

Abstract
We develop a model-based phylogenetic maximum likelihood test for evidence of preferential substitution toward a given residue at individual positions of a protein alignment—directional evolution of protein sequences (DEPS). DEPS can identify both the target residue and sites evolving toward it, help detect selective sweeps and frequency-dependent selection—scenarios that confound most existing tests for selection, and achieve good power and accuracy on simulated data. We applied DEPS to alignments representing different genomic regions of influenza A virus (IAV), sampled from avian hosts (H5N1 serotype) and human hosts (H3N2 serotype), and identified multiple directionally evolving sites in 5/8 genomic segments of H5N1 and H3N2 IAV. We propose a simple descriptive classification of directionally evolving sites into 5 groups based on the temporal distribution of residue frequencies and document known functional correlates, such as immune escape or host adaptation.
Keywords: directional selection, evolution of influenza, maximum likelihood, episodic selection
Introduction
The fundamental role of natural selection in molecular evolution cannot be overstated (for a recent review, see Sabeti et al. [2006]). Comparative molecular studies of the recent 2 decades have devoted a considerable effort to deciphering the nature and quantitative properties of selective forces that have shaped existing sequence diversity. As a result, a multitude of selection analysis methods have been proposed (e.g., see Nei [2005] for a review). Simple and intuitively appealing counting procedures on pairs of aligned sequences, exemplified by the work of Miyata and Yasunaga (1980) and Nei and Gojobori (1986), gave rise to the now ubiquitous dN/dS (Ka/Ks) ratio of nonsynonymous to synonymous substitution rates, which could be compared with the neutral expectation of dN = dS. A conceptually similar test of selection based on within-species polymorphism data combined with between-species fixation data was proposed by McDonald and Kreitman (1991). Statistical shortcomings of counting-based techniques (Muse 1996) were addressed by model-based maximum likelihood estimation techniques (Goldman and Yang 1994; Muse and Gaut 1994), and lack of power to detect selection affecting only a small proportion of sites in a gene was elegantly remedied by the introduction of random effects methods that could estimate site-specific substitution rates efficiently (Nielsen and Yang 1998).
Because these models are based on inferring and comparing site (or branch)-specific substitution rates, they generally require significant sequence divergence to gain power (Anisimova et al. 2002) and could suffer a serious loss of accuracy on extremely sparse data (Suzuki and Nei 2004). This is a potentially serious shortcoming because rapid bursts of positive selection could be localized to substitutions along a single branch in a phylogenetic tree, whereas the remainder of the tree evolves neutrally or even conservatively. Such selective modes, including the important case of selective sweeps, are considerably more difficult to discern. A further improvement to partially address this issue was realized with the introduction of models that permitted substitution rates to vary across lineages (Yang 1998; Guindon et al. 2004; Kosakovsky Pond and Frost 2005b) and the so-called branch-site methods which combined both types of variation (Yang and Nielsen 2002).
A nearly universal assumption for applied codon-based techniques posits that the rate of nonsynonymous substitution is independent of the source and “target” residues—an “equal rates” model. Therefore, positive selection is inferred if an unusually high rate of nonsynonymous replacement (weighted over all residue pairs) relative to the baseline synonymous rate, that is, dN > dS, is inferred for a site, a branch, or both. This assumption stands in stark contrast to the decades-old observation that certain amino acid substitutions are preferred to others—the foundation of the entire class of empirical protein evolution models used for alignment, similarity searching, and comparative sequence analysism, for example, Dayhoff et al. (1972), Jones et al. (1992), Whelan and Goldman (2001), and Yampolsky and Stoltzfus (2005). Despite a number of attempts to incorporate the dependence of nonsynonymous substitution rates on the source and target residues, including several recent innovative papers (Siepel and Haussler 2004; Conant et al. 2007; Doron-Faigenboim and Pupko 2007; Kosiol et al. 2007), such models have not seen wide adoption. Because the appropriate choice of rate residue dependence remains largely unresolved, most existing techniques are best suited to detecting the existence of a specific kind of selection—diversifying (disruptive) or purifying selection, where the actual residues involved are of secondary importance. Directional or frequency-dependent modes of selection may be considerably more difficult to identify due to their more episodic nature. The question of which residues are being selected for or against is rarely raised; and when it is, the answers tend to rely on ad hoc counting techniques that correlate substitutions inferred along a phylogenetic tree with physical and chemical properties of the involved residues (cf. Xia and Li [1998]; McClellan and McCracken [2001]; Ray et al. [2005]). These methods are intuitively appealing but lack rigorous goodness-of-fit measures and do not engage a formal statistical framework for hypothesis testing or model selection.
We propose a straightforward and intuitive method that tests for evidence of accelerated substitutions toward one or more residues, whose identity is inferred in the process, at a subset of sites in a protein alignment in the standard phylogenetic maximum likelihood context. Our directional evolution of protein sequences (DEPS) test builds upon 3 previously published reports. First, the idea of accelerating substitutions toward some characters while retarding substitutions away from these characters has been successfully exploited in the context of modeling evolution of RNA secondary structure (Muse 1995). Second, a method for applying nonreversible models to the detection of evolution toward a fixed residue in the otherwise equal rates setting for pairs of sequences for the analysis of the evolution of drug resistance in the human immunodeficiency virus (HIV) was previously developed by Seoighe et al. (2007). Third, the procedure for efficient maximum likelihood estimation of an organism-specific protein evolution model from multiple genes, which we use for the derivation of the null model, has been previously outlined for HIV Nickle et al. (2007).
We first evaluate the performance of the DEPS test on simulated data and find it to have very low error rates and power of up to 86% in detecting strong effects with alignments of sufficient size and sequence divergence. Next, we apply the DEPS test to 8 genomic fragments of influenza A virus (IAV). IAV is an ideal model organism for our test, first because of a wealth of sequences sampled at different time points, allowing us to detect transient phenomena such as selective sweeps, and second because the hemagglutinin (HA) gene in IAV is a canonical example of directional evolution via antigenic drift (e.g., Bush et al. [1999]). We investigate serotype H3N2 viruses (“Hong-Kong” flu) sampled from human hosts and serotype H5N1 viruses (“avian” flu) sampled from avian hosts.
Methods
Baseline Protein Model
We begin by selecting an empirical model of protein substitution to describe the standard “equilibrium” evolution of the organism under investigation. This continuous time reversible Markov model is defined by a 20 × 20 rate matrix Q, whose (i, j) element corresponds to the instantaneous rate of substituting residue i with residue j (we order the amino acids by their one letter IUPAC code: A, C, D, etc.). We use the standard rate frequency parameterization of the rate matrix: Qij = r(i, j)πj, i ≠ j, where r(i, j) = r(j, i) > 0 is a constant rate multiplier term (used to account for the relative prevalence of substitutions involving residues i and j) and πj(∑jπj = 1) is the stationary frequency of residue j. The r(i, j) are estimated from a curated alignment of 8 genomic regions of IAV protein sequence alignments (serotype H5N1) following the maximum likelihood procedure described previously for models of HIV protein evolution (Nickle et al. 2007). We assume that models derived from a large sample of H5N1 sequences are adequate to describe the baseline evolution of other IAV serotypes. The diagonal entries are defined by Qjj = − ∑ i≠j qji—a standard constraint to ensure that Q is the infinitesimal generator of a Markov process. For identifiability reasons, the r(i, j) are scaled uniformly so that the expected number of substitutions per site per unit time is equal to 1, that is, − ∑jQjjπj = 1. A graphical representation of the H5N1 rate matrix is shown in figure 1; the model is implemented in the HyPhy software package (Kosakovsky Pond et al. 2005). To obtain the probability of substituting residue i with residue j in time t ≥ 0, we compute the transition matrix T(t) = exp(Qt) using a scaled Taylor series approximation of the matrix exponential function (Moler and Van Loan 1978) and look up its (i, j)-th element.
FIG. 1.—

Substitution rates for the empirical H5N1 protein substitution model. Empirical protein matrix was derived from genomic H5N1 influenza sequences. The matrix is symmetric, and the rates are scaled to yield 1 expected substitution per unit time, using empirical H5N1 base frequencies. Shading is proportional to the relative substitution rate; 0 = white (0 rate), 1 = black (for the maximal rate in the entire matrix). Residues are grouped into 4 similarity classes based on the Stanfel (1996) clustering; substitutions within the 4 diagonal blocks are conservative according to this classification. Substitutions that can be realized with a single-nucleotide replacement are marked with circles.
