chemistry. A Hg–Xe lamp of 100 mW cm−2 was employed for at least 8 min before patterning was noted. Lower light intensities (10 mW cm−2) required on the order of 30 min to pattern surfaces. The resolution of the technique is dependent on the height of the spacer, which allegedly allows ROS to diffuse; small gaps of approximately 10 μm result in resolution on the order of 5 μm.12 Lee and Sung13 reported success in patterning silanes down to 500 nm without a spacer, using TiO2 as a photocatalyst and a 450 W Xe lamp for 2 min.
In contrast, we can pattern in just a few seconds using photosensitizer-coated PDMS photomasks and significantly longer wavelength light sources with power densities less than 1 mW cm−2. While lower wavelengths, such as UV, still effectively stimulate the photosensitizers, it is not clear that higher energy sources decrease patterning time. In fact, UV–visible spectrometry indicates that PDMS photomasks show absorption in the UV, and this may increase time necessary to pattern.
Ideally, the photosensitizer film comes into contact with the layer to be patterned to reduce the probability of excited molecule diffusion. However, one must pay close attention to potential contamination issues related to the mask materials. This is why we point out in the Results section that PDMS can result in contamination according to ToF-SIMS results. We are presently working with other mask materials to minimize such complication.
In comparison to photolithographic patterning techniques, which are relatively laborious and require resist deposited across the entire substrate surface, photocatalytic patterning may be accomplished on base chemical layers applied to pristine, well-controlled, pinhole-free surfaces. The process shares some similarities with microcontact printing in that mask materials are brought into close proximity/contact with substrates. However, our process does not rely on mass transfer of the self-assembling monolayer to the surface. For a review on pattern stability via photolithographic, microcontact printing, and SMAP methodologies employing PLL-g-PEG chemistry under cell culture conditions, see Lussi et al.31
5. Conclusions
We have presented an inexpensive, rapid, straightforward, and versatile method of photocatalytically patterning surface chemistry using porphyrins to generate ablative oxygen radicals. This method is flexible with respect to substrate and chemistry. We selectively grafted a nonfouling polymer to remaining unsaturated matrix chemistry. Substrates were analyzed by AFM and ToF-SIMS and tested with fluorescent proteins and cells. Protein adsorption and cell adhesion was restricted to patterned regions, which had been modified with an adhesive chemistry. We intend to exploit and expand this capability to the nanoscale and demonstrate nanometer patterning resolution with large aspect ratio features.
Acknowledgments
This work was partially performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract W-7405-Eng-48 and Contract DE-AC52-07NA27344. We gratefully acknowledge funding from NIH R21 EB003991, NIH R01 EB000462, and 03-ERD-068. We would also like to thank John Reynolds, Chris Orme, Barry Cheung, and Chance Carter for their assistance and insights.
References
- 1.Folch A, Toner M. Annu Rev Biomed Eng. 2000;2:227. doi: 10.1146/annurev.bioeng.2.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Thomas CH, Lhoest JB, Castner DG, McFarland CD, Healy KE. J Biomech Eng. 1999;121(1):40–48. doi: 10.1115/1.2798041. [DOI] [PubMed] [Google Scholar]
- 3.Thomas CH, Collier JH, Sfeir CS, Healy KE. Proc Natl Acad Sci USA. 2002;99(4):1972–1977. doi: 10.1073/pnas.032668799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falconnet D, Koenig A, Assi T, Textor M. Adv Funct Mater. 2004;14(8):749–756. [Google Scholar]
