

Abstract
It has been proposed that occult, disseminated metastatic cells are refractory to chemotherapy due to lack of proliferation. We have shown that p38 activation induces dormancy of squamous carcinoma cells. We now show that p38 signaling in these cells activates a prosurvival mechanism via the up-regulation of the endoplasmic reticulum (ER) chaperone BiP and increased activation of the ER stress–activated eukaryotic translation initiator factor 2α kinase RNA-dependent protein kinase–like ER kinase (PERK) allowing dormant tumor cells to resist drug toxicity. RNA interference and dominant-negative expression studies revealed that both BiP and PERK signaling promote survival and drug resistance of dormant cells, and that BiP up-regulation prevents Bax activation. We propose that stress-dependent activation of p38 via BiP up-regulation and PERK activation protects dormant tumor cells from stress insults, such as chemotherapy.
Introduction
Most patients with inoperable primary cancer, with or without overt metastases, or patients with undetected disseminated disease undergoing surgery for their primary cancer, are not cured by adjuvant chemotherapy. The failure to cure occurs even in patients in which objective shrinkage of the tumor occurs in response to chemotherapy. It is believed that chemotherapy itself may induce resistance to killing (e.g., through the induction of senescence; ref. 1), or that preexisting noncycling dormant cells escape most chemotherapy, because it is aimed at proliferation (2). The mechanisms that cause malignant tumor cells to exit proliferation and activate a survival and G0-G1 arrest “mode” are not well understood, and the assumption that the lack of proliferation of dormant cells is the only reason for their resistance to chemotherapy remains to be proven.
The elucidation of the molecular basis of dormancy is of fundamental interest. We have shown that a rapidly tumorigenic and spontaneously metastasizing human carcinoma (T-HEp3) that is passaged for a prolonged period of time in culture undergoes a nonclonal change reflected in lower extracellular signal-regulated kinase (ERK) activity, increased p38 activity, and a dormant/quiescent phenotype in vivo (D-HEp3; refs. 3, 4). We define tumorigenicity as the ability of cells to promptly grow and form sizeable tumors upon first or serial inoculations in vivo. In contrast, dormancy represents the inability of cells to behave in this manner yet be able to form small nodules in which cells are in a cell cycle arrest or in which the proliferative and apoptotic rates are similar resulting in little overall gain in cell number over time (5, 6). Surprisingly, T-HEp3 and D-HEp3 cells grow at comparable rates in vitro despite their differences in p38 and ERK signaling, suggesting that these differences affect growth only in the in vivo environment (4, 7). We have suggested that this apparent discrepancy may result from inherently different signaling thresholds for in vitro versus in vivo growth (8), and a recent study showed that although inducible activation of p38 in cancer cells can suppress tumorigenicity in vivo, it does not affect in vitro growth (9). In contrast to the in vitro growth rate, which remains unchanged (8), >80% of D-HEp3 cells when inoculated in vivo rapidly arrest in G0-G1 by day 6 after inoculation and remain dormant for several months (6, 8). We also found that activated p38 establishes a negative feedback loop to ERK, and that blocking of p38 by genetic or pharmacologic inhibitors restores ERK activation and interrupts tumor dormancy in vivo (4). From an experimental point of view, the ability to study, in vitro, signaling properties that predict tumor cell behavior in vivo is highly advantageous as the recovery of small numbers of dormant cells from animals is extremely difficult. We have validated the merits of our in vitro model of dormancy by documenting, based on ERK- and p38-regulated green fluorescent protein (GFP) reporter systems, that the signaling differences recorded in vitro reflect the actual signaling properties of tumorigenic and dormant cells in vivo (10).
Based on our studies using in particular HEp3 cells, we have implicated high p38 activity in the induction of dormancy in vivo (4, 7, 10). Although p38 is known to induce growth arrest (11) and/or apoptosis (12), there is also evidence indicating that in some instances, p38 signaling can promote cell survival (13-16). However, knowledge of the proximal targets of p38 that underlie the p38-dependent dormancy program, in particular the balance between cell proliferation and cell death, have not been identified.
A pathway that can induce concomitant growth arrest and survival is that activated by stress to the endoplasmic reticulum (ER), such as the unfolded protein response (UPR; ref. 17). This ER stress pathway induces both G0-G1 arrest and survival when specific transmembrane proteins are activated when the protein folding capacity of this organelle is compromised (18, 19). Viral infections, nutrient deprivation, and hypoxia can all induce an ER stress response (19, 20) that allows for proper protein folding by up-regulating chaperones, such as BiP/Grp78 (21). Under nonstress conditions, BiP is bound to the ER-lumenal domains of at least two transmembrane kinases, RNA-dependent protein kinase–like ER kinase (PERK) and inositol-requiring enzyme 1α (IRE1α), preventing their activation (19). Following excessive accumulation of newly synthesized or unfolded proteins in the ER, BiP preferentially binds to unfolded proteins dissociating from PERK and IRE1α, thereby rendering them active (19). While PERK phosphorylates eukaryotic translation initiator factor 2α (eIF2α) attenuating protein synthesis (21-23), IRE1α splices a 26-nucleotide intron from XBP-1 mRNA, resulting in an active transcription factor required to induce ER stress response genes, such as chaperones (19, 24), thereby promoting survival under stress. In addition, activation of PERK can specifically inhibit the synthesis of cell cycle regulators, such as cyclin D1, which results in a cell cycle arrest (G1; ref. 25). Thus, through PERK and chaperones, such as BiP, cells coordinate growth arrest and survival signals (23, 25, 26). Hence, the ER stress response may thus be said to cause a “dormancy-like state” in which cells are quiescent and survival competent, allowing them to resist stress-induced death.
Because we established that increased p38 activity was a hallmark of in vivo growth arrest in many cancer cells lines (7), we thought to explore whether this stress-activated protein kinase was responsible for a downstream program that resembled ER stress and whether preactivation of ER stress pathway was responsible for decreased sensitivity of dormant cells to chemotherapy and their better survival.
Here, we present evidence that p38-dependent activation of PERK and up-regulation of BiP, two regulators of the stress response, renders these cells resistant to drug-induced apoptosis. Furthermore, we show that BiP up-regulation is required to inhibit Bax activation and chemotherapeutic drug resistance. These findings may provide a mechanistic explanation for the common clinical observation of residual secondary disease following chemotherapy.
