

Abstract
Dicationic Pt(II) complexes containing triphosphine pincer ligands are excellent catalysts for the cycloisomerization of 1,6- and 1,7-dienes into bicyclopropane carbocycles. In analogy to the biosynthetic route to these monoterpene-like compounds, carbocation intermediates are proposed and supported by trapping experiments. Re-activation of the trapped intermediates indicate that cation generation by C-C bond formation is both rapid and reversible.
The generation of carbocations from unsaturated poly-prenoids followed by C-C bond/ring-forming cation-olefin reactions, constitutes the key mechanism for the biosynthesis of most, if not all, of the terpene-derived natural products.1 Under enzyme control, the position of cation generation, the sequence and regioselectivity of the C-C bond forming steps, and the method of cation quenching, all contribute to the skeletal structure and functionality of the cyclization products.
Electrophilic Pt(II) dications supported by a tridentate pincer ligand have recently been shown to catalyze several of the reactions typically associated with terpene biosynthesis (cation generation, cation-olefin, hydride shift, cyclopropanation).2 Key to the activity of these catalysts is their highly electrophilic, but also carbophilic, electronic structures, coupled with the all cis pincer ligands, which block β-hydride elimination in any Pt-alkyl intermediates. The result is a buildup of intermediates that follow pathways not otherwise competitive in catalysts capable of facile β-H elimination. In this communication, we report that a family of 1,5- and 1,6-dienes reacts under Pt-catalysis in a fashion that seemingly parallels the biosynthesis of the [3.1.0] bicyclic monoterpenes. Additionally, we disclose a trapping experiment that supports the intermediacy of organometallic carbocations.
The [3.1.0] bicyclic skeleton is the key structural feature of many monoterpenes (thujane, thujene, sabinene, sabinene hydrate, sabina ketone, etc), which are found in numerous essential oils and consequently find importance in flavors and fragrances.3 Considerable effort has been devoted to the construction of this ring system, and the available methods include both intra-4 and inter-molecular5 addition of carbenoids to alkenes, intramolecular alkylation of enolates,6 a tandem Li-ene cyclization/thiophenoxide elimination,7 a Ti-mediated enyne cyclization,8 and an unusual rearrangement after reaction of silylketene acetals with bromoform-diethylzinc.9 Each of these approaches require multiple steps, and usually lead to oxygenated monoterpenes that are most amenable to the synthesis of the oxidized congeners. 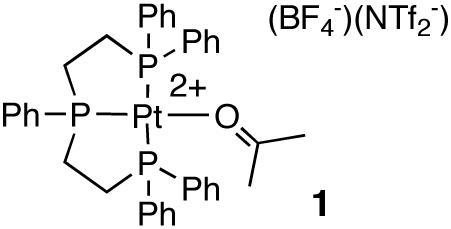
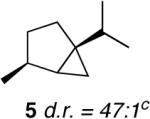
Based on previous cycloisomerization studies with 1, we postulated that it should also be capable of directly providing the [3.1.0]-bicyclic core10 from simple 1-6-dienes. Gratifyingly, a variety of dienes with the substitution pattern shown in Table 1 are converted to the saturated bicyclic products when exposed to 5 mol% of 1. In the case of the 1,6-diene β-citronellene (4), a highly diastereoselective rearrangement occurs and the natural product cis-thujane11 (5) is obtained in 47:1 d.r.
Table 1.
Cycloisomerization of dienes with 5 mol% of 1a
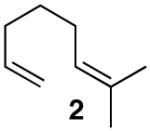
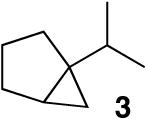
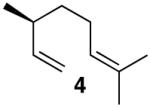

1 generated in situ from [(PPP)PtMe]BF4, acetone (1 equiv), and HNTf2 in MeNO2, 5% catalyst loading, 40°C. See Supporting Information.
isolated.
by GC.
23 °C.
by GC; successive chromatography afforded pure 11 in 5% isolated yield.