This substitution model is fitted to an alignment of protein sequences using the standard maximum likelihood phylogenetic framework to estimate branch lengths (Felsenstein 1981). A 4—bin adaptively discretized beta-gamma distribution is used to account for site-to-site variation in substitution rates (Kosakovsky Pond and Frost 2005a).
Directional Protein Model
Let k = 1…20 be the index of an a priori selected residue that we hypothesize is being selected for at some sites. We encode this feature of the evolutionary process by modifying the baseline rate matrix as follows: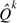 ikk = B × r(i,k)πk, kik = r(i,k) π i/B. B (bias factor) is a random variable that can take 2 values: B = 1 with probability q and B = b ≥ 1 with probability 1 − q. Intuitively, this suggests that a proportion of sites are evolving under the baseline model, whereas the complementary proportion of sites are evolving under a process that accelerates substitutions toward residue k with relative rate b. Alignment-wide parameters b and q are estimated from the data by maximum likelihood in addition to all the parameters of the baseline model, using the standard framework of random effects phylogenetic likelihood models (e.g., Kosakovsky Pond and Frost [2005c]). Our model builds upon a previously published codon-based model used to study the evolution of drug resistance in HIV (Seoighe et al. 2007), where a less realistic null (all amino acid substitutions are occurring at the same rate) and an a priori target residue were postulated.
ikk = B × r(i,k)πk, kik = r(i,k) π i/B. B (bias factor) is a random variable that can take 2 values: B = 1 with probability q and B = b ≥ 1 with probability 1 − q. Intuitively, this suggests that a proportion of sites are evolving under the baseline model, whereas the complementary proportion of sites are evolving under a process that accelerates substitutions toward residue k with relative rate b. Alignment-wide parameters b and q are estimated from the data by maximum likelihood in addition to all the parameters of the baseline model, using the standard framework of random effects phylogenetic likelihood models (e.g., Kosakovsky Pond and Frost [2005c]). Our model builds upon a previously published codon-based model used to study the evolution of drug resistance in HIV (Seoighe et al. 2007), where a less realistic null (all amino acid substitutions are occurring at the same rate) and an a priori target residue were postulated.
The substitution process generated by the rate matrix k is no longer time reversible, and the frequency of residue k will increase over time. The modified process converges to its own equilibrium distribution 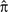 as the evolutionary time is increased to infinity. For finite time t0, we compute δk(t0) = π[exp(k(t0)) − I]—the difference vector between the stationary distribution (assumed at the root of the tree) and the expected distribution of residues after time t0. The kth element of δk(t0), δkk reflects the expected change in the frequency of the target residue and will be positive for b > 0.
as the evolutionary time is increased to infinity. For finite time t0, we compute δk(t0) = π[exp(k(t0)) − I]—the difference vector between the stationary distribution (assumed at the root of the tree) and the expected distribution of residues after time t0. The kth element of δk(t0), δkk reflects the expected change in the frequency of the target residue and will be positive for b > 0.
Because of time irreversibility, the rooting of the phylogenetic tree becomes important; this is to be expected because the evolutionary process now has the component of directionality. An outgroup sequence can be used to establish the appropriate placement of the root on the phylogenetic tree. For computational expediency, we assume that the branch lengths of the directional protein model are proportional to those estimated under the baseline protein model, with the constant of proportionality inferred as a model parameter, except for the 2 immediate descendants of the root node (only the sum can be estimated using a time reversible model), which are estimated by maximum likelihood. It should be noted that the exact computation of branch lengths under the alternative model, measured in expected substitutions per site, is complicated by the fact that the evolutionary process is not in equilibrium. The expected number of substitutions along a branch is affected by the bias and mixing parameters of the alternative models in a nonlinear fashion, and this renders the standard analytical formulas inapplicable. A similar simplification has been validated in the context of codon evolutionary models (Yang 2000; Kosakovsky Pond and Frost 2005c).
Equilibrium Frequency Estimation
Residue frequencies are usually estimated by their observed proportions in the data; however, we found this approach to perform poorly for DEPS. For the null time reversible model, it may be difficult to estimate relatively rare residues, given that 20 frequencies must be tabulated from relatively small alignments. Inaccurate estimates of πj may in turn lead to a false positive signal for directional selection. For example, if πR is underestimated by a factor of 2 by the observed proportion, then the directional model may “restore” the correct frequency by adjusting the bias factor to 2 (because all substitution rates to R have the form πR × B), falsely suggesting that substitutions toward R are accelerated. We estimate 19 frequency parameters by maximum likelihood; on simulated data, this approach results in lower rates of false positives and better power (results not shown), at a trivial computational expense. For the alternative model, stationary frequencies of the mixture process are a function of point estimates for the null model, the bias parameter b and the mixing parameter q.
Testing for Directional Selection
We adopt a 2-step procedure to evaluate evidence for directional evolution toward a given residue at a given protein site, identical in concept to the popular procedure for detecting sites under positive selection (Yang et al. 2000). First, we apply a likelihood ratio test (LRT) to determine whether the directional model for residue k fits the data significantly better than the baseline model. Because the models are nested (e.g., b = 1 reduces the directional model to the baseline model), we can assess significance by using the asymptotic χ32 statistic (the third parameter is the additional branch length estimated under the nonreversible alternative model). This statistic is conservative because not all model parameters are identifiable for all permissible values (e.g., q is not identifiable when b = 1; when q = 1, b is not identifiable), and the appropriate test statistic is a mixture of χ20, χ21, χ22, and χ32 (Self and Liang 1987; Swanson et al. 2003); however, the accurate determination of mixing coefficients is nontrivial in this case and we choose to err on the side of caution. If the P value yielded by the LRT is no greater than a preset value P0 (see below), then an empirical Bayes analysis (EBA) is carried out to decide which sites may be under directional selection. For each site, we evaluate the posterior probability qp = Pr(B = b|data) and compute the empirical Bayes factor (EBF) (Kass and Raftery 1995)
All sites with BF(k) > 100 are reported as evolving preferentially toward residue k.
Lastly, we perform 20 tests with each residue selected to be the target residue. Because the test statistics for individual hypotheses are not necessarily independent, we adopt a conservative multiple testing procedure due to Holm (1979), which bounds the probability of family-wise error (i.e., at least one falsely rejected null hypothesis) at a given significance level α. The Holm procedure requires a list of P values from each individual hypothesis sorted in increasing order, P(1) ≤ P(2)… ≤ P(20). The first hypothesis is rejected P(1) ≤ α/20, the second—if the first was rejected and P(2) ≤ α/19 and so on. We set the family-wise error threshold to α = 0.05.
Testing for Differences between 2 Samples
The directional protein model can be fitted individually to 2 different alignments (alternative model), for example, 2 samples of the same gene from different populations or 2 different genes (with potentially different baseline models), and also jointly, enforcing 1 of 3 types of constraints.
Equal bias: the bias parameter b is shared between 2 alignments; test for significance uses a 1 degree of freedom (df) (χ12) LRT.
Equal proportions: the proportion parameter q is shared between 2 alignments; a 1 df LRT.
Equal distributions: both the bias parameter b and the frequency parameter q are shared between 2 alignments; a 2 df LRT.
Simulations
We evaluated statistical performance of the DEPS test under using several simulation scenarios with 100 replicates each. In all cases, the dimensions of the alignment (566 amino acid sites and 110 sequences), phylogenetic tree (tree length of 0.84 expected substitutions/site/unit time), branch lengths, the distribution of site-to-site substitution rates, and stationary base frequencies were estimated from an alignment of influenza A (H5N1 serotype) HA sequences sampled from avian hosts.
Null Model
This scenario simulates undirected evolution under the null model of the DEPS test and is useful for evaluating the rate of false positives.
Directional Correct Model
Simulated data are generated under the alternative model of the DEPS test and can be used to gauge both the rates of false positives and power of the test. We examined 3 levels of divergence (low, medium, and high) measured by the expected number of amino acid substitutions per site along the tree (under the null model). The expected number of substitutions were 0.84 (the cumulative length of the H5N1 tree), 2.1 (2.5 × the length of the H5N1 tree), and 4.18 (5 × the length of the H5N1 tree), respectively.
