

Abstract
A unified total synthesis is reported to access all of the possible diastereomers of pteriatoxins A-C, with the use of an intramolecular Diels-Alder reaction as the key step to form the carbo-macrocyclic core-structure. The C34/C35-diol protecting groups were found to have significant effects on both the exo/endo-selectivity and the exo facial-selectivity of the intramolecular Diels-Alder process.
Outbreaks of human poisoning due to the ingestion of Pinna shellfish (P. muricata and P. pectinata) were recorded in China and Japan, with typical neurotoxin symptoms such as paralysis, diarrhea, and convulsion. In 1995, Uemura and coworkers isolated pinnatoxin A (PnTX A), one of the major toxic principles responsible for outbreaks of Pinna-shellfish intoxication, from the shellfish Pinna muricata. They elucidated its gross structure and relative stereochemistry, and suggested a biosynthetic pathway.1 Its unique molecular architecture, accompanied by its pronounced biological activity as a Ca2+-channel activator, makes PnTX A an intriguing synthetic target.2 We previously reported the total synthesis of PnTX A, thereby not only confirming its gross-structure and relative stereochemistry, but establishing its absolute configuration as shown in Figure 1.3
Figure 1.
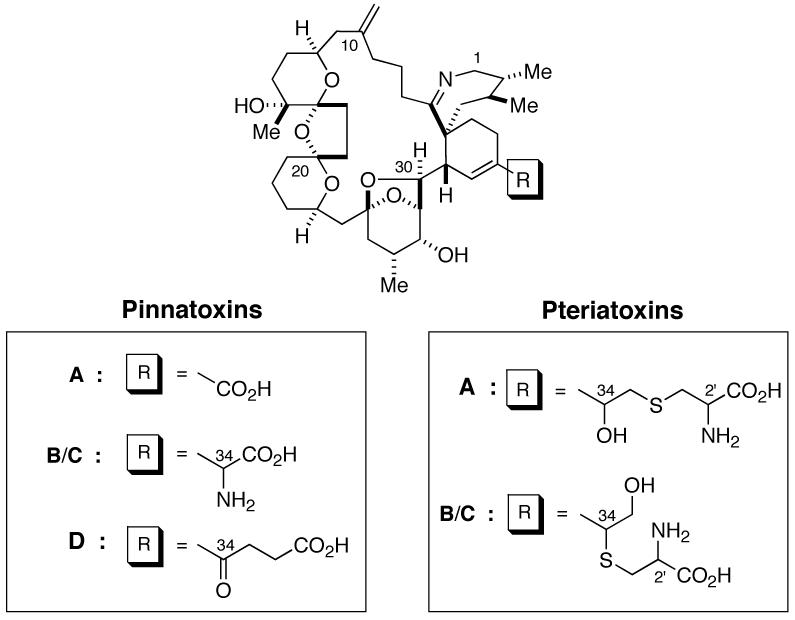
Structure of Pinnatoxins and Pteriatoxins
In 2001, Uemura and coworkers reported two new developments in this area: (1) isolation of PnTXs B and C from the Okinawan bivalve Pinna muricata and (2) isolation of pteriatoxins A, B and C (PtTXs A, B and C) from Pteria penguin.4 These new members were isolated in very minute amounts,5 but were reported to exhibit extremely potent and acute toxicity against mice.6 Considering the similarity in the 1H NMR characteristics of the new toxins with those of PnTX A, Uemura and coworkers suggested the gross-structures shown in Figure 1. In addition, they proposed that these new alkaloids and PnTX A share the same stereochemistry at the macrocyclic core. However, the C34 stereochemistry of PnTXs B/C and the C34 and C2′ stereochemistry of PtTXs A-C were left unassigned.4
Our research interests in this area are two-fold: (1) to establish the complete stereochemistry of PtTXs A-C and PnTXs B/C and (2) to secure an access to stereochemically homogeneous PtTXs A-C7 and PnTXs B/C, thus permitting unambiguous determination of their individual biological profiles. The naturally occurring PtTXs B/C, as well as PnTXs B/C, were isolated as a mixture and shown to be chromatographically inseparable.4 Therefore, the individual biological profile of each toxin was not characterized. In order to achieve our goals, we relied on organic synthesis to access each possible diastereomer in a stereochemically well-defined manner. To synthesize all diastereomers of PtTXs A-C and PnTXs B/C efficiently, we envisioned a unified synthetic plan outlined in Figure 2. This strategy will allow us to synthesize the C34 as well as the C2′ stereoisomers independently, and consequently secure the complete stereochemistry for all the members of the PnTX/PtTX family. In this paper, we report a total synthesis of all members of PtTX class of natural compounds.8
Figure 2.