Materials and Methods
Reagents and antibodies
SB203580 and etoposide were from Calbiochem (Beverly, MA). Thapsigargin and doxorubicin were from Sigma (St. Louis, MO), and tunicamycin was from ICN Biomedicals, Inc. (Aurora, OH). Rabbit anti-phospho-eIF2α (Ser51) was from Biosource (Camarillo, CA); anti-phospho-PERK (Thr980), anti total eIF2α, anti-cleaved caspase-3, and anti-Bax antibodies were from Cell Signaling (Beverly, MA). Anti XBP-1 pAb (anti-XBP-1 and M-186) and anti-PERK (H-300) were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Bax and anti-BiP monoclonal antibodies (mAb) were from BD Biosciences (San Jose, CA; clone 6A7); mouse anti-IgG1 and Alexa 488 conjugated anti-mouse IgG were from Sigma and Molecular Probes (Eugene, OR), respectively. Anti-GADPH was from Ambion (Austin, TX). Horseradish peroxidase (HRP)–conjugated anti-mouse IgG antibody and mounting media were from Vector Laboratories (Burlingame, CA). Anti-rabbit IgG antibody conjugated with HRP was from Chemicon International (Temecula, CA).
Cell lines, cell proliferation, and viability assays
Tumorigenic (T-HEp3) and “spontaneous” dormant (D-HEp3) human epidermoid carcinoma HEp3 cell (3), D-HEp3-neo, and D-HEp3-p38 DN cell lines were described previously (4). Proliferation studies were done as described previously (8). For viability experiments, cells were plated (7.5 × 104 to 2 × 104 per well) and cultured overnight. After 24 hours, cells were treated with either tunicamycin or thapsigargin for 16 hours or etoposide for 40 hours or doxorubicin for 30 hours. The percentage of viable cells was estimated by trypan blue staining.
Proteomics
T-HEp3 cells were grown in serum-free medium, or D-HEp3 cells were treated with or without SB203580 (5 μmol/L) for 48 hours in serum-free medium. Protein extraction for proteomic analysis was done for each cell line to produce “cytosolic” (F1) and “membrane/nuclear” (F2) fractions (Ready Prep Bio-Rad, Hercules, CA) as per vendor’s instruction. These fractions were separated in nonequilibrium pH gradients for the first dimension, and then these gradients were separated by SDS-PAGE (second dimension) as reported (27). Gels were stained simultaneously with a mass spectrometry (MS)–compatible silver staining protocol (http://cpmcnet.columbia.edu/dept/protein/), and to exclude artificial differences in spot intensity among the different gels, protein extracts were run at least in triplicate two-dimensional gels. Software-based analysis (Melanie II and NIH image) of the two fractions revealed ~1,000 protein spots, which mostly represent medium and high abundance proteins (data not shown). We identified ~60 proteins spots that were changing in abundance upon inhibition of p38 signaling in D-HEp3 cells and between T-Hep3 and D-HEp3 cells in both F1 and F2 fractions. These changes were highly reproducible among three independent experiments, and protein spots that changed (increase/decrease) reproducibly were excised from the gels and stored at −80°C for further matrix-assisted laser desorption/ionization (MALDI)-MS analysis. All spots were reduced and alkylated with iodoacetamide, digested with 5:1 endoproteinase Lys-C/trypsin, and analyzed by MALDI-MS. Peptide masses were analyzed using ProFound and MS-digest software querying NCBInr and TrEMBL databases (Table 1) done in the Columbia University Protein Core and DNA Sequencing Facility/HHMI. All protein spot changes were normalized to the spot corresponding to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was identified by MALDI-MS and was unaffected by p38 inhibition.
Table 1.
Targets of p38 signaling identified through proteomics
| Protein matched | Accession no. | Protein mass (kDa)/pI* | Regulation by active p38 (fold change) | % Tryptic peptides matched | % Sequence coverage of peptides |
|---|---|---|---|---|---|
| BiP/GRP78 | 6470150† | 71/5.22 | Increase (3.5), F1 | 17.3 | 28.6 |
| PDI (ER60) | 1085373† | 56.7/6.1 | Increase (2.5), F1 | 12 | 21 |
| Cyclophilin B | Q9VK5‡ | 23.7/9.42 | Increase (2.5), F2 | 19.3 | 34.25 |
| HSP47 | 8574445† | 46.4/8.75 | Increase (3), F1 | 37.8 | 41.8 |
NOTE: MALDI/MS and protein ID from two-dimensional gel spots was done with NS-Fit and Pro-Found algorithm. F1 = cytosolic fraction; F2 = nuclear/membrane fraction.
Calculated values.
NCBInr database.
TrEMBL database.
Transient cDNA and small interfering RNA transfections
Cells were transiently transfected with either pCAX-F-XBP1-venus plasmid expressing XBP1-venus fusion protein (28) or pEGFP alone. In addition cells were also transfected with pGADD153-EGFP reporter (a gift from Dr. S. Howell, University of California, San Diego) alone or together with one of the following plasmids: pFLAG-CMV-2-GADD34, pcDNA-PERK C-term-myc, pcDNA-PERK WT-myc (kindly provided by Dr. David Ron, New York University; ref. 29), MMK6b(E) active mutant, or DN-p38 mutant (7) using FuGene transfection reagent (Roche, Indianapolis, IN). GFP expression was detected 48 hours after transfection by fluorescence-activated cell sorting (FACS). Transfection of pGEM3-BiP-Luc reporter construct was carried out as described (7). For small interfering RNA (siRNA) transfections, 24 hours before transfection, D-HEp3 cells were seeded at a density of 5 × 105 (50-60% confluence) per 6-cm dish, and 24 hours later, cells were transfected with 100 nmol/L of GAPDH or BiP siRNA or no siRNA as a control using siPORT (Ambion) reagent. Cells were retransfected 3 days later as indicated above and harvested 2 days later to extract total protein as described below.
FACS analysis
In all experiments, GFP fluorescence derived from reporter constructs was quantitated using Epics ALTRA flow cytometer (Beckman Coulter, Chaska, MN). Data analysis was done as previously described (30). Conformational changes in Bax was detected as previously described (31). Apoptosis was detected using a terminal deoxytransferase–mediated nick-end labeling assay kit (TUNEL, Apo-Direct) from BD PharMingen (San Diego, CA). Briefly, cells were fixed and labeled with FITC-dUTP followed by staining with propidium iodide following the recommended protocol. The cell populations positive for FITC-dUTP–labeled DNA breaks were quantitated in propidium iodide versus FITC density plots. Data analysis was done using the Expo-32 software (Beckman Coulter).
Immunoblotting
Cells were washed with PBS and lysed in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors and were then analyzed by Western blot as described previously (4).
Immunofluorescence microscopy
Analysis of Bax activation and active caspase-3 by immunofluorescence was done as previously described (4, 7). In all cases, images were captured using a Zeiss Axioskop epifluorescence microscope (Oberkochen, Germany) using Plan-Neofluar ×40 and ×100 (numerical aperture, 1.5 oil) lenses (Zeiss). Images were digitalized with an Axiocam (Zeiss) digital camera using a fixed gain and aperture time at room temperature. Raw images were visualized and analyzed using Adobe Photoshop 6.0 and ImageJ software.