Several categories of directionally evolving sites were introduced in each data replicate. The choice of target residues was largely arbitrary, with the sole requirement that the rates of substitution toward a target residue not be uniformly high or low (cf. fig. 1).
Because a reasonable expectation is that most sites in a biological data set will not be evolving toward any particular residue, 536 (94.7)% sites in each replicate were generated under the null model. Such sites are useful for measuring rates of false positives.
In all, 15 (2.6)% sites exhibiting somewhat accelerated evolution (B = 5) toward arginine; expected increase in arginine frequency δRR = 14.9%, 25.2%, 35.8% (for the 3 divergence levels).
In all, 10 (1.8%) sites were accelerated toward isoleucine (B = 10, δII = 26.0%, 38.1%, 50.3%).
In all, 5 (0.9%) sites were strongly (B = 100, δTT = 55.3%, 70.0%, 81.8%) accelerated toward threonine.
Directional Incorrect Model
We reanalyzed the data generated under the directional (H5N1 baseline) model using the JTT (Jones et al. 1992) matrix, derived primarily from mammalian protein sequences, to define the baseline model in order to investigate the performance of the DEPS test when the baseline protein substitution model is misspecified.
Influenza A Alignments
We collected alignments representing 8 genomic segments of serotypes H3N2 (human hosts), sampled between 1968 and 2005 and H5N1 (avian hosts) IAV sampled between 1996 and 2006. H3N2 sequence alignments were previously analyzed for diversifying and purifying selection (Suzuki 2006) and were kindly provided to us by Dr Suzuki. H5N1 sequences were downloaded from the National Center for Biotechnology Information (NCBI) Influenza virus resource (Bao et al. 2007), automatically aligned using the HyPhy package (Kosakovsky Pond et al. 2005) and adjusted by hand to exclude sequences tagged as nonfunctional or containing potential sequencing errors. Size and divergence levels for each alignment are shown in tables 1 and 2. Each alignment was screened for evidence of discordant phylogenetic signal using the single breakpoint likelihood method (Kosakovsky Pond et al. 2006); none of the alignments produced a significant recombination signal, consistent with the evidence that intrasegment homologous recombination in IAV is rare (Nelson and Holmes 2007). We reconstructed phylogenetic trees for each segment using Neighbor-Joining (Saitou and Nei 1987) coupled with the TN93 genetic distances (Tamura and Nei [1993]) computed on nucleotide sequences and rooted them on the oldest sequences in each sample: a sequence sampled from a goose in China in 1996 (GenBank accession numbers AF144300–AF144307) for H5N1 alignments and the most recent common ancestor of oldest sequences in H3N2 alignments. The convention of rooting IAV trees on the oldest sequence has seen wide use in literature (e.g., Bush et al. [1999]; Nelson et al. [2007, 2008]).
Table 1.
Summary of Selection Analyses on Genomic Segments of H5N1 Avian Influenza A Sequences
| Gene/Segment | Summary |
Directional Selection |
FEL Selection (no. of sites) |
|||||||
| Sequences | Sites | Tree L | Residue | P Value | Bias | Proportion (%) | No. of Sites | Positive | Negative | |
| PB2/1 | 286 | 759 | 0.68 | M | 0.0006 | 59.79 | 1.10 | 2 | 0 | 423 |
| PB1/2 | 286 | 757 | 0.57 | C | 0.0006 | 358.30 | 3.14 | 3 | 1 | 367 |
| S | 0.0028 | 29.62 | 1.27 | 1 | ||||||
| V | 0.0002 | 11.46 | 4.32 | 1 | ||||||
| PA/3 | 287 | 716 | 0.85 | F | 0.0000 | 4885.68 | 0.27 | 1 | 1 | 367 |
| Q | 0.0000 | 30.97 | 2.55 | 2 | ||||||
| HA/4 | 288 | 568 | 1.20 | E | 0.0000 | 71.21 | 0.97 | 1 | 9 | 184 |
| I | 0.0000 | 10.90 | 9.90 | 1 | ||||||
| K | 0.0001 | 22.37 | 2.02 | 3 | ||||||
| L | 0.0000 | 25.80 | 2.76 | 2 | ||||||
| P | 0.0000 | 11.09 | 5.07 | 2 | ||||||
| T | 0.0012 | 3.24 | 11.85 | 1 | ||||||
| NP/5 | 288 | 498 | 0.55 | None | 0 | 269 | ||||
| NA/6 | 287 | 469 | 1.20 | E | 0.0000 | 61.27 | 1.21 | 1 | 3 | 167 |
| I | 0.0002 | 14.77 | 2.92 | 1 | ||||||
| M | 0.0015 | 13.18 | 6.02 | 2 | ||||||
| MP/7 | 286 | 252 | 0.51 | None | 2 | 101 | ||||
| NS1/8 | 288 | 230 | 2.35 | None | 9 | 61 | ||||
NOTE.—PA, polymerase acidic; NP, nucleoprotein; MP, matrix protein; and NS1, nonstructural protein 1. Directional and traditional selection analyses of avian H5N1 influenza A genomic segments, referenced by their standard abbreviations. For each gene/segment, we report the number of sequences and sites in the alignment, total tree length measured in expected substitutions per site, and all residues which were directionally selected for according to DEPS along with the total number of codon sites detected as being subject to positive (diversifying) or negative selection by FEL at P = 0.05. For each residue detected by DEPS, we list the P value for the DEPS test, the maximum likelihood estimates for the bias term and proportion of directionally evolving sites in the directional protein model, and the number of sites detected by EBA (EBF of 100 or more) as being under directional selection.
Table 2.
Summary of Selection Analyses on Genomic Segments of H3N2 Human Influenza A Sequences
| Gene/Segment | Summary |
Directional Selection |
FEL Selection (no. of sites) |
|||||||
| Sequences | Sites | Tree L | Residue | P Value | Bias | Proportion (%) | No. of Sites | Positive | Negative | |
| PB2/1 | 259 | 759 | 0.27 | None | 0 | 197 | ||||
| PB1/2 | 256 | 757 | 0.25 | I | 0.0011 | 21.13 | 3.96 | 3 | 0 | 175 |
| PA/3 | 268 | 716 | 0.29 | None | 0 | 171 | ||||
| HA/4 | 284 | 566 | 0.77 | F | 0.0015 | 89.79 | 1.97 | 1 | 7 | 107 |
| I | 0.0011 | 36.55 | 3.00 | 3 | ||||||
| K | 0.0003 | 7.64 | 12.41 | 2 | ||||||
| T | 0.0026 | 117.97 | 2.08 | 3 | ||||||
| NP/5 | 246 | 498 | 0.38 | None | 2 | 128 | ||||
| NA/6 | 345 | 469 | 1.11 | F | 0.0023 | 1813.03 | 0.90 | 2 | 3 | 133 |
| K | 0.0025 | 21.70 | 5.67 | 2 | ||||||
| Q | 0.0020 | 14.32 | 5.44 | 2 | ||||||
| M1/7 | 173 | 252 | 0.26 | I | 0.0006 | 827.77 | 1.11 | 1 | 0 | 61 |
| M2/7 | 113 | 97 | 0.89 | None | 0 | 7 | ||||
| NS1/8 | 164 | 230 | 0.69 | I | 0.0018 | 201.44 | 1.03 | 1 | 0 | 17 |
NOTE.—Directional and traditional selection analyses of human H3N2 influenza A genomic segments, referenced by their standard abbreviations. See table 1 for an explanation of column headings.
Software Implementation and Performance
Biological and simulated sequence alignments used in this study, in NEXUS format (Maddison et al. 1997), are available from http://www.hyphy.org/pubs/DEPS. The DEPS test has been parallelized to take advantage of message passing interface distributed computing environments, distributing the fitting of alternative models with different target residues to separate cluster nodes, but the computational complexity of the test is sufficiently low to make it feasible for running on a single desktop. A complete analysis of the 110 sequence 566 site HA alignment, for example, took approximately 30 min on a dual quad-core Xeon 3 GHz Mac Pro (OS X 10.5, HyPhy version 1.00β using 6 threads). An implementation of DEPS has been included in the HyPhy distribution as a standard analysis: DirectionalREL.bf located under the positive selection rubrik.