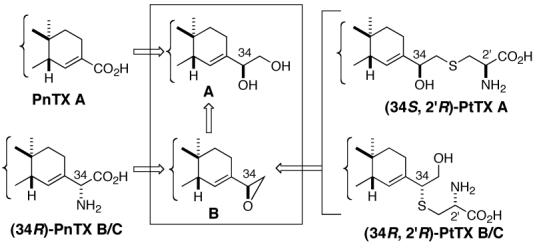
Unified Synthesis of the PnTX/PtTX-Class of Marine Natural Products
PtTXs are envisioned to be assembled from three building blocks: dithiane 1, vinyl iodide 2, and alkyl iodide 3 (Figure 3).9 It is worthwhile noting that 1 and 2 are the building blocks used for our PnTX A synthesis, but it is necessary to develop a synthetic route to the building block 3. The synthesis of the C33-C35 segments 7a (C34-β series) and 7b (C34-α series) is summarized in Scheme 1. Hydrolysis of the racemic vinyl bromide diacetate 110 with Amano lipase PS800 furnished a mixture of optically active 5 (optically purity: > 96%) and 6 (optically purity: >96%). Based on the literature precedent,11 their absolute configuration was assigned as indicated, which was further confirmed via chemical correlation with D-(+)- and L-(-)-solketals.12 This route secured access to antipodes 7a and 7b in multi-gram quantities.
Figure 3.
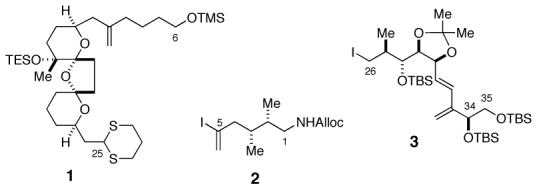
Three Building Blocks of PtTXs A-C
Scheme 1.
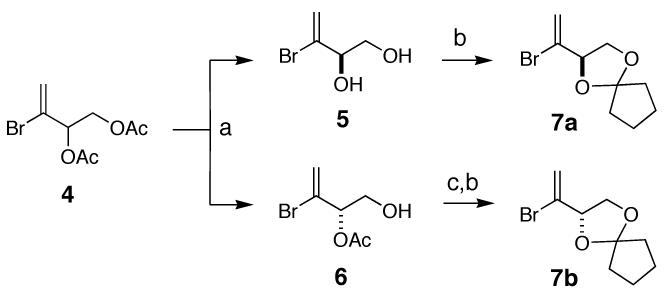
Reagents. a. Amano lipase PS800 (5: 46%; 6: 41%). b. cyclopentanone, p-TsOH (79%). c. LiOH (97%).
The optically active vinyl bromide 7a was then elaborated to the C26-C35 segment 3a (Scheme 2). In this synthesis, the C32-C33 bond was formed via Ni/Cr-mediated coupling3,13,14 of 7a and 8,3 to furnish a diasteromeric mixture of allylic alcohols, which, upon acylation and Pd-mediated elimination, afforded diene 9a. At this stage it was necessary to convert the C29/C30 acetonide to the more labile orthoester protecting group (3a) due to our inability to deprotect the acetonide at later stages in the synthesis.
Scheme 2.