Results
Proteomic identification of p38 targets in D-HEp3 cells
Previous studies from our laboratory have shown the importance of p38 signaling to the maintenance of the dormancy of HEp3 cells in vivo (4, 7), but the downstream targets of this pathway are not yet characterized. Thus, we compared the proteomes of parental T-HEp3 human squamous carcinoma cells with low p38 activity, which form rapidly growing tumors in vivo to spontaneously dormant HEp3 (D-HEp3) cells, which have high p38 activity, arrest in G0-G1 48 to 72 hours after inoculation, and form small dormant tumor nodules in vivo (4, 7). In addition, we also compared the proteomes of control D-HEp3 cells with those treated for 48 hours with a p38 inhibitor SB203580 (5 μmol/L), or D-HEp3 clones stably transfected with an “empty” vector (D-Neo) with clones stably expressing dnp38α (D-dnp38). Our analysis revealed ~60 protein spots representing medium and high abundance proteins that changed in intensity upon inhibition of p38 in D-HEp3 cells. Spots for which the mean absorbance changed significantly (P < 0.05) upon p38 inhibition were analyzed by MALDI-MS. We identified four of these spots as BiP (ATPase-chaperone), ER60 (disulfide isomerase), HSP47 (collagen chaperone), and cyclophilin B (isoprolyl-isomerase; Table 1; Fig. 1A); all molecular chaperones induced during an unfolded protein response. Furthermore, we found that these proteins were significantly down-regulated in parental tumorigenic T-HEp3 cells compared with D-HEp3 cells (Fig. 1A). Thus, high p38 activity is associated with high ER chaperone protein levels.
Figure 1. Proteomic identification of targets of p38 signaling.

A, quantitation of protein spot abundance normalized to GAPDH values by densitometry. Two-dimensional gel spots corresponding to BiP, ER60, HSP47, and cyclophilin B from representative gels were processed as described in Materials and Methods. Columns, mean absorbance obtained using NIH image software and statistically validated using t test (GraphPad Prism, software); bars, SD. P is indicated on the columns corresponding to D-HEp3 and compared with T-HEp3 and D-HEp3 cells treated for 48 hours with 5 μmol/L SB203580. From at least triplicate independent gels. B, Western blot showing that BiP is up-regulated in D-Hep3 versus T-HEp3 cells. ERK was used as a loading control. Numbers on top, normalized BiP/ERK absorbance for each BiP band. C, BiP promoter activity in T-Hep3 versus D-HEp3 cells. Cells were transfected with a pGEM-3 construct encoding firefly luciferase under the control of −350 to +50 of the BiP promoter. Firefly luciferase activity was normalized to Renilla luciferase activity driven by a cytomegalovirus promoter. Columns, mean of triplicate determinations; bars, SD. D, Western blot showing that BiP is down-regulated in D-HEp3 cells by 5 μmol/L SB203580 treatment for 24, 48, and 72 hours, respectively. GAPDH was used as a loading control.
Western blot analysis confirmed that BiP was indeed up-regulated in D-HEp3 cells than in T-HEp3 cells (Fig. 1B). Furthermore, the activity of a BiP promoter-driven luciferase reporter construct was ~3-fold higher in D-HEp3 relative to T-HEp3 cells, suggesting that the higher levels of BiP in D-HEp3 cells result in part from higher transcription rates (Fig. 1C). In addition, this up-regulation was increasingly inhibited 24 to 72 hours following inhibition of p38 by 5 μmol/L SB203580 (Fig. 1D). The basal expression of BiP in T-HEp3 cells was unaffected by treatment with SB203580, indicating that basal p38 activity is dispensable for BiP expression in T-HEp3 cells (data not shown). Taken together with the proteomic data, these results show that there is a strong association between high p38 activity and high protein expression of BiP, a prototypical stress-induced ER chaperone.
Phosphorylation of PERK and eIF2α in D-HEp3 versus T-HEp3 cells
We next tested whether classic transducers of ER stress signals (i.e., PERK and eIF2α) were active in T-Hep3 and D-HEp3 cells. The activity of the PERK signaling pathway was tested using phosphospecific antibodies to PERK and eIF2α and normalized to total PERK and eIF2α levels, respectively. Basal PERK activation was ~3-fold greater in D-HEp3 relative to T-HEp3 cells (Fig. 2A, compare lanes 1 and 4), which correlated with increased phosphorylation of eIF2α at Ser51 in D-HEp3 cells (Fig. 2B, compare lanes 1 and 3). Treatment of D-Hep3 and T-HEp3 cells with tunicamycin for 4 hours resulted in a 1.6- and 3-fold increase, respectively, in normalized phospho-PERK levels (Fig. 2A). Although such a treatment had no effect on phospho-eIF2α levels in D-HEp3 cells, it resulted in a ~2.5-fold increase in phospho-eIF2α in T-HEp3 cells (Fig. 2B). In addition, PERK activation was p38 dependent as expression of dnp38, or treatment with SB203580, decreased phospho-PERK levels (Fig. 2C). Our results show that D-HEp3 cells have a p38-dependent increase in PERK signaling. Increased PERK phosphorylation in D-HEp3 seems to result in higher eIF2α phosphorylation compared with T-HEp3 cells.
Figure 2. Detection of PERK and eIF2α phosphorylation.

A, T-Hep3 and D-HEp3 cells were treated for the indicated time points with 5 μg/mL tunicamycin (Tn) and then lysed with radioimmunoprecipitation assay lysis buffer and analyzed by Western blot for phospho-Thr980 (p-Thr980)-PERK and for PERK, which served as a loading control. Absorbance (O.D.) numbers are indicated below the PERK blot and are the result of normalizing phospho-PERK (p-PERK) to PERK.B, control and tunicamycin-treated cells were lysed with radioimmunoprecipitation assay lysis buffer and analyzed by Western blot for phospho-Ser51 (p-Ser51)-eIF2α and eIF2α levels, which served as a loading control. C, lysates prepared with radioimmunoprecipitation assay lysis buffer were Western blotted for phospho-PERK from the indicated cell lines (top) or for phospho-PERK and ERK (middle and bottom) in D-HEp3 cells treated with or without 5 μmol/L SB203580 for 24 hours. ERK was used as a loading control. T, T-HEp3; D-neo and Ddnp38, D-Hep3 cells stably expressing an “empty” vector or a vector encoding a dominant-negative p38α. D and E, T-Hep3 and D-HEp3 cells were transiently cotransfected with the GADD153-EGFP reporter plasmid and the indicated plasmids (GADD34 or PERKΔC) (D) or were transfected with pEGFP alone (E). GADD153-induced EGFP was analyzed 48 hours after transfection by fluorescence microscopy (D, left) or FACS, where a total 5 × 104 events were captured and the GFP fluorescence in FLH2>10 was plotted in a histogram (D, right). Constitutive EGFP expression was also analyzed by fluorescence microscopy as before, and the percentage of EGFP-expressing cells in FLH2>10 was plotted (D) Bar, 180 μm. F, cells cotransfected as in (D) with DNp38 or MKK6Eb plasmids were processed for FACS analysis. Columns, mean number of GFP-positive events in FLH2>10; bars, SD.