Results
Simulation Results
Accuracy
For the data that conformed to the null (equilibrium) model of character evolution, the application of DEPS resulted in low rates of false positives. For the low level of divergence, there were 0/100 replicates with evidence of directional evolution toward any residue; 1/100 replicates was falsely identified as having sites evolving directionally (toward residue S) in the medium diversity simulation and 1/100 (toward residue M) in the high divergence case. Overall, the test appears to be conservative when the baseline model is correctly specified.
Power
DEPS can reliably detect strong (i.e., selective sweep) directional selection (B = 100 for T) for even relatively low levels of sequence divergence, returning positive results in 69%, 85%, and 86% of cases for the 3 levels of sequence divergence. The power to detect individual sites under directional selection was adequate (45.2%, 55.5%, 57.2%). Because site-to-site rate variation was variation modeled by a gamma distribution with α = 0.2, the fact that the proportion of detected sites does not increase more dramatically when the mean divergence level is quintupled is not surprising, given that Pr{Γ(X, 0.2) ≤ 0.1} = 0.496, suggesting that on average, half of the directionally evolving sites have low divergence levels.
Intermediate level directional selection (B = 10 for I) was considerably more difficult to detect, with only 3 positive results for low sequence divergence, but rising to 41 for intermediate and to 64—for high divergence. However, only 20% of individual sites simulated under directional selection were found by our empirical Bayes procedure. For weak directional selection (B = 5 for R), there was very little power, even for divergent sequences, both at the LRT level (0%, 1%, 16%) and for individual sites (0%, 6.7%, 14.2%). The rate of false positives was very low in all cases (≤1%).
Estimated proportions of directionally selected sites and rate biases tended to be biased slightly upward. For example, for high divergence levels, the mean estimated proportion of sites of under selection was 1.45% (standard deviation 0.66%), whereas the true proportion was 0.88%; the bias term had sampled mean of 163.2 (true value 100) and high variance (standard deviation 757.51).
The Effect of Tree Estimation Errors
To assess the effect of potentially inaccurate topological reconstruction, we reanalyzed the replicates from the high divergence simulation scenario using trees inferred by Neighbor-Joining (corrected p distance) instead of using the correct tree. This approximation had very little effect on the performance of the method. In all, 0/100 (1/100 for the true tree) alignments showed LRT false positives. Simulated weak directional evolution toward R was recovered in 10 (vs. 11 for the correct tree) cases, with 16% (vs. 14.2%) of individual sites under directional selection found by the EBA. For medium-strength evolution toward I, 64/100 (vs. 64/100) replicates were significant under LRT and 19.7% of individual sites (vs. 16%) were detected with EBA. Finally, high-strength directional evolution toward T was recalled in 85/100 (vs. 86/100) cases, with 63% (vs. 57.2%) of individual sites correctly identified.
The Effect of Model Misspecification
When the JTT (Jones et al. [1992]) model was used as the incorrect baseline model for the analysis of high divergence directional data simulated under the H5N1 empirical model, we found that the power of the test was largely unaffected. The test found evidence of directional evolution toward R in 21/100 replicates, 61/100 toward I, and 83/100 toward T. However, the number of replicates where at least one of the identified target residues was incorrect (not I, R, T) increased dramatically to 21/100. The proportion of individual sites that were identified as directionally selected toward an incorrect target residue remained small at 0.3%. Most of the false positive signal for the LRT derived from scenarios when a large proportion of residues (e.g., >50%) were identified to be evolving under weak (R < 5) directional selection toward an incorrect residue, suggesting that the directional model was simply attempting to compensate for the global under- or overestimate of the empirical substitution rate resulting from model misspecification.
If the appropriate empirical model is not available, then one can estimate all 189 rates in the general reversible protein model (REV) and use those rate estimates for baseline and directionally biased models. This approach is clearly more computationally challenging but not insurmountably so. With the use of REV, the rate of false positives dropped to 8/100 and the power of the test declined by about 25%.
Evidence for Directional Evolution in Influenza A Genes
Directional selection tests found evidence for at least one directionally evolving residue in 5/8 genomic segments of avian H5N1 influenza (table 1) and in 4/8 genomic segments of human H3N2 influenza (table 2). H3N2 alignments had less sequence variability than their H5N1 counterparts, as measured by the total length of the phylogenetic tree, implying less statistical power to detect directional selection in H3N2 sequences. The largest numbers of target residues and sites under directional selection were inferred for HA and neuraminidase (NA)—the 2 most divergent genomic segments. Extreme modes of directional bias, such as evolution toward phenylalanine (F) in H3N2 NA that is subject to a very strong bias (B = 1791.9, profile likelihood 95% confidence intervals [CIs] 220.1–5687.6) but affects only a few sites (0.96%, 0.12–3%) and evolution toward threonine (T) in H5N1 HA that experiences to a weak bias (B = 3.24, 1.97–5.24) but affects a substantial estimated proportion of sites (11.85%, 2.0–27.7%), as well as a number of intermediate modes are represented in the results.
Using a stringent EBF cutoff of 100, only a small (<3%) proportion of individual sites were identified as evolving directionally for each genomic segment. These proportions were comparable with the proportions of sites inferred to be under diversifying positive selection by a fixed effects likelihood (FEL) method. FEL and most other current methods for detecting positive selection are based on a codon substitution model and adopt the equal rates model for amino acid residue replacement. Consequently, positive selection is detected if the average (over all residue pairs) nonsynonymous substitution rate at a site exceeds synonymous rate at the same site. FEL estimates the 2 rates directly at each site and tests for their equality using an LRT (for details, see Kosakovsky Pond and Frost [2005c]).
Perhaps the most important observation to be gleaned from examining site-by-site results (tables 3 and 4) is that directionally selected sites are overwhelmingly not the same as sites found to be under diversifying selection, with a few exceptions in HA. Moreover, there are several sites that appear to be under purifying selection based on a traditional dN/dS analysis but are nonetheless evolving directionally. This suggests that DEPS is geared toward detecting types of selection not handled by a traditional approach and hence can lead to qualitatively new insights into the nature of evolutionary forces. Clearly, DEPS and FEL make a number of different modeling assumptions, and difference in results could be due to any of the assumptions. We do not attempt to identify the exact cause for such differences and provide FEL results as a reference because the test typifies widely used analytical tools for selection analysis.
Table 3.