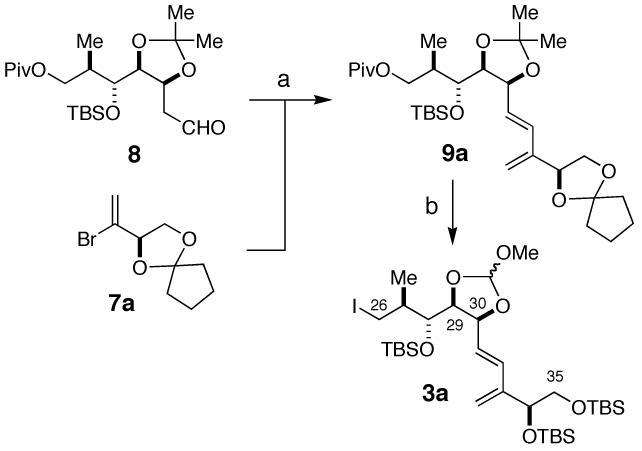
Reagents. a. 1. 7a, NiCl2, CrCl2, 86%. 2. Ac2O, Py. 93%, 3. Pd(OAc)2, CaCO3, 97%. b. 1. TFA, H2O, CH2Cl2, 91%, 2. Ac2O, Py. 99%. 3. TFA, H2O, CH2Cl2, 93%. 4. CH(OMe)3, PPTS. 5. K2CO3, MeOH. 6. TBSCl, DMAP, 98% over 3 steps. 7. DIBAL, 82%. 8. MeI, DEAD, PPh3, 88%.
Coupling of iodide 3a with dithiane 1 then furnished the C6-C35 portion of the PtTXs in high yield (Scheme 3). Elaboration to the requisite Diels-Alder precursor 12a commenced with removal of the orthoester and dithiane deprotection with concomitant formation of the C25-C30 bicyclo-ketal. Oxidation of the C6 hydroxyl then permitted installation of the C1-C5 moiety via a Ni/Cr-mediated coupling.3,13 Alternatively, introduction of the C1-C5 moiety, followed by the C25-C30 bicyclo-ketal formation also afforded 12a, but the material throughput via the sequence of reactions shown in Scheme 3 proved superior.15
Scheme 3.
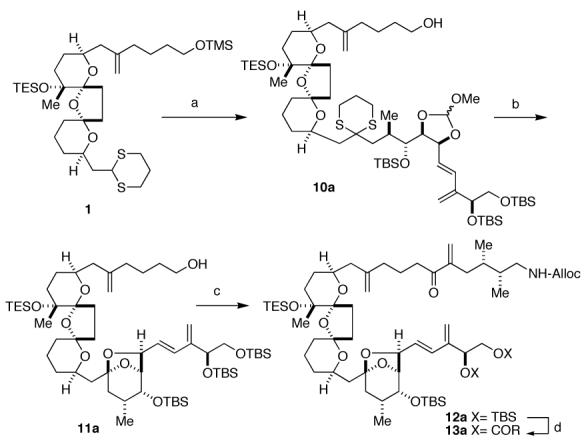
Reagents. (a). 1. 3a, t-BuLi, HMPA. 2. K2CO3, MeOH, 73% over 2 steps. (b). 1. PPTS. 2. K2CO3, MeOH, 73% over 2 steps. 3. PIFA, 74%. (c) 1. SO3·Py, DMSO, 83%. 2. 2, NiCl2, CrCl2, 84%. 3. Dess-Martin oxidation, 79%. (d). 1. HF·Py, Py. 2. RCOCl, NEt3.
The next stage of synthesis was the crucial intramolecular Diels-Alder reaction to form the carbo-macrocycle. Previously, we successfully relied on an intramolecular Diels-Alder to construct the macrocycle of PnTX A.3 However, it is worthwhile noting that the diene used in that series was conjugated with a tert-butyl ester, which exhibited a high tendency for self [4+2] cycloaddition and therefore it was necessary to handle the diene only as a dilute solution. To the contrary, we anticipated, and indeed found, that the diene in the current series shows no tendency of self [4+2] cycloaddition.