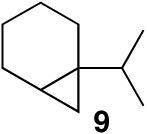
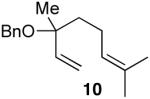
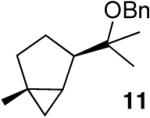
In keeping with the high carbophilicity of these strong Lewis acids, ester-containing compounds did not poison the catalyst and the expected product was obtained (entry 3). The [4.1.0] bicyclic compound 9 could also be obtained but at a decreased rate (entry 4). A [3.1.0] bicyclic product was also obtained from the cycloisomerization of O-Bn linalool 10 (entry 5). In this case, the major product resulted from allylic isomerization to a mixture of O-Bn geraniol/nerol, however a single diastereomer of 11 was also obtained in low yield. A skeletal rearrangement has obviously taken place that will require additional study and optimization. Despite the moderate isolated yields,12 these one-step reactions represent the most efficient syntheses of the [3.1.0] bicyclic core from simple acyclic starting materials.
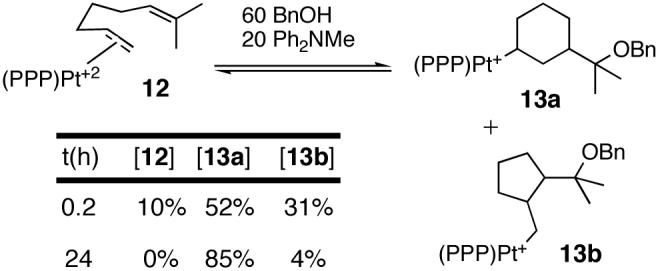
The initiating step in the cycloisomerization reactions was presumed to involve electrophilic activation of the terminal alkene and intramolecular addition of the trisubstituted alkene to cyclogenerate intermediate carbocations that were stable to β-H elimination.2 These putative Pt(II)-alkyl carbocations had not been observed directly, but we hoped that a trapping nucleophile could intercept them for characterization. In the event, excess benzyl alcohol and Ph2NMe rapidly converted 12, the catalysts resting state,13 to a mixture of endo- and exocyclic Pt-alkyls.14,15,16 The composition of this mixture changed with time to ultimately favor the endocyclic product 13a (see inset, Scheme 1).17 Characterization of the organic fragment was achieved by reductive removal with NaBH4.15,18,19
Scheme 1.
Trapping of carbocationic intermediates
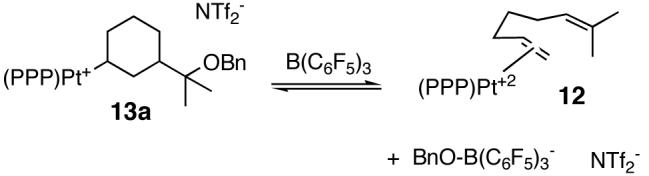
The data in Scheme 1 indicate that C-C bond formation was not only reversible but that both endo- and exocyclic regiochemistries were kinetically viable. When 13a was treated with one equiv of the strong Lewis acid B(C6F5)3, benzyloxide abstraction occurred concomitant with retro-cyclization to 12 (eq 1) (13a:12 ∼3:1 at early times).20 This mixture is eventually driven to 3 and (PPP)Pt(OBn)+.21 The observation that 13a returns to the catalyst resting state (12) upon -OBn abstraction, and the time-dependent ratio of 13a/13b together suggest that cyclization is rapid and reversible and that a post-cyclization step (H- shift or cyclopropanation) is turnover-limiting.
 |
(1) |
Since both endo- and exo- cyclization geometries are demonstrably viable, two plausible mechanisms for the cycloisomerization of 2 to 3 must be considered (Scheme 2). The two scenarios differ in the timing of ring construction, with the 6-endo pathway (b) most closely following the sequence of thujane biosynthesis.3 Distinguishing the two mechanisms may require computational help, but in both cases the cyclopropanation event is proposed to proceed via a key carbocation that is γ to the metal (from a 1,2-hydride shift of the kinetic cation). Related, stereospecific, double inversion processes are known for Sn,22 Fe,23 and Ti24 alkyls with γ-carbocations.25
Scheme 2.