Sites Found to Be under Directional Selection in Avian H5N1 Influenza A Genomic Segments
| Site | Composition | Root | Inferred Substitutions | DEPS EBF | FEL dN/dS | FEL P | Class |
| PB2/segment 1 | |||||||
| 339 | K150T129M4Q3 | K | K→1Q, K→1T, T→1K, T→4M | >105(M) | 0.3566 | 0.0357 | HPS |
| 727 | G283M2R1 | G | G→2M, G→1R | 3217.9(M) | 0.4876 | 0.2681 | RRS |
| PB1/segment 2 | |||||||
| 14 | V239A46 | A | A→8V, V→3A | >105(V) | 0.9406 | 0.9115 | SSS |
| 80 | S284C2 | S | S→2C | >105(C) | 0.1738 | 0.0380 | RRS |
| 384 | L229S53I4 | S | L→1I, L→5S, S→1L | >105(S) | 3.1310 | 0.1157 | RSS |
| 490 | F284C1L1 | F | F→1C, F→1L | 276.9(C) | 0.2552 | 0.1350 | RRS |
| 492 | F285C1 | F | F→1C | 309.5(C) | Infinite | 0.3865 | RRS |
| PA/segment 3 | |||||||
| 3 | D223F10E2 | D | D→1E, D→9F, E→1D | >105(F) | Infinite | 0.3715 | RSS |
| 68 | P279Q8 | P | P→5Q | >105(Q) | 0.0000 | 0.0000 | RSS |
| 261 | L206M58Q18F3V2 | L | L→1F, L→1M, L→3Q, L→1V, M→1V | 1232.0(Q) | 0.3838 | 0.3487 | HPS |
| HA/segment 4 | |||||||
| 52 | T282K5A1 | T | T→1A, T→5K | >105(K) | 2.1119 | 0.4542 | RSS |
| 139 | S268P18A2 | S | S→1A, S→6P | 1342.1(P) | 2.2425 | 0.2765 | RSS |
| 145 | S189L97A2 | S | S→1A, S→6L | >105(L) | 1.2137 | 0.7794 | RSS |
| 154 | Q213L54H13N8 | H | H→1L, H→3N, H→1Q, L→3Q, Q→6L | 1638.6(L) | 14.0226 | 0.0003 | HPS |
| 157 | S195P93 | S | S→8P | >105(P) | Infinite | 0.0073 | CRS |
| 172 | T155A126S4K2I1 | A | A→1I, A→2S, A→13T, T→3A, T→1K | 4037.1(T) | 7.4929 | 0.0078 | HPS |
| 200 | A244E44 | A | A→5E | >105(E) | 0.2405 | 0.0095 | RSS |
| 429 | K286Q2 | Q | Q→1K | 103.0(K) | 0.0659 | 0.0011 | SSS |
| 515 | K229N59 | N | K→1N, N→3K | 337.9(K) | Infinite | 0.0959 | CRS |
| 527 | I236M50V2 | M | I→1V, M→6I, M→1V | 1141.2(I) | 0.7172 | 0.8662 | CRS |
| NA/segment 6 | |||||||
| 211 | I248M38V1 | I | I→3M, M→4I, M→1V | >105(I) | 0.2674 | 0.1488 | HPS |
| 338 | V265M22 | V | V→5M | 6618.7(M) | 1.8231 | 0.5620 | RSS |
| 382 | G168E118 | G | G→8E | >105(E) | 0.9612 | 0.9542 | HPS |
| 389 | V271L9M5A1 | V | V→1A, V→2L, V→4M | 141.0(M) | 0.9782 | 0.9748 | HPS |
NOTE.—Sites found to be under directional selection in avian H5N1 influenza A HA. Site coordinates are in terms of the AAD51927 (A/Goose/Guangdong/1/96 [H5N1]) reference strain. Sites that were also detected as positively selected by the FEL method (at P ≤ 0.05) have their indices shown in italic; for sites that were detected as negatively selected by FEL, the indices are shown in bold. For each site, we report the observed residue composition, the inferred root state and substitutions (using maximum likelihood joint ancestral state reconstruction under the appropriate alternative protein model), the DEPS EBF, the target residue, the synonymous/nonsynonymous (dN/dS) ratio estimated at that site by FEL, the P value for nonneutral evolution at the site returned by FEL, and notes on known or putative function/relevance of the site. Classes of sites are described in the text, with the following abbreviations: SSS—selective sweep site, CRS—consensus replacement site, RSS—repeated substitutions site, RRS—rare residue substitution, and HPS—highly polymorphic site.
Table 4.
Sites Found to Be under Directional Selection in Human H3N2 Influenza A Genomic Segments
| Site | Composition | Root | Inferred Substitutions | DEPS EBF | FEL dN/dS | FEL P | Class |
| PB1/segment 2 | |||||||
| 111 | M244I12 | M | M→6I | >105(I) | 0.0017 | 0.0048 | RSS |
| 298 | L253I3 | L | L→2I | 140.3(I) | 1.0747 | 0.9531 | RRS |
| 753 | L254I2 | L | L→2I | 136.1(I) | Infinite | 0.1755 | RRS |
| HA/segment 4a | |||||||
| 10 | I279F4T1 | I | I→3F, I→1T | >105(F) | 2.0430 | 0.4976 | RRS |
| 61 | S265I14N5 | S | S→2I, S→2N | 337.9(I) | 1.4582 | 0.7281 | RSS |
| 151 | T269G10K4E1 | G | G→1E, G→1K, K→1T | 2109.7(T) | Infinite | 0.1173 | SSS |
| 161 | K237N40S7 | S | K→1N, N→4K, N→1S, S→1N | 1936.6(K) | Infinite | 0.0434 | HPS |
| 171 | H164T116Y4 | T | H→1T, T→1Y, Y→2H | 3047.0(T) | 3.1920 | 0.2254 | HPS |
| 174 | K267E13G4 | G | E→2K, G→2E | 212.2(K) | 1.1638 | 0.8913 | SSS |
| 245 | R269K6I6G1S1Q1 | R | R→1G, R→4I, R→6K, R→1Q, R→1S | >105(I) | 4.1979 | 0.0990 | RRS |
| 264 | T278N6 | N | N→1T | 111.9(T) | 0.1101 | 0.0343 | SSS |
| 347 | I279L5 | L | L→1I | 101.9(I) | Infinite | 0.6259 | SSS |
| NA/segment 6 | |||||||
| 42 | C300F43W2 | C | C→2F, C→2W | >105(F) | 2.3485 | 0.4107 | SSS |
| 77 | I334M5V3K2T1 | I | I→2K, I→2M, I→1T, I→3V | 174.6(K) | Infinite | 0.2199 | RRS |
| 267 | T139P108Q44L22K15S14A3 | P | P→1K, P→1L, P→3Q, P→1S | 103.5(Q) | 4.8550 | 0.0574 | HPS |
| P→3T, T→2A, T→1K | |||||||
| 308 | K329E16 | E | E→2K | 2138.8(K) | 0.3327 | 0.2839 | SSS |
| 338 | L275R62Q5W3 | R | R→1L, R→5Q, R→1W | >105(Q) | 1.0216 | 0.9697 | HPS |
| 467 | M344F1 | M | M→1F | 1173.0(F) | 0.0000 | 0.1283 | RRS |
| MP/segment 7 | |||||||
| 77 | R170I3 | R | R→3I | >105(I) | Infinite | 0.1476 | RRS |
| NS1/segment 8 | |||||||
| 176 | N160I4 | N | N→3I | >105(I) | Infinite | 0.2584 | RRS |
NOTE.—Sites found to be under directional selection in human H3N2 influenza A HA. Site coordinates are in terms of the Q91MA7 (A/Hong Kong/1/68 [H3N2]) reference strain. Sites that were also detected as positively selected by the FEL method (at P ≤ 0.05) have their indices shown in italic; for sites that were detected as negatively selected by FEL, the indices are shown in bold. See table 3 for an explanation of column headings and site class abbreviations.
Our HA site coordinates are shifted by +16 relative to the numbering in Smith et al. (2004).
Classes of Directionally Evolving Sites
To assist in interpreting evolutionary patterns at a given site, we inferred the most likely root ancestral state using maximum likelihood (Yang et al. 1995) under the influenza-specific substitution model biased toward the appropriate residue and counted inferred substitutions at a given site, based on the most likely joint ancestral state reconstruction (Pupko et al. 2000) under the same model. A closer scrutiny of inferred evolutionary histories at directionally evolving sites suggests 5 broad patterns. Our list is not meant to be an exhaustive enumeration of all possible selection regimes but rather a simple classification aid.
Selective Sweep Sites
A selective sweep occurs when a given residue found at high frequency in sequences from early time points is completely replaced by a different residue at later time points. Formally, we require that the target residue is a minority residue (<50% frequency at the earliest time point) and that it increases in frequency to 100% and remains fixed from then on. A clear-cut example of this behavior can be found at site 429 in H5N1 HA, where ancestral glutamine is completely replaced with lysine (see fig. 2) between 1997 and 1999 along a single tree branch. However, following this fixation, a large number of synonymous substitutions had accumulated, driving an overall strong signal of negative selection at the site (α = 0.24, β = 3.75, P = 0.001 for ω = β/α < 1 as estimated by FEL and LRT) when analyzed using a traditional method for selection detection. A rapid selective sweep followed by a long period of purifying selection in serially sampled sequences is precisely the situation that confounds traditional selection techniques, which zero in on the abundance of synonymous evolution (indicative of purifying selection) following fixation; but a selective sweep is easily detected by the DEPS test. Other examples of selective sweeps are site 14 in H5N1 PB1 RNA polymerase and sites 151, 174, 264, 347 in H3N2 HA. Sites in highly variable regions, for example in antigenic sites of IAV HA, may undergo repeated selective sweeps. For example, residue 161 in H3N2 HA that is located in an experimentally characterized antigenic site (Wilson et al. [1981]) appears to have undergone at least 2 selective sweeps (Smith et al. [2004]). First, an S→N substitution induced a transition between 2 antigenic clusters of sequences, the first comprising sequences sampled between 1968 and 1972 and the second including sequences sampled between 1972 and 1975. The second selective sweep was effected by an N→K substitution between 1987 and 1995 and resulted in a marked shift in antibody-binding profiles. DEPS is capable of detecting certain types of multiple selective sweeps by identifying the target residue of the most recent one. However, as shown in the next section, repeated selective sweeps can also result in a site being detected as positively selected by FEL but not directionally evolving by DEPS.