To monitor downstream signaling from PERK and p38, we examined whether these signals resulted in activation of ER stress response element (ERSE) containing GADD153 promoter. To this extent, D-Hep3 and T-HEp3 cells were transfected with a GFP reporter driven by GADD153 promoter, and the number of GFP expressing cells was recorded by FACS analysis. D-HEp3 cells displayed significantly higher GADD153 promoter activity than T-HEp3 cells (Fig. 2D). This differential activation was not due to a difference in transfection efficiency between T-Hep3 and D-HEp3 cells, as it is also maintained following normalization to the cotransfected dsRED protein expression in these cells (Fig. 2E). Overexpression of either GADD34, the regulatory subunit of the eIF2α phosphatase PP1, or PERK-ΔC, a COOH-terminal deletion mutant of PERK that lacks kinase activity and functions as a dominant-negative, down-regulated GADD153 promoter activation (Fig. 2D). The above results confirm that signals activated by PERK can lead to GADD153 promoter activation in D-HEp3 cells. That GADD34 reduces this activation suggests that it is dependent on eIF2α phosphorylation. Moreover, inhibition or hyperactivation of p38 signaling pathway by the coexpression of a dnp38α or an active mutant of MKK6 (MKK6Eb) resulted in a strong inhibition or activation, respectively, of GADD153 promoter activity (Fig. 2F). These experiments further support that an ERSE-containing promoter, such as GADD153, can be activated in D-HEp3 cells by an MKK6-p38 signal.
Chemotherapy resistance of D-HEp3 and T-HEp3 cells
Tumor dormancy has been associated with chemotherapy resistance, a phenomenon that is mainly attributed to proliferative quiescence (2). However, the possibility that other mechanisms, specifically survival mechanisms, may contribute to resistance has not been explored to date. We therefore tested whether our dormancy cell models exhibited chemotherapy resistance and, if so, whether this resulted from a failure of chemotherapy-induced apoptosis. To test for chemotherapy resistance, cell cultures were treated with 10 to 80 μmol/L etoposide (topoisomerase II inhibitor) or with 1 to 10 μg/mL doxorubicin, an intercalating agent and topoisomerase II inhibitor, for 20 to 40 hours, and the percentage of viable cells was determined using a trypan blue exclusion test. D-HEp3 cells proved to be highly resistant to drug-induced cell death relative to T-HEp3 cells (Fig. 3A-C). D-Hep3 and T-HEp3 cells had an IC50 of 80 and 20 μmol/L, respectively, for etoposide, and an IC50 of 22 and 4.5 μg/mL, respectively, for doxorubicin (Fig. 3C). In contrast to the differential effect of doxorubicin on T-Hep3 and D-HEp3 cell viability, doxorubicin inhibited cell proliferation to a similar extent (Fig. 3B). This, added to the fact that T-Hep3 and D-HEp3 cells proliferate at similar rates in culture (see Introduction and refs. 3, 4, 10), strongly suggests that the differential resistance of T-Hep3 and D-HEp3 cells to chemotherapy is dependent on a differential ability to resist drug-induced apoptosis. TUNEL assays unequivocally supported this hypothesis. Thus, whereas untreated T-Hep3 and D-HEp3 cells showed similar low levels of DNA breaks, treatment with doxorubicin (1 μg/mL) or etoposide (40 μmol/L) for 20 hours induced a strong increase in apoptosis only in T-HEp3 cells (Fig. 3D; data not shown).
Figure 3. T-HEp3 and D-HEp3 cell resistance to chemotherapy-induced apoptosis.
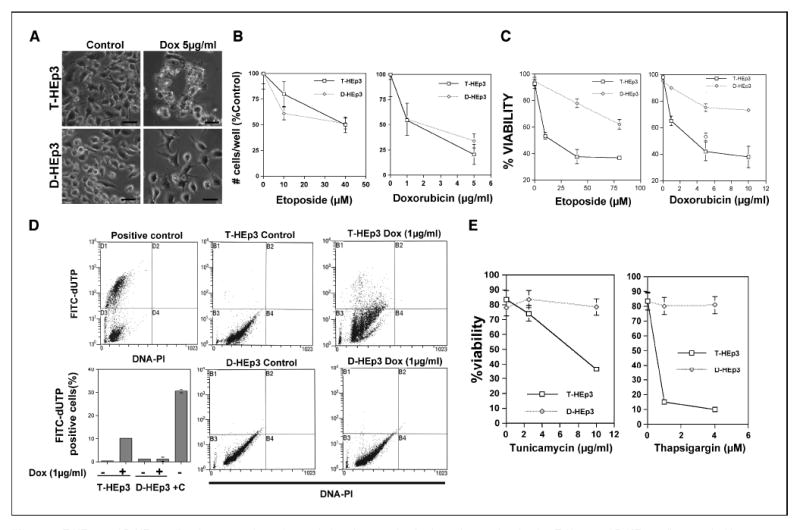
A, photomicrographs showing T-Hep3 and D-HEp3 cells treated with or without doxorubicin (Dox) for 30 hours in serum containing media. Bar, 160 μm. B, T-HEp3 and D-HEp3 cells treated with or without the indicated concentrations of doxorubicin or etoposide for 30 hours in serum containing media were detached with trypsin/EDTA and counted using a Coulter counter. C, cell viability assays using trypan blue exclusion test for T-HEp3 (□), D-HEp3 (◇) treated with or without the indicated concentrations of doxorubicin or etoposide for 30 hours. D, quantitation of DNA breaks as measured using the TUNEL assay Apo-Direct kit. Cells were counterstained with propidium iodide (PI) to detect all DNA content, and a positive control provided by the kit is shown in the top left dot plot. Apoptotic cells with labeled DNA breaks accumulate in the top left quadrant (D1, positive control or B1 in samples). Points, mean of % FITC-dUTP–positive cells in each sample (bottom left); bars, SD. +C, positive control. E, T-HEp3 and D-HEp3 cells were treated with the indicated concentrations of tunicamycin or thapsigargin for 16 hours, and the percentage of viable cells was determined by trypan blue exclusion assay as in (B).
Overexpression of multidrug resistance protein-1 (MDR-1/ABCB1) is not likely to be the mechanism mediating chemoresistance of D-HEp3 cells because surface expression of MDR-1 was higher in T-HEp3 than in D-HEp3 cells (data not shown). Furthermore, Affymetrix gene array comparison of D-HEp3 cells treated with or without 5 μmol/L SB203580 for 48 hours showed no statistically significant difference in the expression of the ABC transporter genes implicated in chemotherapeutic drug resistance (e.g., ABCB1-10, ABCC1-6, ABCC8-11, and ABCG1-8; ref. 32; data not shown).