- 5.Kane RS, Takayama S, Ostuni E, Ingber DE, Whitesides GM. Biomaterials. 1999;20(23–24):2363–2376. doi: 10.1016/s0142-9612(99)00165-9. [DOI] [PubMed] [Google Scholar]
- 6.Jeon NL, Choi IS, Whitesides GM, Kim NY, Laibinis PE, Harada Y, Finnie KR, Girolami GS, Nuzzo RG. Appl Phys Lett. 1999;75(26):4201–4203. [Google Scholar]
- 7.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Biotechnol Prog. 1998;14(3):356–363. doi: 10.1021/bp980031m. [DOI] [PubMed] [Google Scholar]
- 8.Tourovskaia A, Barber T, Wickes BT, Hirdes D, Grin B, Castner DG, Healy KE, Folch A. Langmuir. 2003;19(11):4754–4764. [Google Scholar]
- 9.Lussi JW, Michel R, Reviakine I, Falconnet D, Goessl A, Csucs G, Hubbell JA, Textor M. Prog Surf Sci. 2004;76(3–5):55–69. [Google Scholar]
- 10.Michel R, Lussi JW, Csucs G, Reviakine I, Danuser G, Ketterer B, Hubbell JA, Textor M, Spencer ND. Langmuir. 2002;18(8):3281–3287. [Google Scholar]
- 11.Tatsuma T, Kubo W, Fujishima A. Langmuir. 2002;18(25):9632–9634. [Google Scholar]
- 12.Kubo W, Tatsuma T, Fujishima A, Kobayashi H. J Phys Chem B. 2004;108(9):3005–3009. [Google Scholar]
- 13.Lee JP, Sung MM. J Am Chem Soc. 2004;126(1):28–29. doi: 10.1021/ja038769+. [DOI] [PubMed] [Google Scholar]
- 14.Bearinger JP, Castner DG, Golledge SL, Rezania A, Hubchak S, Healy KE. Langmuir. 1997;13(19):5175–5183. [Google Scholar]
- 15.Lopez GP, Biebuyck HA, Frisbie CD, Whitesides GM. Science. 1993;260(5108):647–649. doi: 10.1126/science.8480175. [DOI] [PubMed] [Google Scholar]
- 16.Elbert DL, Hubbell JA. Chem Biol. 1998;5(3):177–183. doi: 10.1016/s1074-5521(98)90062-x. [DOI] [PubMed] [Google Scholar]
- 17.Sawhney AS, Hubbell JA. Biomaterials. 1992;13(12):863–870. doi: 10.1016/0142-9612(92)90180-v. [DOI] [PubMed] [Google Scholar]
- 18.Bearinger JP, Hiddessen AL, Wu KJJ, Christian AT, Dugan LC, Stone G, Camarero J, Hinz AK, Hubbell JA. In: Laudon M, Romanowicz B, editors. Biomolecular patterning via photocatalytic lithography; NSTI Nanotech 2005; Anaheim, CA. May 8–12, 2005; Cambridge, MA: Nano Science and Technology Institute Publishing Office; 2005. [Google Scholar]
- 19.Bearinger JP, Terrettaz S, Michel R, Tirelli N, Vogel H, Textor M, Hubbell JA. Nat Mater. 2003;2(4):259–264. doi: 10.1038/nmat851. [DOI] [PubMed] [Google Scholar]
- 20.Napoli A, Tirelli N, Kilcher G, Hubbell JA. Macromolecules. 2001;34(26):8913–8917. [Google Scholar]
- 21.Camou S, Fujita H, Fujii T. Lab Chip. 2003;3(1):40–45. doi: 10.1039/b211280a. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Wang WS, Fang J, Varahramyan K. J Micromech Microeng. 2004;14(5):675–680. [Google Scholar]
- 23.Agarwall M, Gunasekaran RA, Coane P, Varahramyan K. J Micromech Microeng. 2004;14(12):1665–1673. [Google Scholar]
- 24.Csucs G, Kunzler T, Feldman K, Robin F, Spencer ND. Langmuir. 2003;19(15):6104–6109. [Google Scholar]
- 25.Hiddessen AL, Bearinger JP, Pallavicini MG, Colvin ME. Biophys J. 2005;88(1):523A–523A. [Google Scholar]
- 26.Dolphin D. Porphyrins Physical Chemistry Part A. Academic Press; New York: 1978. [Google Scholar]
- 27.Moan J, Peng Q. Anticancer Res. 2003;23(5A):3591–3600. [PubMed] [Google Scholar]
- 28.Moan J. Photochem Photobiol. 1986;43(6):681–688. doi: 10.1111/j.1751-1097.1986.tb05647.x. [DOI] [PubMed] [Google Scholar]
- 29.Moan J, Berg K. Photochem Photobiol. 1991;53(4):549–553. doi: 10.1111/j.1751-1097.1991.tb03669.x. [DOI] [PubMed] [Google Scholar]
- 30.Nikjoo HONP, Goodhead DT, Terrissol M. Int J Radiat Biol. 1997;71:467–483. doi: 10.1080/095530097143798. [DOI] [PubMed] [Google Scholar]
- 31.Lussi JW, Falconnet D, Hubbell JA, Textor M, Csucs G. Biomaterials. 2006;27(12):2534–2541. doi: 10.1016/j.biomaterials.2005.11.027. [DOI] [PubMed] [Google Scholar]