FIG. 2.—

Residue frequencies plotted over time at several example sites. The vertical axis shows the proportion of each residue (see legend for shading-to-residue correspondence) at a given yearly time point (shown along the horizontal axis). Each bar is further labeled with the total number of samples available for that year. The target residue detected by DEPS at that site is shown in black, and its frequency in all samples is indicated as the horizontal dashed line; other residues are shown in varying shades of gray and labeled in the legend.
Consensus Replacement Sites
A substantial increase in the frequency of a residue, often with the concomitant drop in the frequency of the ancestral residue, describes another common evolutionary pattern for directionally evolving sites. Formally, the target residue must be a minority residue (<50% frequency at the earliest time point) and increase in frequency (not necessarily monotonically) until it reaches 50% or more, and the frequency must not dip below 50% at any later time point. Consider site 527 in H5N1 HA (see fig. 2), where the frequency of methionine drops from 100% in 1996–2000 to <10% following 2004 and isoleucine becomes the most frequent residue. Neutral evolution (ω = 0.71) is suggested when using FEL; intuitively, a fairly large number of inferred nonsynonymous substitutions (8, see table 3) are counterbalanced by synonymous substitutions in isoleucine. However tempting, it would be fruitless, however, to speculate whether or not isoleucine may approach fixation in avian H5N1 samples or whether multiple residues will be maintained in the population without sampling additional sequences in later years; a change in the selective environment can also exert evolutionary pressures favoring a different residue.
Repeated Substitution Sites
Another category of sites under directional selection are those where the proportion of sequences with the target residue is relatively small, but substitutions toward the target occur along multiple branches; these sites also tend to have fluctuating frequencies of the target residue and could reflect maintenance of a minority variant or weak directional selection. We define repeated substitutions sites as those that do not fall in the previous 2 categories but maintain the same majority (>50% frequency) residue at every time point, have at least 2.5% of sequences with the target residue (summed over all time points), and multiple inferred substitutions toward that residue take place at the site. For instance, site 139 in H5N1 HA exhibits a small but persistent proportion of proline (see fig. 2), whereas the ancestral serine remains the dominant residue. Because sequences with prolines in this position are not monophyletic, repeated substitutions toward proline (6 were inferred by maximum likelihood) are necessary to explain this behavior. Convergent evolution in a low-frequency selective environment, for example, a specific immune profile of the host, is one possible cause.
Rare residue substitutions
If the source and the target residues have a low substitution rate in the baseline model, a few substitutions may be sufficient to detect a site as directionally selected, although such findings may be sensitive to baseline model misspecification and should be treated with caution. RRS sites have no more than 2.5% of sequences with the target residue over all time points and maintain the same majority residue over all time points. Site 176 in H3N2 nonstructural 1 protein is a good example—only 3 substitutions of asparagine with isoleucine (a low-rate substitution, see fig. 1) are inferred at that site. An even more extreme case is site 467 in H3N2 NA, where a single low-rate substitution from methionine to phenylalanine appears sufficiently significant. Another type of sites that can be allocated to this category are those where the substitutions are biochemically conservative, but there is a measurable proportion of weakly directionally evolving sites, which together provide the signal of selection. For example, residues 298 and 753 in H3N2 PB1 have only 2 conservative L→I substitutions, but the gene as a whole has an estimated 4% of weakly-to-moderately (B = 21) directionally selected sites.
Highly Polymorphic Sites
In several cases (e.g., positions 154 and 172 in H5N1 HA), sites with a high degree of amino acid polymorphism have been classified as directionally evolving by DEPS. Highly polymorphic site is a catchall category that includes those sites that do not belong in the 4 previously defined classes. These sites are often also found to be under diversifying selection by FEL and have complex evolutionary dynamics, often with fluctuating residue frequencies. This evolutionary pattern is a reminder that directional evolution toward a residue and diversifying selection are not mutually exclusive. Alternatively, the proportion of sequences with the target residue can fluctuate quite dramatically, as is the case for site 171 in H3N2 HA (fig. 2)—a pattern suggestive of frequency-dependent selection.
Potential Functional Significance of Sites Involved in Directional Evolution
For some of the sites detected by DEPS (tables 3 and 4), it is possible to propose a plausible selective force driving adaptation. For example, surface genes of influenza A (HA and NA) have been studied extensively, and functional importance of various protein components, selective pressures, and escape mechanisms have been characterized. N-linked glycosylation in HA is a well-known mechanism whereby antibody-binding sites can be masked, for instance due to steric hindrance (e.g., Munk et al. [1992]). For 3 sites in HA (site 172 in H5N1; sites 151 and 264 in H3N2), the substitution of the ancestral residue with the residue detected by DEPS resulted in the acquisition of a new potential N-linked glycosylation site, encoded by a 4-residue N-(not P)-S/T-(not P) sequence motif. In all 3 cases, the substitution targeted the third residue in the motif. Site 347 is located in the membrane fusion peptide of the HA2 subunit of the H3N2 HA (Harter et al. [1989]).
HA and NA are also known to acquire substitutions within B-cell epitopes that prevent or cripple recognition and binding by host antibodies. In avian H5N1 HA, site 157 was shown to develop S→P mutations in response to selection by a monoclonal antibody in mice (Kaverin et al. 2007) and sites 154 and 145 reside in antigenic sites 1 and 3, respectively (Stevens et al. 2006). In human H3N2 HA, residues 161 and 171 are located within antigenic sites A and B, respectively, whereas residue 151 is within a receptor-binding pocket and residue 245 is adjacent to another receptor-binding domain (Wilson et al. 1981; Lindstrom et al. 1996). H3N2 NA was shown to acquire an E→K substitution at residue 308 in response to passaging in presence of an NA-specific monoclonal antibody, whereas residue 338 is a part of an antigenic site (Colman et al. 1983).
A previous detailed analysis of correlation between genetic and phenotypic (antigenic) distances in H3N2 HA (Smith et al. 2004) revealed a number of amino acid substitutions that could be involved in the antigenic evolution of influenza A. Of 9 directionally evolving residues identified by DEPS (table 4), positions 161, 171, and 174 (145, 155, and 157 in the coordinates of table 1 in Smith et al. [2004]) have been associated with fixed differences between antigenic clusters of sequences.
Five sites detected by DEPS in H3N2 HA were also identified as undergoing potentially adaptive residue frequency shifts in another recent study of temporal evolutionary patterns (Shih et al. 2007). Sites 151, 161, 171, 174, and 264 (our coordinates are shifted by +16) were identified as having undergone one or more frequency switches by Shih et al. (2007).
The T-cell-mediated branch of the human immune system is another potential selective force; mutations within epitopes targeted by the major histocompatibility complex of a host can thwart T-cell binding and prevent subsequent cell killing by cytotoxic T-lymphocytes. A large number of T-cell epitopes have been characterized throughout the viral genome; for example, Suzuki (2006) collated many known epitopes for H3N2 serotype virus. Among directionally selected sites, residues 61 and 264 in HA are located within previously characterized T-cell epitopes. However, it should be noted that although CTL epitope coverage of the HA gene is potentially rather extensive, epitope mapping studies have commenced only recently (e.g., Wang et al. [2007]). Therefore, it is not clear to what extent CTL-mediated selection may be responsible for directional evolution of individual sites.
Comparing FEL and DEPS Results on H5N1 HA
An FEL analysis of H5N1 HA revealed 9 sites (3, 11, 99, 154, 156, 157, 171, 172, and 403) under the influence of positive selection (P < 0.05), 3 of which (154, 157, and 172) were also detected by DEPS (table 3 and fig. 3). Several evolutionary scenarios could plausibly explain the situations when FEL reports positive selection and DEPS does not.
FIG. 3.—

Residue frequencies over time at those sites in H5N1 HA that were detected by FEL as positively selected but not detected as directionally evolving by DEPS. The vertical axis shows the proportion of each residue (see legend for shading-to-residue correspondence) at a given yearly time point (shown along the horizontal axis). Each bar is further labeled with the total number of samples available for that year. The ancestral residue at that site, inferred by maximum likelihood using the baseline model, is shown in black, and its frequency in all samples is indicated as the horizontal dashed line; other residues are shown in varying shades of gray and labeled in the legend. Each site is annotated with substitutions inferred under the baseline.