We next determined whether the sensitivity of T-Hep3 versus D-HEp3 cells to chemotherapy was also evidenced in response to other proapoptotic stimuli that induce cell death independent of cell proliferation. In contrast to T-HEp3 cells, D-HEp3 cells also exhibited enhanced resistance to cell death induced by ER stressors, such as tunicamycin (a glycosylation inhibitor), thapsigargin (an ER-calcium pump blocker; Fig. 3E), or to that induced by glucose deprivation (data not shown). We conclude from our model system studies that the chemotherapy resistance that is characteristically associated with tumor dormancy is in good part a consequence of dormancy-related survival mechanisms rather than that of proliferation.
Does inhibition of BiP and PERK render dormant cells susceptible to doxorubicin-induced apoptosis?
Based on our data showing an active ER stress response in D-HEp3 cells (Fig. 2), we hypothesized that their resistance to doxorubicin-induced apoptosis was due to ER stress response–related survival mechanisms. More specifically, we hypothesized that the up-regulation of BiP and/or the activation of PERK in D-HEp3 cells may function as an antiapoptotic mechanism(s) that render these cells resistant to chemotherapy relative to T-HEp3 cells.
To test the role of BiP in resistance to doxorubicin-induced apoptosis, we down-regulated BiP protein levels in D-HEp3 cells using a siRNA (BiP-si; target exons 2-3 of human BiP mRNA). Western blot analysis showed that BiP-si resulted in >3-fold reduction in total BiP protein content (Fig. 4A). A siRNA to GAPDH had no effect on BiP expression in D-HEp3 cells but down-regulated GAPDH protein levels, indicating specificity of BiP-si (Fig. 4A). Down-regulation of BiP resulted in a statistically significant (~2-fold) increase in D-HEp3 sensitivity to doxorubicin (5 μg/mL, 28 hours)–induced cell death as measured by trypan blue exclusion test (Fig. 4B); a relative increase in basal cell death was also seen but the absolute value of this increase was very small. Cell viability following doxorubicin treatment, however, was unaffected by the down-regulation of GAPDH (data not shown). BiP-si resulted in increased apoptosis as evidenced by a strong increase in the percentage of D-HEp3 cells positive for DNA breaks measured by TUNEL assay when exposed to 5 μg/mL doxorubicin for 8 hours (Fig. 4C and D). However, cell survival upon genotoxic stress in T-HEp3 cells was unaffected by BiP down-regulation (verified by Western blot analysis; data not shown; Fig. 4D), suggesting that in the T-HEp3 cell model, the already low basal expression of BiP (Fig. 1) does not provide a survival signal in the context of doxorubicin-induced stress. These results suggest that the specific p38-dependent up-regulation of BiP in D-HEp3 cells is functionally linked to the resistance of these cells to drug-induced apoptosis.
Figure 4. Functional role for BiP and PERK in dormant cell drug resistance.

A, Western blot analysis of siRNA-mediated down-regulation of BiP in D-HEp3 cells. siRNA to BiP caused a >3-fold down-regulation in BiP protein; no change was observed on GAPDH levels except when an siRNA to this gene was used, which did not affect BiP expression levels. B and C, control and BiP siRNA–transfected cells were treated with or without 5 μg/mL doxorubicin (Dox) for 24 hours and analyzed for overall cell death by trypan blue exclusion test (B) or by TUNEL assay (C). B, columns, mean; bars, SD. *, P < 0.01. C, % cells in each quadrant. PMT4, PI staining; PMT2, FITC-dUTP staining. Note the increase in the percentage of apoptotic cells in the upper right quadrant following BiP-si and doxorubicin treatment. D, quantitation of DNA breaks as measured using the TUNEL assay Apo-Direct kit. Control and BiP siRNA–transfected cells treated with or without 2 μg/mL (T-HEp3) or 5 μg/mL (D-HEp3) of doxorubicin were analyzed for apoptosis by TUNEL assay as in (C). Columns, mean of % FITC-dUTP–positive cells in each sample; bars, SD. E, representative fields of D-HEp3 cells transiently cotransfected with EGFP and empty vector, wt-PERK, PERK-ΔC, or GADD34, respectively. After transfection (48 hours), cells were treated overnight with doxorubicin, and then cells were fixed in 70% ethanol and stained with Hoescht 33342 (20 mmol/L). Nonspecific nuclear fluorescence due to doxorubicin accumulation in cells crossed to the GFP channel. Bar, 60 μm. Open arrowheads, left, GFP-negative apoptotic cells. Arrow, GFP-positive nonapoptotic cells. Closed arrowhead, right, GFP-positive apoptotic cells. F, quantitation of apoptotic bodies as measured using Hoescht 33342 in (E). At least 150 cells positive for GFP were scored for the presence of apoptotic bodies characterized by nuclear DNA condensation. Columns, mean; bars, SD (E). #, P < 0.05, EGFP + doxorubicin versus GADD34 + doxorubicin. *, P < 0.01, EGFP + doxorubicin versus PERK-ΔC + doxorubicin, as determined by ANOVA test. G, representative fields of D-HEp3 cells transiently cotransfected with EGFP and empty vector, PERK-ΔC, or GADD34 vectors, respectively. After transfection (48 hours), cells were treated overnight with etoposide and were fixed and stained with α-cleaved caspase-3 antobody (see Materials and Methods) and analyzed for caspase-3 activation by immunofluorescence. Arrows, b and f, EGFP-negative and caspase-3–positive cells. Arrows, d and h, EGFP-positive and caspase-3–positive cells. Bar, 40 μm. H, quantitation of caspase-3 activation as measured using immunofluorescence in (G). D-HEp3 cells transiently cotransfected with EGFP and PERK-ΔC or GADD34, respectively, were treated, fixed, and stained as described in Materials and Methods. More than two hundred cells positive for GFP were scored for caspase-3 activation. Columns, mean; bars, SD. #, P < 0.001, GADD34 versus GADD34 + etoposide or PERK-ΔC versus PERK-ΔC + etoposide. *, P < 0.005, EGFP + etoposide versus GADD34 + etoposide or PERK-ΔC + etoposide as determined by Mann-Whitney test.
We next explored the functional contribution of PERK signaling to chemoresistance in D-HEp3 cells by transiently cotransfecting cells with expression vectors encoding enhanced GFP (EGFP) and either wtPERK, PERK-ΔC, or GADD34 (EGFP and empty vector as control). After 24 hours, cells were treated with or without 5 μg/mL doxorubicin for 13 hours, fixed, and stained with Hoechst to detect apoptotic nuclei characterized by DNA condensation and cell shrinkage (Fig. 4E). The percentage of apoptotic GFP-positive cells in the 4′,6-diamidino-2-phenylindole channel was quantitated under a fluorescence microscope. In control D-HEp3 cells, modulation of PERK resulted in slight differences in the extent of basal apoptosis, but these were not statistically significant (Fig. 4F). However, upon doxorubicin challenge, inhibition of PERK signaling by PERK-ΔC or GADD34 increased the rate of apoptosis by 2- to 3-fold, effects that were statistically significant (Fig. 4F), whereas this was unaffected by the overexpression of wtPERK (Fig. 4E). In support of the Hoechst staining data, immunofluorescence analysis of caspase-3 activation, a hallmark of apoptosis, following etoposide treatment showed that PERK inhibition by PERK-ΔC or GADD34 significantly increased the levels of active caspase-3 by 2- to 3-fold (Fig. 4G-H). We conclude from these experiments that in agreement with our hypothesis, activation of the PERK pathway and phosphorylation of eIF2α mediate a survival signal that protects D-HEp3 cells from drug-induced cell death.