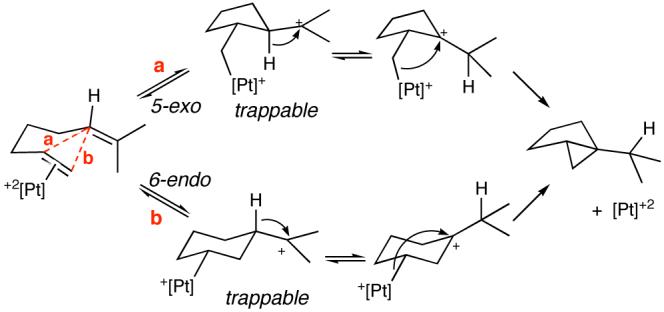
We have described the cycloisomerization of dienes to [3.1.0]- and [4.1.0] bicyclic compounds, including the one-step synthesis of the natural product cis-thujane. The catalyst for these reactions, a Lewis-acidic Pt(II) pincer complex, activates alkenes for cation-olefin cyclizations as evidenced by the trapping of intermediate organo-platinum carbocations.
Supplementary Material
Acknowledgment
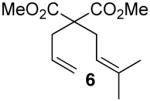
We thank Mr. Aujin Kim for synthesizing 6, Dr. Gary Glish for performing CI MS and gratefully acknowledge the NIGMS (GM60578) for funding. W.D.K thanks Glaxo-Smith-Kline for a graduate fellowship. M.R.G. is a Camille-Dreyfus Teacher-Scholar.
References
- (1).Porter JW, Spurgeon SL, editors. Biosynthesis of Isoprenoid Compounds. Vol. 1. John Wiley & Sons; New York: 1981. [Google Scholar]
- (2)(a).Hahn C, Cucciolito ME, Vitagliano A. J. Am. Chem. Soc. 2002;124:9038. doi: 10.1021/ja0263386. [DOI] [PubMed] [Google Scholar]; (b) Kerber WD, Koh JH, Gagné MR. Org. Lett. 2004;6:3013. doi: 10.1021/ol048780u. [DOI] [PubMed] [Google Scholar]
- (3)(a).Croteau R. Chem. Rev. 1987;87:929–954. [Google Scholar]; (b) Croteau R. Proc. of the Int. Haarman Reimer Symp., 3rd; Recent Dev. Flavor Fragrance Chem; 1993.p. 263. [Google Scholar]
- (4)(a).Hodgson DM, Chung YK, Paris JM. J. Am. Chem. Soc. 2004;126:8664. doi: 10.1021/ja047346k. [DOI] [PubMed] [Google Scholar]; (b) Harrak Y, Blaszykowski C, Bernard M, Cariou K, Mainetti E, Mouries V, Dhimane A-L, Fensterbank L, Malacria M. J. Am. Chem. Soc. 2004;126:8656. doi: 10.1021/ja0474695. [DOI] [PubMed] [Google Scholar]; (c) Barberis M, Perez-Prieto J. Tetrahedron Lett. 2003;44:6683. [Google Scholar]; (d) Mori K, Ohki M, Matsui M. Tetrahedron. 1970;26:2821. doi: 10.1016/s0040-4020(01)92926-6. [DOI] [PubMed] [Google Scholar]; (e) Vig OP, Bhatia MS, Gupta KC, Matta KL. J. Ind. Chem. Soc. 1969;46:991. [Google Scholar]
- (5)(a).Galopin CC. Tetrahedron Lett. 2001;42:5589. [Google Scholar]; (b) Cheng D, Kreethadumrongdat T, Cohen T. Org. Lett. 2001;3:2121. doi: 10.1021/ol016086y. [DOI] [PubMed] [Google Scholar]; (c) Fanta WI, Erman WF. J. Org. Chem. 1968;33:1656. [Google Scholar]; (d) Guha PC, Krishnamurthy S. Chem. Ber. 1937;70B:2112. [Google Scholar]
- (6)(a).Hamon DPG, Shirley NJ. Aust. J. Chem. 1987;40:1321. [Google Scholar]; (b) Gaoni Y. Tetrahedron. 1972;28:5525. [Google Scholar]; (c) Alexandre C, Rouessac F. Bull. Chem. Soc. Jpn. 1972;45:2241. [Google Scholar]; (d) Nelson NA, Mortimer GA. J. Org. Chem. 1957;22:1146. [Google Scholar]
- (7).Cheng D, Knox KR, Cohen T. J. Am. Chem. Soc. 2000;122:412. [Google Scholar]
- (8).Urabe H, Suzuki K, Sato F. J. Am. Chem. Soc. 1997;119:10014. [Google Scholar]
- (9).Rousseau G, Slougui N. J. Am. Chem. Soc. 1984;106:7283. [Google Scholar]
- (10)(a).Ene-yne cycloisomerization with PtCl2 and Au(III) catalysts can also generate (through different mechanisms) related bicyclopropane structures. See for example:Harrak Y, Blaszykowski C, Bernard M, Cariou K, Mainetti E, Mouries V, Dhimane A-L, Fensterbank L, Malacria M. J. Am. Chem. Soc. 2004;126:8656. doi: 10.1021/ja0474695.Mamane V, Gress T, Krause H, Fürstner A. J. Am. Chem. Soc. 2004;126:8654. doi: 10.1021/ja048094q.Fürstner A, Hannen P. Chem. Commun. 2004:2546. doi: 10.1039/b412354a.Nieto-Oberhuber C, Munoz MP, Bunuel E, Nevado C, Cardenas DJ, Echavarren AM. Angew. Chem., Int. Ed. 2004;43:2402. doi: 10.1002/anie.200353207.Luzung MR, Markham JP, Toste FD. J. Am. Chem. Soc. 2004;126:10858. doi: 10.1021/ja046248w.