Maintenance of Multiple Variants
Sites 3, 99, 171, and 403 experience multiple phylogenetically independent substitutions from the root/majority state to several target residues. Temporal profiles of residue frequencies suggest that multiple allelic variants may persist in the population. This is consistent with diversifying selection, detection of which is the strength of FEL and conceptually similar approaches.
Exchangeable Residues
Site 11 appears to toggle between several highly exchangeable residues (such as V and I, see fig. 1). These types of substitutions would be traditionally called “conservative” and can be accommodated by high individual rates already included in the baseline model.
Successive (partial) Selective Sweeps
Temporal frequency profiling of site 156 suggests successive selective sweeps involving multiple residues at complex polymorphic sites. Targets of selection that change from time to time are not accommodated by the alternative model of the DEPS test, and further stratification of sequence samples into shorter time periods could help resolve this situation.
We note that the above categories are neither exhaustive nor definitive. For instance, site 171 could be an instance of either multiple allelic variants or successive selective sweeps.
H5N1 and H3N2 Gene-by-Gene Comparison
For every target residue, we tested (as described in Methods) each of the 8 genomic segments in H5N1 against its counterpart in H3N2 to determine if either the bias term (b) and/or the proportion of directionally evolving sites (q) differed between serotypes. Only 6/320 pairwise comparisons were potentially different (P < 0.05), 4 of them in HA (the bias term for I, K, and T and the proportion of T), 1 in PB1 (the bias term for G), and 1 in NS1 (the proportion of I). For instance, directional evolution toward T in H5N1 is weaker than in H3N2 but affects a greater proportion of sites.
Predicting Evolutionary Targets
To illustrate the relation between inferred target residues and observed frequency change patterns, we downloaded all avian H5N1 (1,054) and human H3N2 (2,072) near full-length (≥1,500 nt) HA sequences from the NCBI influenza virus resource (Bao et al. [2007]) and tabulated sampled frequencies of target residues identified by DEPS as evolving directionally. Figure 4 shows 4 distinct possibilities for residue frequency trends in a larger sample, where the residues are a subset of those in figure 2: site 527 in H5N1 shows residue fixation, site 172 in H5N1 shows an increase in target residue frequency followed by a decrease in frequency, fluctuation in frequency, and persistent low frequency. Trend detection is complicated by the fact that sample sizes vary drastically from year to year; we visually represented sampling variability by the width of corresponding CIs on estimated residue frequencies.
FIG. 4.—

DEPS target residue frequency variation at several HA sites in large samples of H3N2 and H5N sequences. Frequency of the target residue predicted by DEPS based on a database sample of H5N1 and H3N2 HA sequences. Sampling years and number of samples per year are shown along the horizontal axes. The observed frequency of the target residue at a given codon is shown by notched black horizontal lines, whereas gray bars represent sampling uncertainty estimated by 95% CIs on the appropriate parameter of the multinomial distribution of residue frequencies from a given year, using the method of Quesenberry and Hurst (1964).
Discussion
We have presented a maximum likelihood approach to finding evidence of directional evolution, as measured by elevated substitution rates toward a specific residue, from multiple alignments of protein sequences in the phylogenetic framework. The method incorporates organism-specific residue frequencies and baseline pairwise residue substitution rates and permits site-to-site variation in baseline substitution rates. An LRT is used to evaluate deviations from organism-wide mean substitution patterns at a proportion of sites in a gene, where evolution is accelerated toward a particular residue. Target residues of directional evolution and sites evolving toward these targets are inferred, although the test could be easily modified to test for convergent evolution toward an a priori target residue or sequence. The test appears conservative on simulated data, unless the baseline model is severely misspecified, but the use of a general time reversible model as an additional control step reduces false positive error rates to near-nominal levels. The power of the test to detect strong effects is moderate to excellent, depending on the level of sequence divergence, and weaker effects can be identified as more sequence variability is included in the sample.
The analysis of influenza A genes revealed a number of directionally evolving sites, covering a wide range of apparent evolutionary patterns. Our method appears to be especially adept at locating sites that have undergone rapid selective sweeps or consensus residue replacement, which confound a traditional dN/dS based FEL technique. Evolutionary patterns indicating target residue frequency increase, maintenance of a minority variant, and frequency-dependent selection have also been detected; many of these were missed by FEL or even strongly classified as being under purifying selection (note that the 2 are not mutually exclusive); this demonstrates that DEPS zeroes in on the components of the evolutionary process not well modeled by the traditional techniques.
We also demonstrated how to look for differences in strengths of directional selection at the level of a gene, for instance, when samples of the same gene from 2 distinct populations or selective environments are available. Many of the directionally evolving sites could be assigned to functional categories where directional evolution is expected, for instance immunogenic sites of surface proteins or potential glycosylation sites, and it may prove worthwhile to investigate computationally identified residues experimentally.
Our method has a number of limitations that we make explicit in order to give a practicing bioinformatician the data needed to make an informed choice as to whether or not the method should be applied to their data. An outgroup must be available to establish the direction of evolution, and serially sampled data are needed to perform post hoc classification of identified sites. Small (or low divergence) samples (e.g., <50 sequences) are likely to reveal only the strongest effect of directional selection; larger samples may be needed to detect more subtle selection. Many existing samples are often biased toward a specific region, strain, or clade; this should be kept in mind when the results are interpreted, for example, a putative selective sweep may be incomplete because of low frequency or lack of sampling of sequences with the original residue. The method relies on the accurate specification of the null model; we strongly encourage using organism- or gene-specific empirical substitution matrices or the general reversible model to reduce the risk of false positives. The current test could be misled by recombination. However, it can be easily augmented, for example, as described in Kosakovsky Pond et al. (2006) in the context of FEL, to account for phylogenetic discordance in the sample.
DEPS provides a formal statistical framework to test for evidence of accelerated substitutions to a particular residue at a specific site (relative to the baseline null model); it does not attempt to predict the evolutionary path at any given site. We strongly caution against equating a positive DEPS result with the deduction that the identified target residue at a given site is the ultimate evolutionary target. First, as we discussed previously, a number of possible evolutionary scenarios not necessarily leading to a selective sweep can give rise to a positive finding. Second, many organisms, including IAV, are shaped by complex and changing dynamic selective forces and population-level processes that could easily alter the fitness landscape in the future, rendering all predictions based on the past and present states of the system irrelevant. For example, Shih et al. (2007) identified multiple frequency shifts at antigenically relevant sites in H3N2 HA, many of which failed to reach fixation, likely due to ongoing selection and changing fitness landscapes. Third, inference is based on finite and often biased samples of sequences, with a strong potential to alter the results of any evolutionary analysis. Fourth, even if a residue eventually becomes fixed in the population, the timescale for this process is highly variable, as suggested by the transition time analysis in Shih et al. (2007) that encompassed the range from 4 to 32 years. It may therefore be beneficial to “zoom in” at a particular time interval to obtain better resolution of transient selective dynamics.
Evolutionary dynamics of the population from which the sequences are drawn play a very important role in directional selection patterns. For instance, the lack of local persistence in H3N2 human IAV (Nelson et al. 2007) with possible seasonal “reseeding” of the epidemic (Russell et al. 2008) may give rise to frequent oscillations in residue frequencies, thereby diluting the signal for directional selection and complicating interpretation. On the other hand, directional evolution in a relatively stronger geographical separation among avian H5N1 IAV clades (e.g., Webster and Govorkova [2006]) may result in an environment where minority alleles are maintained in a global population, and this can be detected by DEPS.
In conclusion, the new test for DEPS fills in a methodological gap in the modern evolutionary toolbox for the analysis of natural selection by complementing the weakness of popular existing dN/dS based methods in detecting transient and frequency-dependent selection and can help identify functionally important residues in sequences sampled over time.
Acknowledgments
This research was supported in part by the National Institutes of Health (AI43638, AI47745, and AI57167), the University of California Universitywide AIDS Research Program (grant number IS02-SD-701), and by a University of California, San Diego Center for AIDS Research/NIAID Developmental Award to S.D.W.F. and S.L.K.P. (AI36214).