We first studied the intramolecular Diels-Alder reaction on substrate 12a in the C34 β-series. Under thermal conditions (dodecane, 170 °C), 12a slowly cyclized, to give a 0.8:1.0:0.8 mixture of the intramolecular Diels-Alder products in 78% combined yield (Table 1). The two exo-products were chemically correlated with the two exo-products in the previous synthesis,3,12 thereby establishing their stereochemistry. However, the stereochemistry of endo-product(s) remains to be established. Upon changing the C34/C35 protecting groups from TBS to acyl groups, the cycloaddition took place noticeably faster.16,17 Interestingly, the exo/endo-selectivity was found to depend sharply on the acyl protecting group, with the benzoate giving the best ratio. The facial selectivity of exo-addition was found also to depend on the protecting group, with the p-methoxybenzoate giving the best ratio. On the balance of the two selectivities, we chose the p-methoxybenzoate substrate for preparative purpose and obtained the desired product 14a-iii in 51% isolated yield.
Table 1.
Intramolecular Diels-Alder Reactions
 | ||||||
|---|---|---|---|---|---|---|
| Temp | A | B | C | D | E | |
| 12a | 170 °C | 0.8 : 1.0 : 0.8 | 78% | 24% | 2.3 : 1.0 | 0.8 : 1.0 |
| 13a-i | 160 °C | 1.4 : 1.0 : 0.3 | >90% | NA | 8.0 : 1.0 | 1.4 : 1.0 |
| 13a-ii | 160 °C | 1.6 : 1.0 : 0.4 | 73% | 37% | 6.5 : 1.0 | 1.6 : 1.0 |
| 13a-iii | 160 °C | 5.2 : 1.0 : 1.2 : 2.0 | 88% | 51% | 1.9 : 1.0 | 5.2 : 1.0 |
| 13a-iv | 160 °C | 3.6 : 1.0 : 1.5 | 50% | NA | 3.1 : 1.0 | 3.6 : 1.0 |
| 13a-v | 160 °C | 1.9 : 1.0 : 1.1 : 0.3 | 93% | NA | 2.1 : 1.0 | 1.9 : 1.0 |
| 13a-vi | 160 °C | 1.3 : 1.0 : 0.6 | 97% | 41% | 3.8 : 1.0 | 1.3 : 1.0 |
| 13a-vii | 160 °C | 1.0 : 1.0 : 1.0 | >90% | NA | 2.0 : 1.0 | 1.0 : 1.0 |
| 13a-viii | 160 °C | 1.0 : 1.0 : 1.0 | >90% | NA | 2.0 : 1.0 | 1.0 : 1.0 |
 | ||||||
|---|---|---|---|---|---|---|
| Temp | A | B | C | D | E | |
| 12b | 185 °C | 1.1 : 1.0 : 0.8 | 52% | 20% | 2.6 : 1.0 | 1.1 : 1.0 |
| 13b-i | 160 °C | 3.6 : 1.0 : 1.0 | 48% | 30% | 4.6 : 1.0 | 3.6 : 1.0 |
| 13b-ii | 160 °C | 1.0 : 1.1 : 3.1 | >90% | 20% | 0.7 : 1.0 | 0.9 : 1.0 |
| 13b-iii | 160 ° | 2.4 : 1.3 : 1.0 | 80% | 36% | 3.7 : 1.0 | 1.8 : 1.0 |
| ” | 130 °C | 3.5 : 2.1 : 1.0 | NA | NA | 5.6 : 1.0 | 1.7 : 1.0 |
| 13b-iv | 160 °C | 1.4 : 1.5 : 1.0 | 76% | NA | 2.9 : 1.0 | 0.9 : 1.0 |
A: product ratio (exo-desired:exo-undesired:endo-1:endo-2). B: combined yield. C: isolated yield of desired exo product. D: exo/endo ratio. E: exo-facial selectivity (desired:undesired).
The overall trend of the intramolecular Diels-Alder reaction in the C34 α-series was similar to that in the β-series, except that the chemical yield in the α-series was lower than the corresponding β-series. In this series, the temperature effect on the exo/endo-selectivity and the facial selectivity of exo-products was tested on the acetate substrate 13b-iii. Interestingly, on lowering the reaction temperature, the exo/endo-selectivity was noticeably improved, whereas the facial selectivity of exo-products remained virtually unchanged.
