

Abstract
A model of unlinked diallelic loci affecting the risk of a complex inherited disease is explored. The loci are equivalent in their effect on disease risk and are in Hardy–Weinberg and linkage equilibrium. The goal is to determine what assumptions about dependence of disease risk on genotype are consistent with data for diseases such as schizophrenia, bipolar disorder, autism, and multiple sclerosis that are relatively common (0.1–2% prevalence) and that have high concordance rates for monozygotic twins (30–50%) and high risks to first-degree relatives of affected individuals (risk ratios exceeding 4). These observations are consistent with a variety of models, including generalized additive, multiplicative, and threshold models, provided that disease risk increases rapidly for a narrow range of numbers of causative alleles. If causative alleles are in relatively high frequency, then the combined effects of numerous causative loci are necessary to substantially elevate disease risk.
COMPLEX inherited diseases are, by definition, affected by more than one genetic locus. Although many alleles associated with higher risks of complex diseases have been identified, almost nothing is known about interactions among them or whether there is any commonality to the genetic architecture of different diseases. In this article, I examine a class of models of complex inherited diseases in which all loci increasing disease risk are equivalent in their effects. The goal is to find general properties of such models in randomly mating populations.
I am particularly concerned with diseases that have prevalences in the range 0.1–2% and that are highly heritable, meaning that the concordance probability for monozygotic (MZ) twins is in the range 30–50% and the risk ratio for first-degree relatives [Risch's (1990) λ1] is in the range 4–10. Such diseases are regarded as common because their prevalence is much higher than monogenic diseases and because they constitute a major burden on health care systems in developed countries. Several diseases, including autism (Szatmari et al. 2007), schizophrenia (Sullivan et al. 2003), bipolar disorder (Smoller and Finn 2003), multiple sclerosis (Oksenberg and Barcellos 2005), and type 1 and type 2 diabetes (Permutt et al. 2005), are of this type. Other diseases, including congenital heart disease (Romano-Zelekha et al. 2001) and most cancers (Amundadottir et al. 2004), are as prevalent but have substantially lower concordance rates for MZ twins and smaller risk ratios.
I show that evidence of high heritability requires that there be a large variance in risk among individuals. Consequently, risk considered as a function of the number of causative alleles has to be steeply increasing in the narrow range of genotypes found in appreciable frequency in a population.
Most recent analyses of complex diseases have been based on a model of multiplicative interactions across loci. The use of the multiplicative model is traceable to Risch (1990), who showed it provides a better fit to estimates of recurrence risk in first-, second-, and third-degree relatives than do models of additive and heterogeneous interactions. Specifically, Risch (1990) showed that, under the multiplicative model, estimated recurrence risks for schizophrenia (and by implication other complex diseases) decreased more rapidly with the degree of relationship than is predicted by an additive model. He also showed that a model of genetic heterogeneity (Morton et al. 1970), in which each of several causative loci separately increases risk, is similar to an additive model and hence also inconsistent with the schizophrenia data.
The multiplicative model largely replaced two models from classical quantitative genetics, the threshold model, in which an underlying liability is treated as a normally distributed quantitative character, and the major-gene model, in which the risk conferred by a single locus is affected by many modifier loci of small effect. Both these models have been extensively analyzed (Smith 1971) and for some purposes they represent extremes of the range of possible quantitative genetic models (Curnow and Smith 1975). Neither model depends on the number of loci, on the frequencies of alleles at each locus, or on explicit assumptions about interactions within and between loci. Instead they assume normality of underlying genetic and environmental effects and are parameterized in terms of variance components. Edwards (1960) showed that it is very difficult to distinguish between the threshold and major-gene models by using recurrence risk data.
Risch's (1990) multiplicative model differs from the two quantitative genetic models because it makes explicit assumptions about the effects of each locus. Its popularity derives in part from its mathematical simplicity. The recurrence risk attributable to each locus is calculated separately and then the overall recurrence risk is obtained by multiplying across loci. The multiplicative model has served as the basis for estimating the number of causative loci (Farrall and Holder 1992; Schliekelman and Slatkin 2002; Lindsey 2005) and has been used in other theoretical studies, including those addressing the question of whether rare or common alleles are primarily responsible for complex diseases (Pritchard 2001; Reich and Lander 2001; Peng and Kimmel 2007).