References
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19(6):950–958. doi: 10.1093/oxfordjournals.molbev.a004152. [DOI] [PubMed] [Google Scholar]
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the national center for biotechnology information. J Virol. 2007;82(2):596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the evolution of human influenza A. Science. 1999;286(5446):1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Colman PM, Varghese JN, Laver WG. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature. 1983;303(5921):41–44. doi: 10.1038/303041a0. [DOI] [PubMed] [Google Scholar]
- Conant GC, Wagner GP, Stadler PF. Modeling amino acid substitution patterns in orthologous and paralogous genes. Mol Phylogenet Evol. 2007;42(2):298–307. doi: 10.1016/j.ympev.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Dayhoff MO, Eck EV, Park CM. A model of evolutionary change in proteins. In: Dayhoff MO, editor. Atlas of protein sequence and structure. Vol. 5. Washington (DC): National Biomedical Research Foundation; 1972. pp. 89–99. [Google Scholar]
- Doron-Faigenboim A, Pupko T. A combined empirical and mechanistic codon model. Mol Biol Evol. 2007;24(2):388–397. doi: 10.1093/molbev/msl175. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11(5):725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Guindon SA, Rodrigo G, Dyer KA, Huelsenbeck JP. Modeling the site-specific variation of selection patterns along lineages. Proc Natl Acad Sci USA. 2004;101(35):12957–12962. doi: 10.1073/pnas.0402177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter C, James P, Bachi T, Semenza G, Brunner J. Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the “fusion peptide”. J Biol Chem. 1989;264(11):6459–6464. [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Jones D, Taylor W, Thornton J. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8(3):275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Kass R, Raftery A. Bayes factors. J Am Stat Assoc. 1995;90(430):773–795. [Google Scholar]
- Kaverin NV, Rudneva IA, Govorkova EA, Timofeeva TA, Shilov AA, Kochergin-Nikitsky KS, Krylov PS, Webster RG. Epitope mapping of the hemagglutinin molecule of a highly pathogenic H5N1 influenza virus by using monoclonal antibodies. J Virol. 2007;81(23):12911–12917. doi: 10.1128/JVI.01522-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. A simple hierarchical approach to modeling distributions of substitution rates. Mol Biol Evol. 2005a;22:223–234. doi: 10.1093/molbev/msi009. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol Biol Evol. 2005b;22(3):478–485. doi: 10.1093/molbev/msi031. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005c;22(5):1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21(5):676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006;23(10):1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Kosiol C, Holmes I, Goldman N. An empirical codon model for protein sequence evolution. Mol Biol Evol. 2007;24(7):1464–1479. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- Lindstrom S, Sugita S, Endo A, Ishida M, Huang P, Xi SH, Nerome K. Evolutionary characterization of recent human H3N2 influenza A isolates from Japan and China: novel changes in the receptor binding domain. Arch Virol. 1996;141(7):1349–1355. doi: 10.1007/BF01718836. [DOI] [PubMed] [Google Scholar]
- Maddison DR, Swofford DL, Maddison WP. NEXUS: an extensible file format for systematic information. Syst Biol. 1997;46(4):590–621. doi: 10.1093/sysbio/46.4.590. [DOI] [PubMed] [Google Scholar]
- McClellan DA, McCracken KG. Estimating the influence of selection on the variable amino acid sites of the cytochrome b protein functional domains. Mol Biol Evol. 2001;18(6):917–925. doi: 10.1093/oxfordjournals.molbev.a003892. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351(6328):652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Miyata T, Yasunaga T. Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J Mol Evol. 1980;16(1):23–36. doi: 10.1007/BF01732067. [DOI] [PubMed] [Google Scholar]
- Moler C, Van Loan C. Nineteen dubious ways to compute the exponential of a matrix. Siam Rev. 1978;20:801–836. [Google Scholar]
- Munk K, Pritzer E, Kretzschmar E, Gutte B, Garten W, Klenk HD. Carbohydrate masking of an antigenic epitope of influenza virus haemagglutinin independent of oligosaccharide size. Glycobiology. 1992;2(3):233–240. doi: 10.1093/glycob/2.3.233. [DOI] [PubMed] [Google Scholar]
- Muse SV. Evolutionary analyses of DNA sequences subject to constraints on secondary structure. Genetics. 1995;139:1429–1439. doi: 10.1093/genetics/139.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse SV. Estimating synonymous and nonsynonymous substitution rates. Mol Biol Evol. 1996;13(1):105–114. doi: 10.1093/oxfordjournals.molbev.a025549. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Nei M. Selectionism and neutralism in molecular evolution. Mol Biol Evol. 2005;22(12):2318–2342. doi: 10.1093/molbev/msi242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nelson MI, Holmes EC. The evolution of epidemic influenza. Nat Rev Genet. 2007;8(3):196–205. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- Nelson MI, Simonsen L, Viboud C, Miller MA, Holmes EC. Phylogenetic analysis reveals the global migration of seasonal influenza A viruses. PLoS Pathog. 2007;3(9):1220–1228. doi: 10.1371/journal.ppat.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MI, Viboud C, Simonsen L, et al. (12 co-authors) Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog. 2008;4(2):e1000012. doi: 10.1371/journal.ppat.1000012. (12 co-author) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickle DC, Heath L, Jensen MA, Gilbert PB, Mullins JI, Kosakovsky Pond SL. HIV-specific probabilistic models of protein evolution. PLoS ONE. 2007;2:e503. doi: 10.1371/journal.pone.0000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang ZH. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupko T, Pe'er I, Shamir R, Graur D. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol Biol Evol. 2000;17:890–896. doi: 10.1093/oxfordjournals.molbev.a026369. [DOI] [PubMed] [Google Scholar]
- Quesenberry C, Hurst D. Large-sample simultaneous confidence intervals for multinomial proportions. Techonometrics. 1964;6:191–195. [Google Scholar]
- Ray SC, Fanning L, Wang X-H, Netski DM, Kenny-walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201(11):1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CA, Jones TC, Barr IG, et al. (28 co-authors) The global circulation of seasonal influenza A (H3N2) viruses. Science. 2008;320(5874):340–346. doi: 10.1126/science.1154137. (12 co-author) [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. Positive natural selection in the human lineage. Science. 2006;312(5780):1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Self SG, Liang KY. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc. 1987;82(398):605–1310. [Google Scholar]
- Seoighe C, Ketwaroo F, Pillay V, et al. (11 co-authors) A model of directional selection applied to the evolution of drug resistance in HIV-1. Mol Biol Evol. 2007;24(4):1025–1031. doi: 10.1093/molbev/msm021. [DOI] [PubMed] [Google Scholar]
- Shih AC-C, Hsiao T-C, Ho M-S, Li W-H. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc Natl Acad Sci USA. 2007;104(15):6283–6288. doi: 10.1073/pnas.0701396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305(5682):371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Stanfel L. A new approach to clustering the amino acids. J Theor Biol. 1996;183(2):195–205. doi: 10.1006/jtbi.1996.0213. [DOI] [PubMed] [Google Scholar]
- Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312(5772):404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. Natural selection on the influenza virus genome. Mol Biol Evol. 2006;23(10):1902–1911. doi: 10.1093/molbev/msl050. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nei M. False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Mol Biol Evol. 2004;21(5):914–921. doi: 10.1093/molbev/msh098. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Nielsen R, Yang Q. Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003;20(1):18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Wang M, Lamberth K, Harndahl M, et al. (15 co-authors) CTL epitopes for influenza A including the H5N1 bird flu; genome-, pathogen-, and hla-wide screening. Vaccine. 2007;25(15):2823–2831. doi: 10.1016/j.vaccine.2006.12.038. [DOI] [PubMed] [Google Scholar]
- Webster RG, Govorkova EA. H5N1 influenza—continuing evolution and spread. N Engl J Med. 2006;355(21):2174–2177. doi: 10.1056/NEJMp068205. [DOI] [PubMed] [Google Scholar]
- Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289(5796):366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Xia X, Li WH. What amino acid properties affect protein evolution? J Mol Evol. 1998;47(5):557–564. doi: 10.1007/pl00006412. [DOI] [PubMed] [Google Scholar]
- Yampolsky LY, Stoltzfus A. The exchangeability of amino acids in proteins. Genetics. 2005;170(4):1459–1472. doi: 10.1534/genetics.104.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kumar S, Nei M. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics. 1995;141:1641–1650. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155(1):431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH. Maximum likelihood estimation on large phylogenies and analysis of adaptive evolution in human influenza virus A. J Mol Evol. 2000;51:423–432. doi: 10.1007/s002390010105. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]