We next planned to introduce the cysteine moiety via epoxide 15a, which was in turn prepared from the C34/C35-diester 14a-iii. Considering the allylic nature of epoxide, also supported by literature precedents,18 we anticipated that ring-opening could be achieved preferentially at the secondary center in an SN2 fashion, i.e., 15a→16. Experimentally, treatment of 15a with the anion derived from N-Boc-L-Cys(SH)-OCHPh2 yielded a 2:1 mixture of (34R,2′R)-16 and (34S,2′R)-17 in an approximately 85% combined yield. Critically, both 16 and 17 were found to be free from contamination of the corresponding C34- or C2′-stereoisomers,19 thereby demonstrating that the epoxide-ring opening took place in an SN2 fashion and therefore their stereochemistry was assigned as indicated. Similarly, employing the anion derived from N-Boc-D-Cys(SH)-OCHPh2, 15a afforded a 2:1 mixture of two products corresponding to (34R,2′S)-16 and (34S,2′S)-17 in comparable yield. Once again, both products were found stereochemically homogeneous.19
In the pinnatoxin A synthesis, the Diels-Alder product was converted to the natural product in 3 steps, i.e., (1) Pd(PPh3)4, AcOH (deprotection of the Alloc group), (2) 200 °C, 1×10-2 mmHg (imine-cyclization), and (3) TFA/CH2Cl2 (deprotection of the t-Bu ester).3 For the present series, the Alloc-deprotection smoothly took place under the same conditions, to give the desired amino-ketone. Disappointingly, the resultant amino-ketone did not survive under the thermolysis conditions. In our earlier work we found imine-formation under traditional, weakly acidic conditions to be unsuccessful. For example, in the PnTX A synthesis, reaction in the presence of AcOH and Et3N did not promote imine-cyclization at room temperature, whereas undesired N-acetylation was observed at elevated temperatures. However, the instability of amino-ketone in this series to the original thermolysis conditions led us to revisit the imine-cyclization under weakly acidic conditions. Specifically, we searched for weakly acidic conditions under which the undesired N-acylation might be avoided or suppressed; in particular, we focused on combinations of sterically congested carboxylic acids and tertiary amines and eventually found that 2,4,6-(i-Pr)3C6H2CO2H20/Et3N salt meets our needs.
To complete the total synthesis, the only remaining task was to remove the protecting groups of the cysteine-moiety. For two specific reasons, we chose N-Boc-Cys-OCHPh2. First, our previous work3 demonstrated that all the functional groups present in PnTX A, including the seven-membered imine, survive under the TFA/CH2Cl2 (deprotection of the t-Bu ester) conditions. Second, the combination of these two specific protecting groups ensured that the protecting group of the carboxylic acid is cleaved prior to that of the amine.21 Upon treatment with TFA in CH2Cl2 at room temperature, both of the protecting groups were smoothly removed. Finally, preparative LC allowed separation and isolation of pure synthetic (34S,2′R)-PtTX A and (34R,2′R)-PtTX B/C.22 Overall, the epoxide 15a furnished two out of the four possible stereoisomers at C34 and C2′ for both the PtTX A and PtTX B/C series.
Applying the same synthetic sequence on the building block 7b, we were able to synthesize the remaining stereosiomers at C34 and C2′ for both the PtTX A and PtTXs B/C series. However, comparison of NMR spectroscopic data between the synthetic and natural samples did not lead us to the conclusion. In the accompanying paper, we report our efforts to establish the stereochemistry of natural PtTXs A-C, thereby demonstrating that the availability of all possible stereoisomers is essential to rigorously address the problem.23
Supplementary Material
Scheme 4.