In this article, I first review the population genetics of multiple loci in randomly mating populations and then define three exchangeable models (additive, multiplicative, and threshold) in which loci are equivalent in their effect on disease risk. I show that when allele frequencies are the same at each locus, the results of the three classes of models are similar provided that risk increases steeply in a narrow range of numbers of causative alleles. Finally, the threshold model is explored to illustrate how different features of the risk model affect the frequencies of causative alleles and the pattern of recurrence risk.
THEORY
Multilocus Mendelism:
Throughout, the genotypes of each locus are assumed to be in their Hardy–Weinberg (HW) frequencies and all loci are assumed to be in linkage equilibrium (LE), assumptions summarized as HWLE (Barton and Turelli 2004). Each locus has only two alleles, + and −, where + tends to increase disease risk. There are L loci and the frequency of + at locus j is pj.
Under HW, the probabilities that an individual has genotypes +/+, +/−, and −/− at locus j are 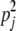 , 2pj(1 − pj), and (1 − pj)2. If the loci are in LE, the joint probability that a randomly chosen individual has i1 +/+ loci, i2 +/− loci, and i3 −/− loci (i1 + i2+ i3 = L) is obtained by taking the convolution of L distributions with these probabilities. If all pj are equal, that distribution is a trinomial with sample size L.
, 2pj(1 − pj), and (1 − pj)2. If the loci are in LE, the joint probability that a randomly chosen individual has i1 +/+ loci, i2 +/− loci, and i3 −/− loci (i1 + i2+ i3 = L) is obtained by taking the convolution of L distributions with these probabilities. If all pj are equal, that distribution is a trinomial with sample size L.
The joint probability of pairs of genotypes at each locus in relatives with relationship R depends on two quantities, θ, the probability that the relatives share exactly one allele identical by descent (IBD), and γ, the probability that they share two alleles IBD. In an outbred population, 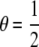 and
and 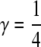 for full siblings, θ = 1 and γ = 0 for parents and offspring, and so on. The joint probability of genotypes at a locus is computed from Mendelism combined with the HW frequencies
for full siblings, θ = 1 and γ = 0 for parents and offspring, and so on. The joint probability of genotypes at a locus is computed from Mendelism combined with the HW frequencies
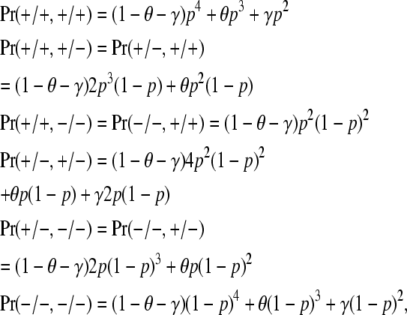 |
(1) |
where the subscript j is omitted for notational convenience. Equation 1 is adapted from Table 5 of Liu and Weir (2005). In Risch's (1990) notation cR = θ/2 + γ and uR = γ.
TABLE 5.