- (11).Rees JC, Whittaker D. Org. Mag. Res. 1981;15:363. [Google Scholar]
- (12).The balance of material in these reactions is usually alkene-containing isomers that appear to originate from the consumption of cyclopropane.
- (13).The trapping reactions are complete within minutes as observed by 31P NMR.
- (14).This nomenclature refers to the position of the electrophilic alkene in the C-C bond forming transition state.
- (15).Both 6-exo and 6-endo cyclization modes were observed in stoichiometric cascade cyclization of poly-enes. See:Koh JH, Gagné MR. Angew. Chem., Int. Ed. 2004;43:3459–3461. doi: 10.1002/anie.200453913.
- (16)(a).For catalytic asymmetric alkoxycyclization of enynes with Pt(II) and Au(I), see:Charruault L, Véronique M, Taras R, Gladiali S, Genêt J-P. Chem. Commun. 2004:851. doi: 10.1039/b400908h.Muñoz MP, Adrio J, Carretero JC, Echavarren AM. Organometallics. 2005;24:1293.
- (17).The balance of the material was two Pt-alkyls that remain uncharacterized.
- (18).Hahn C, Morvillo P, Vitagliano A. Eur. J. Inorg. Chem. 2001:419. [Google Scholar]
- (19).A single diastereomer was obtained from reductive cleavage of 13b. Although trans relative stereochemistry seems most likely, this has not been conclusively determined.
- (20).A similar -OBn abstraction likely mediates the isomerization of 10 to O-Bn geraniol/nerol (entry 5, Table 1).
- (21).As 2 is consumed, 1 abstracts alkoxide from boron to generate [(PPP)PtOBn]+.
- (22)(a).Davis DD, Johnson HT. J. Am. Chem. Soc. 1974;96:7576. [Google Scholar]; (b) Fleming I, Urch CJ. Tetrahedron Lett. 1983;24:4591. [Google Scholar]; (c) McWilliam DC, Balasubramanian TR, Kuivila HG. J. Am. Chem. Soc. 1978;100:6407. [Google Scholar]; (d) Lambert JB, Salvador LA, So JH. Organometallics. 1993;12:697. [Google Scholar]
- (23)(a).Casey CP, Smith LJ. Organometallics. 1989;8:2288. [Google Scholar]; (b) Casey CP, Smith Vosejpka LJ. Organometallics. 1992;11:738. [Google Scholar]; (c) Brookhart M, Liu Y, Goldman EW, Timmers DA, Williams GD. J. Am. Chem. Soc. 1991:927. [Google Scholar]; (d) Brookhart M, Liu Y. J. Am. Chem. Soc. 1991:939. [Google Scholar]
- (24).Casey CP, Strotman NA. J. Am. Chem. Soc. 2004;126:1699. doi: 10.1021/ja030436p. [DOI] [PubMed] [Google Scholar]
- (25).An alternative possibility is direct metallation to form a Pt(IV) metallacyclobutane followed by reductive elimination. Evidence disfavoring this mechanism include the observation of cyclopropane products with related Pd(II) catalysts, which would implicate the involvement of a Pd(IV) intermediate, see ref. 2b. However, this possibility has not been rigorously disproven.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.