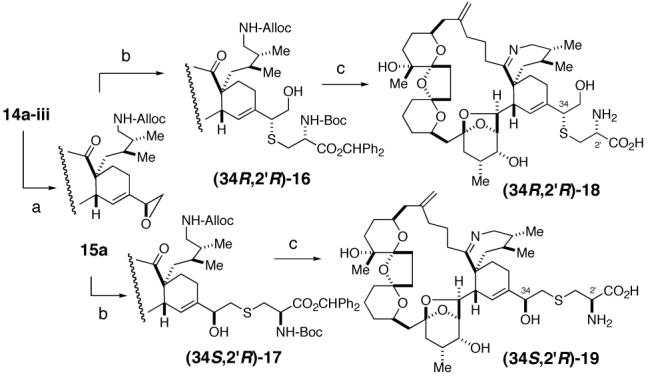
Reagents. (a) 1. K2CO3. 2. HF·Py, Py, 74% over 2 steps. 2. TsCl. 3. K2CO3, 78% over 2 steps. (b) N-Boc-L-Cys(SH)-OCHPh2 (an inseparable mixture of 16 and 17), 85%. (c) 1. Pd(PPh3)4, AcOH, 72%. 2. 1,3,5-(i-Pr)3C6H2CO2H/Et3N salt, 80 °C, xylene. 3. TFA, CH2Cl2, followed by HPLC separation of 18 and 19.
Acknowledgements
Financial support from the National Institutes of Health (NS12108) is gratefully acknowledged. R.P. and S.S.H. thank the German Academic Exchange Service (DAAD) and the National Institutes of Health (NIH NRSA GM19950), respectively, for a postdoctoral fellowship.
References
- 1(a).Uemura D, Chuo T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chen H. J. Am. Chem. Soc. 1995;117:1155. [Google Scholar]; (b) Chuo T, Kamo O, Uemura D. Tetrahedron Lett. 1996;37:4023. [Google Scholar]; (c) Chou T, Haino T, Kuramoto M, Uemura D. Tetrahedron Lett. 1996;37:4027. [Google Scholar]
- 2.For the synthetic work on PnTX, see: (a) Hirama group:Sakamoto S, Sakazaki H, Hagiwara K, Kamada K, Ishii K, Noda T, Inoue M, Hirama M. Angew. Chem. Int. Ed. 2004;43:6505. doi: 10.1002/anie.200461802.Wang J, Sakamoto S, Kamada K, Nitta A, Noda T, Oguri H, Hirama M. Synlett. 2003:891.Ishiwata A, Sakamoto S, Noda T, Hirama M. Synlett. 1999:692.Nitta A, Ishiwata A, Noda T, Hirama M. Synlett. 1999:695.Noda T, Ishiwata A, Uemura S, Sakamoto S, Hirama M. Synlett. 1998:298.(b) Hashimoto group:Nakamura S, Inagaki J, Kudo M, Sugimoto T, Obara K, Nakajima M, Hashimoto S. Tetrahedron. 2002;58:10353.Nakamura S, Inagaki J, Sugimoto T, Ura Y, Hashimoto S. Tetrahedron. 2002;58:10375.Nakamura S, Inagaki J, Sugimoto T, Kudo M, Nakajima M, Hashimoto S. Org. Lett. 2001;3:4075. doi: 10.1021/ol0168364.(c) Murai group:Ishihara J, Horie M, Shimada Y, Tojo S, Murai A. Synlett. 2002:403.Ishihara J, Tojo S, Kamikawa A, Murai A. Chem. Commun. 2001:1392.Sugimoto T, Ishihara J, Murai A. Synlett. 1999:541.Ishihara J, Sugimoto T, Murai A. Synlett. 1998:603.Sugimoto T, Ishihara J, Murai A. Tetrahedron Lett. 1997;38:7379.(d) Kitching group:Suthers BD, Jacobs MF, Kitching W. Tetrahedron Lett. 1998;39:2621.(e) Zakarian group:Pelc MJ, Zakarian A. Org. Lett. 2005;7:1629. doi: 10.1021/ol050321l.