Up-regulation of BiP by p38 is required to inhibit Bax activation in dormant cells
Activation of Bax, a proapoptotic protein that favors cytochrome c release from mitochondria, has been shown to mediate apoptosis triggered by signals from both the intrinsic and the ER stress–mediated apoptotic pathways. Thus, we tested whether T-Hep3 and D-HEp3 cells displayed differential activation of Bax in response to doxorubicin or etoposide treatment and whether p38 or BiP function had any influence on this proapoptotic signal. Bax activation was measured using an anti-human Bax 6A7 antibody, which binds to an epitope in the NH2-terminal region (amino acids 13-19) that is exposed only upon activation (31, 33). Although T-Hep3 and D-HEp3 cells showed similar levels of total Bax protein (Fig. 5A), treatment of these cells with doxorubicin (1 μg/mL, 24 hours) or etoposide (80 or 40 μmol/L, 24 hours) caused a 3- to 4-fold increase in Bax activation as measured by stronger binding of 6A7 mAb to T-HEp3 than D-HEp3 cells as determined by immunofluorescence (Fig. 5B) and FACS analysis (Fig. 5C, red line).
Figure 5. p38 and BiP inhibit Bax activation.
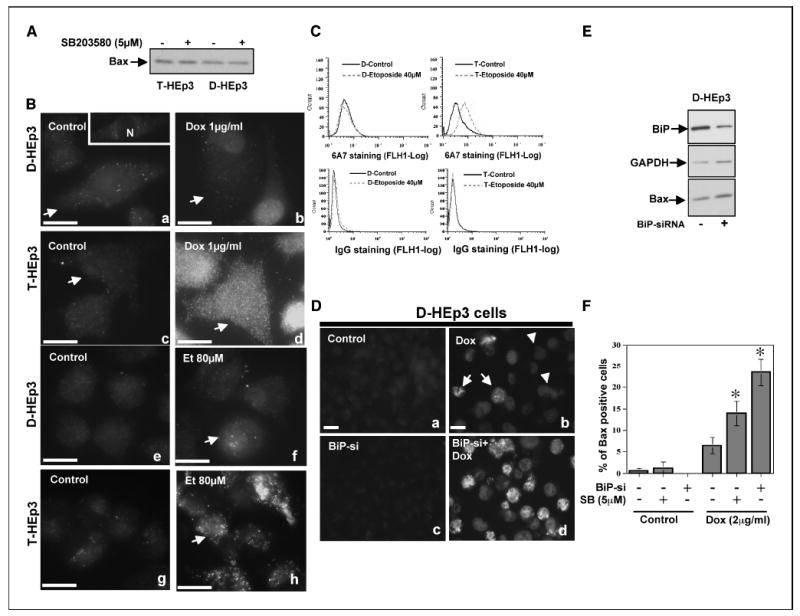
A, Western blot analysis of Bax levels in T-Hep3 and D-HEp3 cells treated with SB203580 for 48 hours. B, the indicated cells treated with or without doxorubicin (Dox, 1 μg/mL) or etoposide (Et, 80 μmol/L) for 24 hours were fixed and stained with α-Bax 6A7 antibody (see Materials and Methods) and analyzed for Bax activation by immunofluorescence. Bax staining seemed punctate, characteristic of ER and mitochondrial localization (31). The slight nuclear staining was due to doxorubicin emitting a low signal that crossed into the FITC channel (b). a, inset, plane of focus that sectioned the nucleus, to illustrate that the Bax signal is excluded from the nucleus. Bar, 40 μm. C, FACS analysis of T-Hep3 or D-HEp3 cells treated with 40 μmol/L etoposide for 24 hours and then stained with 6A7 mAb (top) or an isotype-matched IgG (bottom). The signal was developed using an Alexa-488–conjugated goat anti-mouse antibody and quantitated through FACS. D, Bax activation in control or BiP siRNA–transfected cells treated with or without 2 μg/mL doxorubicin was analyzed by immunofluorescence as in B (D, top). Nonspecific nuclear fluorescence due to doxorubicin accumulation in cells crossed to the FITC channel (arrowheads). Bar, 40 μm (D, bottom). E, Western blot analysis showing that total Bax levels are unaffected following siRNA mediated down-regulation of BiP. GAPDH was used as a loading control. F, quantitation of Bax activation in SB-treated or BiP siRNA–transfected cells treated with or without doxorubicin (2 μg/mL). Columns, mean of % Bax-positive cells in each sample; bars, SD. *, P < 0.001, for the indicated treatments versus doxorubicin treatment alone
As Hsp70 was previously found to inhibit Bax activation (34), we hypothesized that BiP, a Hsp70 family member with prosurvival functions (35), may have a similar effect on Bax activity in D-HEp3 cells. To test this hypothesis, control D-HEp3 cells with high BiP levels or the same cells in which BiP was down-regulated by BiP-si (Fig. 5D) were treated with or without doxorubicin (2 μg/mL) for 20 hours, and the cells were processed for immunofluorescence as in Fig. 5B. Down-regulation of BiP by itself had no effect on basal Bax activation or total Bax protein levels (Fig. 5D and E). However, after doxorubicin treatment, ~25% of the cells in which BiP was down-regulated showed a strong activation of Bax, whereas only 6% of control cells treated with doxorubicin showed positive staining for active Bax (Fig. 5D). Although total Bax levels were unaffected by the pretreatment of D-HEp3 cells with SB203580 for 24 hours (Fig. 5A), it resulted in a larger proportion of D-HEp3 cells with active Bax staining, albeit the increase was not as dramatic as with BiP down-regulation (Fig. 5F). Simultaneous inhibition of p38 signaling and BiP expression, however, did not result in an larger increase in Bax activation (BiP-si + SB203580 = 18.4 ± 1.1% versus BiP-si = 23.4 ± 3.2; not significant, Mann-Whitney test). Enhanced survival of D-HEp3 cells did not seem to depend on Akt activation because there was ~2-fold more phospho-Akt in T-HEp3 than in D-HEp3 cells (data not shown). Taken together, our results suggest that apoptosis of T-HEp3 cells in response to doxorubicin treatment follows a classic Bax-dependent apoptotic pathway. In contrast, in D-HEp3 cells BiP up-regulation inhibits, through a yet to be determined mechanism, Bax activation rendering these cells refractory to drug-induced apoptotic signals.
Discussion
Mechanisms that can protect growing tumors from drug toxicity (36) by preventing the entry of drugs into tumor cells (32) or that circumvent the induction of apoptosis are usually studied in the context of proliferating tumor cells. In contrast, very little is known about the mechanisms that make nondividing or slow-dividing cells present for example in minimal residual disease, resistant to drug treatment. In an effort to understand the molecular mechanisms that specifically protect dormant cells from chemotherapy, we have taken advantage of our model of tumorigenic versus dormant HEp3 carcinoma cells in which in vitro behavior predicts the in vivo phenotype (7). Using this model, we have identified a novel antiapoptotic mechanism mediated by p38, PERK, and BiP proteins that strikingly confers drug resistance selectively to dormant tumor cells. Our results show that in dormant cells the p38-dependent activation of PERK and the up-regulation of BiP, which by inhibiting Bax activation and subsequent initiation of the apoptotic signal protects these cells from drug-induced apoptosis. In contrast, the absence of survival signals mediated by BiP and PERK in the tumorigenic cells renders them sensitive to drug-induced apoptosis. Underscoring the importance of our results, we found that drug resistance of dormant cells was independent of proliferation or expression of MDR genes.
What signals could trigger an ER stress response, such as the UPR, that would protect disseminated dormant cells from chemotherapeutic treatment? Studies have shown that hypoxic/anoxic areas of solid tumors activate the UPR, which enables them to adapt to the hypoxic stress and survive (35). We propose that in the case of cells that have separated from the primary tumor and undergone stress imposed by dissemination, stress signaling via p38 predominates over mitogenic ERK signaling (10). In this context, activation of the UPR and growth arrest dependent on p38 could restrict immediate metastatic growth. Most importantly, under these conditions, the survival signals provided by BiP and PERK might protect these cells from different types of stress, such as chemotherapy.
Our results show that two signals, one dependent on BiP and another propagated by PERK, serve as survival signals in drug-resistant dormant cells. Hypoxia can induce BiP in tumor cells and in a wide range of primary tumors, which invariably have hypoxic regions, BiP is also found to be up-regulated (18). Subsequent studies showed that forced overexpression of BiP conferred drug resistance to various tumor cell types (37). Furthermore, recently published results in breast carcinoma cells showed that BiP is up-regulated in chemotherapy-resistant clones and that suppression of BiP expression could sensitize these cells to etoposide-mediated cell death (38). We recently showed (7) that some of the drug-resistant cells analyzed in the above mentioned study (38), such as MCF-7, have high p38 activation (7). It is possible, although unknown, that p38 signaling in these cells is responsible for higher BiP levels and chemoresistance. We found that high p38 activation in dormant cells associated with increased BiP-luc promoter activation. Although unknown in our system, it is possible that as reported previously, p38-dependent activation of the transcription factor ATF6 results in BiP up-regulation (39).
Analysis of the mechanisms linking p38 activation and higher BiP levels to drug resistance in D-Hep3 versus T-HEp3 cells revealed that in the latter doxorubicin treatment favored Bax activation and apoptosis. In contrast, in D-HEp3 cells, their resistance to further stress was due to lack of Bax activation in response to doxorubicin or other (tunicamycin, thapsigargin, low glucose, etc.) treatments. The inability of D-HEp3 cells to activate Bax was attributed to the p38-dependent up-regulation of BiP levels as down-regulation of BiP with an siRNA caused a 4-fold increase in Bax activation following doxorubicin treatment. In comparison, inhibition of p38 alone resulted in a less impressive but still significant increase in Bax activation in D-HEp3 cells. Inhibition of p38 following BiP down-regulation was not additive or synergistic, possibly because down-regulation of BiP is sufficient to reach a maximal effect under these experimental conditions. These results show that the high p38 activity in these dormant cells orchestrates a survival response via the up-regulation of BiP and subsequent downstream inhibition of Bax activity. The mechanism by which BiP inhibits Bax activation is presently unknown. There is evidence that the cytoplasmic family member of BiP, Hsp70, and DnaJ/Hsp40 can directly bind Bax and prevent its activation in response to apoptotic signals (34). However, because BiP resides in the ER lumen, we speculate that BiP might interfere with Bax activation by an alternative mechanism. Other mechanisms have proposed that BiP overexpression can inhibit activation of caspase-3 and caspase-7 or that BiP can span the ER membrane and interact with caspase-12, preventing its processing by caspase-7 (37). We are currently exploring if such mechanisms protect D-HEp3 cells from drug toxicity.
Activation of PERK was also found to signal for survival in dormant tumor cells. Because both PERK-ΔC mutant and GADD34 sensitized these cells to doxorubicin treatment, it is possible that proteins expressed in the presence of phospho-eIF2α may mediate survival. As expression of BiP is dependent on ATF4, which is translated only when eIF2α is phosphorylated (40, 41), it is possible that PERK-induced survival is an indirect consequence of BiP up-regulation. However, we cannot rule out the possibility that other phospho-eIF2α–independent mechanisms activated by PERK, such as activation of the transcription factor Nrf2, shown to mediate survival following ER stress (42), could protect D-HEp3 cells from drug-induced apoptosis. The mechanisms by which p38 activates PERK are currently under study. Collectively, our results already provide insight into a novel link among p38, BiP, and PERK signaling with a phenotypic shift (tumorigenicity to dormancy) that selectively equips cells predicted to become dormant in vivo with a drug resistance mechanism. Thus, we believe our model and results provide a unique opportunity to further dissect the mechanisms that protect dormant cells from chemotherapy.
Compared with T-HEp3 cells, D-HEp3 cells displayed a higher and p38-dependent splicing of XBP-1 mRNA, a hallmark of active ER stress that occurs exclusively following an increase in the ER protein load (Supplementary Fig. 1A and B). It is therefore possible, although unknown, that increased XBP-1 is a consequence of an increase in load of proteins in the ER in dormant D-HEp3 cells. However, other mechanisms may be operational because cells null for the protein tyrosine phosphatase PTP-1B display a strong decrease in p38 activation and reduced XBP-1 splicing (43). Although our results implicate a p38-dependent up-regulation of XBP-1 in D-HEp3 cells, it is still unclear how this may contribute to survival and/or dormancy of these cells. We presume up-regulation of XBP-1 may have a survival function because its expression is increased in hypoxic tumors (44).
To summarize, we have found that in dormant HEp3 squamous carcinoma cells BiP and PERK play a key role in coordinating a survival response against drug-induced apoptosis by preventing Bax activation. Our findings have revealed a previously unrecognized survival mechanism that selectively protects dormant but not tumorigenic cells from chemotherapy. Although the exact nature of the mechanisms that propel malignant cells into dormancy are unknown, we propose that treatments that inhibit the survival mechanisms induced by p38 during the dormancy period may allow conventional or new chemotherapy approaches to eradicate dormant cells.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
The University at Albany/State University of New York Startup Funds, Samuel Waxman Cancer Research Foundation, NIH/National Cancer Institute grant CA109182 (J.A. Aguirre-Ghiso), and Ruth L. Kirschstein National Research Service Award (NIH/National Cancer Institute) Fellowship (A.C. Ranganathan).