- 3.McCauley JA, Nagasawa K, Lander PA, Mischke SG, Semones MA, Kishi Y. J. Am. Chem. Soc. 1998;120:7647. [Google Scholar]
- 4(a).Takada N, Umemura N, Suenaga K, Chou T, Nagatsu A, Haino T, Yamada K, Uemura D. Tetrahedron Lett. 2001;42:3491. [Google Scholar]; (b) Takada N, Umemura N, Suenaga K, Uemura D. Tetrahedron Lett. 2001;42:3495. [Google Scholar]
- 5.A 1:1 mixture (0.3 mg) of PnTXs B/C was isolated from 21 kg of the viscera of Pinna muricata. PtTX A (20 μg) and PtTX B/C (8 μg as a 1:1 mixture) were isolated from 82 kg of the viscera of Pteria penguin.
- 6.Reported LD99 in Ref. 4: PnTXs B/C=22 μg/kg; PtTX A=100 μg/kg; PtTXs B/C=8 μg/kg).
- 7.The stereochemical homogeneity of PtTX A is discussed in the accompanying paper.
- 8.A total synthesis of PnTXs B and C from the Diels-Alder products 14a-iii and 14b-iii, respectively, will be reported elsewhere.
- 9.The building blocks dithiane 1 and vinyl iodide 2 were reported in Ref. 3. We adopted the reported synthesis for preparation of vinyl iodide 2. However, we have made substantial modifications on the synthesis of dithiane 1; for details, see Supporting Information.
- 10(a).Petrow AA. Zh. Obshch. Khim. 1940;10:1013.(b)5 is known, see:Ohgiya T, Nishiyama S. Heterocycles. 2004;63:2349.
- 11.Faber K. Biotransformations in Organic Chemistry. Springer Verlag; Heidelberg: 2004. [Google Scholar]
- 12.For details, see Supporting Information.
- 13(a).For selected reviews on Cr-mediated reactions, see:Fürstner A. Chem. Rev. 1999;99:991. doi: 10.1021/cr9703360.Wessjohann LA, Scheid G. Synthesis. 1999:1.Nozaki H, Takai K. Proc. Japan Acad. 2000;76:123.Saccomano NA. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 1. Pergamon; Oxford: 1991. p. 173.
- 14.A Ni/Cr-mediated coupling similar to the present case is known:Taylor RE, Ciavarri JP. Org. Lett. 1999;1:467. doi: 10.1021/ol026356s.
- 15.The alternative sequence of reactions for transformation from 10a to 12a were: (a). 1. PIFA. 2. TPAP, NMO. 3. NiCl2, CrCl2, 2. 4. Dess-Martin oxidation, 27% over 4 steps. (b) 1. PPTs. 2. K2CO3, MeOH. 3. PPTs, 63% over 3 steps.
- 16.The substrates 12 and 13 did not survive in the presence of Lewis acids or in the absence of Sumilizer.
- 17.The intramolecular Diels-Alder reaction of 12a and 12b was not observed below the indicated temperature.
- 18(a).Brittain J, Gareau Y. Tetrahedron Lett. 1993;34:3363. [Google Scholar]; (b) Yadav JS, Reddy BVS, Baishya G. Chem. Lett. 2002:906. [Google Scholar]; (c) Fan R-H, Hou X-L. J. Org. Chem. 2003;68:726. doi: 10.1021/jo025983s. [DOI] [PubMed] [Google Scholar]
- 19.This was evident because these stereoisomers were not detected in the corresponding C34 α-series or vice versa.
- 20.Fuson RC, Horning EC. J. Am. Chem. Soc. 1940;62:2962. [Google Scholar]
- 21.We assumed, and indeed observed, that acid-promoted deprotection of the t-Bu ester requires much more forcing conditions, once the amino-protecting group is hydrolyzed.
- 22.Column: YMC-Pack ODS-A, 250×10 mm, Solvent: H2O/CH3CN=3/1 (v/v) containing 0.1% TFA, Flow rate: 2 mL/min, isocratic, Detection: 216 nm.
- 23.Hao J, Matsuura F, Kishi Y, Kita M, Uemura D, Asai N, Iwashita T. J. Am. Chem. Soc. 2006;128 doi: 10.1021/ja061893j. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.