We thank Dr. Linda Hendershot (St. Jude Children’s Hospital) and Dr. David Ron (New York University) for helpful comments and sharing reagents, Dr. Masayuki Miura (University of Tokyo) for the pCAX-F-XBP1-venus plasmid, Guy Russo (CFG, University at Albany, NY) for assisting us with plasmid preparations, Dr. Hernan Farina and Sarah Rosenbaum (Mount Sinai School of Medicine, NY) for help with some experiments, Dr. Serafin Piñol Roma (City University of New York) for expert advice on the nonequilibrium pH gradient/two-dimensional gels, Mary Ann Gawinowicz (Columbia University) for advice on MALDI-MS sample preparation, and Drs. Liliana Ossowski and Rafael Mira y Lopez (Mount Sinai School of Medicine) for critical reading of the article.
References
- 1.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–15. [PubMed] [Google Scholar]
- 2.Naumov GN, Townson JL, MacDonald IC, et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 3.Ossowski L, Reich E. Changes in malignant phenotype of a human carcinoma conditioned by growth environment. Cell. 1983;33:323–33. doi: 10.1016/0092-8674(83)90414-2. [DOI] [PubMed] [Google Scholar]
- 4.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–79. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 6.Yu W, Kim J, Ossowski L. Reduction in surface urokinase receptor forces malignant cells into a protracted state of dormancy. J Cell Biol. 1997;137:767–77. doi: 10.1083/jcb.137.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–95. [PubMed] [Google Scholar]
- 8.Aguirre-Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Timofeev O, Lee TY, Bulavin DV. A subtle change in p38 MAPK activity is sufficient to suppress in vivo tumorigenesis. Cell Cycle. 2005;4:118–20. doi: 10.4161/cc.4.1.1342. [DOI] [PubMed] [Google Scholar]
- 10.Aguirre-Ghiso JA, Ossowski L, Rosenbaum SK. Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res. 2004;64:7336–45. doi: 10.1158/0008-5472.CAN-04-0113. [DOI] [PubMed] [Google Scholar]
- 11.Molnar A, Theodoras AM, Zon LI, Kyriakis JM. Cdc42Hs, but not Rac1, inhibits serum-stimulated cell cycle progression at G1/S through a mechanism requiring p38/RK. J Biol Chem. 1997;272:13229–35. doi: 10.1074/jbc.272.20.13229. [DOI] [PubMed] [Google Scholar]
- 12.Li SP, Junttila MR, Han J, Kahari VM, Westermarck J. p38 Mitogen-activated protein kinase pathway suppresses cell survival by inducing dephosphorylation of mitogen-activated protein/extracellular signal-regulated kinase kinase1,2. Cancer Res. 2003;63:3473–7. [PubMed] [Google Scholar]
- 13.Weldon CB, Parker AP, Patten D, et al. Sensitization of apoptotically-resistant breast carcinoma cells to TNF and TRAIL by inhibition of p38 mitogen-activated protein kinase signaling. Int J Oncol. 2004;24:1473–80. [PubMed] [Google Scholar]
- 14.Vega MI, Huerta-Yepaz S, Garban H, et al. Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: pivotal role of p38 MAPK in drug resistance. Oncogene. 2004;23:3530–40. doi: 10.1038/sj.onc.1207336. [DOI] [PubMed] [Google Scholar]
- 15.Zechner D, Craig R, Hanford DS, et al. MKK6 activates myocardial cell NF-κB and inhibits apoptosis in a p38 mitogen-activated protein kinase-dependent manner. J Biol Chem. 1998;273:8232–9. doi: 10.1074/jbc.273.14.8232. [DOI] [PubMed] [Google Scholar]
- 16.Carvalho H, Evelson P, Sigaud S, Gonzalez-Flecha B. Mitogen-activated protein kinases modulate H(2)O(2)-induced apoptosis in primary rat alveolar epithelial cells. J Cell Biochem. 2004;92:502–13. doi: 10.1002/jcb.20070. [DOI] [PubMed] [Google Scholar]
- 17.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–98. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–8. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–99. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman RJ, Scheuner D, Schroder M, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3:411–21. doi: 10.1038/nrm829. [DOI] [PubMed] [Google Scholar]
- 22.Yan W, Frank CL, Korth MJ, et al. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci U S A. 2002;99:15920–5. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000;97:12625–30. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urano F, Bertolotti A, Ron D. IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci. 2000;113(Pt 21):3697–702. doi: 10.1242/jcs.113.21.3697. [DOI] [PubMed] [Google Scholar]
- 25.Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci U S A. 1999;96:8505–10. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niwa M, Walter P. Pausing to decide. Proc Natl Acad Sci U S A. 2000;97:12396–7. doi: 10.1073/pnas.250476097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 1988;2:215–27. doi: 10.1101/gad.2.2.215. [DOI] [PubMed] [Google Scholar]
- 28.Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med. 2004;10:98–102. doi: 10.1038/nm970. [DOI] [PubMed] [Google Scholar]
- 29.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguirre-Ghiso J, Ossowski L, Rosenbaum S. GFP tagging of ERK and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res. 2004;64:7336–45. doi: 10.1158/0008-5472.CAN-04-0113. [DOI] [PubMed] [Google Scholar]
- 31.Zong WX, Li C, Hatzivassiliou G, et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szakacs G, Annereau JP, Lababidi S, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. 2004;6:129–37. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 33.Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–83. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 34.Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- 35.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 36.Waxman DJ, Schwartz PS. Harnessing apoptosis for improved anticancer gene therapy. Cancer Res. 2003;63:8563–72. [PubMed] [Google Scholar]
- 37.Reddy RK, Mao C, Baumeister P, et al. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 38.Dong D, Ko B, Baumeister P, et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res. 2005;65:5785–91. doi: 10.1158/0008-5472.CAN-05-0754. [DOI] [PubMed] [Google Scholar]
- 39.Luo S, Lee AS. Requirement of the p38 mitogen-activated protein kinase signalling pathway for the induction of the 78 kDa glucose-regulated protein/immunoglobulin heavy-chain binding protein by azetidine stress: activating transcription factor 6 as a target for stress-induced phosphorylation. Biochem J. 2002;366:787–95. doi: 10.1042/BJ20011802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278:37375–85. doi: 10.1074/jbc.M303619200. [DOI] [PubMed] [Google Scholar]
- 41.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 42.Cullinan SB, Zhang D, Hannink M, et al. Nrf 2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu F, Nguyen DT, Stuible M, et al. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J Biol Chem. 2004;279:49689–93. doi: 10.1074/jbc.C400261200. [DOI] [PubMed] [Google Scholar]
- 44.Romero-Ramirez L, Cao H, Nelson D, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–7. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).