Dependence of population-attributable risk (PAR) on SNP frequencies in three studies of type II diabetes (Scott et al. 2007; Sladek et al. 2007; Zeggini et al. 2007)
| SNP | Source | pcontrol | pcase | PAR |
|---|---|---|---|---|
| rs1801282 | Scott et al. (2007) | 0.823 | 0.848 | 0.16447 |
| rs4402960 | Scott et al. (2007) | 0.304 | 0.341 | 0.056146 |
| rs7754840 | Scott et al. (2007) | 0.36 | 0.387 | 0.044046 |
| rs13266634 | Scott et al. (2007) | 0.609 | 0.649 | 0.11396 |
| rs10811661 | Scott et al. (2007) | 0.85 | 0.872 | 0.17187 |
| rs1111875 | Scott et al. (2007) | 0.522 | 0.546 | 0.052863 |
| rs7903146 | Scott et al. (2007) | 0.181 | 0.227 | 0.059508 |
| rs5219 | Scott et al. (2007) | 0.464 | 0.489 | 0.048924 |
| rs9300039 | Scott et al. (2007) | 0.892 | 0.924 | 0.42105 |
| rs8050136 | Scott et al. (2007) | 0.381 | 0.406 | 0.042088 |
| rs7923837 | Sladek et al. (2007) | 0.623 | 0.665 | 0.11141 |
| rs7480010 | Sladek et al. (2007) | 0.301 | 0.336 | 0.050072 |
| rs3740878 | Sladek et al. (2007) | 0.728 | 0.76 | 0.11765 |
| rs11037909 | Sladek et al. (2007) | 0.729 | 0.76 | 0.11439 |
| rs1094369 | Zeggini et al. (2007) | 0.319 | 0.361 | 0.060957 |
Data from Scott et al. are from their Table S1; data from Sladek et al. are from their Table 1; data from Zeggini et al. are from their Table S2. When the same SNP was found in two or more studies, data from only one study were used. PAR is computed using Equation 4 in the text, with pcase = pC and pcontrol = p.
For unlinked loci, the joint probabilities of the nine pairs of genotypic configurations are obtained by taking the L-fold convolution of the probabilities in Equation 1. If p is the same at every locus, the result is a multinomial with nine categories and sample size L.
Exchangeable models of risk:
In the sense used here, a model is exchangeable if the identities of the loci can be exchanged and leave the risk unchanged. In that case, the overall risk depends on i1, the number of +/+ loci, i2, the number of +/− loci, and i3, the number of −/− loci (i1 + i2 + i3 = L). That usage is consistent with the meaning of “exchangeable” in probabilistic models such as that of Cannings (1974). I consider four exchangeable models in this article, the unconstrained multiplicative, the constrained multiplicative, the additive, and the threshold models. The unconstrained multiplicative and additive models were analyzed by Risch (1990). The threshold model is from classical quantitative genetics and is also a generalization of the model of genetic heterogeneity analyzed by Risch (1990).
In the unconstrained multiplicative model, the risk attributable to locus j is fj: 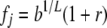 if locus j is +/+,
if locus j is +/+,  if locus j is +/−, and
if locus j is +/−, and 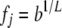 if locus j is −/−. With this parameterization, b (the background risk) is the risk to an individual homozygous for − at all L loci, h is the degree of dominance, and 1 + r is the ratio of the risk to an individual with +/+ at a locus to an individual with −/− at that locus (the odds ratio). The overall risk is obtained by multiplying across loci, weighting each locus by the probability of its genotype. For a given set of parameter values, the risks assigned to some genotypes by the multiplicative model may exceed 1. In the unconstrained multiplicative model, risks >1 are allowed, and in the constrained multiplicative model the risk is set to 1 if the computed risk is >1.
if locus j is −/−. With this parameterization, b (the background risk) is the risk to an individual homozygous for − at all L loci, h is the degree of dominance, and 1 + r is the ratio of the risk to an individual with +/+ at a locus to an individual with −/− at that locus (the odds ratio). The overall risk is obtained by multiplying across loci, weighting each locus by the probability of its genotype. For a given set of parameter values, the risks assigned to some genotypes by the multiplicative model may exceed 1. In the unconstrained multiplicative model, risks >1 are allowed, and in the constrained multiplicative model the risk is set to 1 if the computed risk is >1.
In the additive model, the contribution of each locus to overall risk is the same as in the multiplicative model. The difference is that overall risk is the sum of the contributions from each locus: 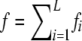 . The risk for the additive model is defined to be
. The risk for the additive model is defined to be 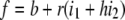 . Here, the additive model is constrained so that 0 ≤ f ≤ 1.
. Here, the additive model is constrained so that 0 ≤ f ≤ 1.
The threshold model comes from the theory of quantitative genetics (Falconer 1981; Chap. 18). The model assumes an underlying liability, x, which is the sum of a genetic component g = i1 + hi2 and an independent environmental component e: x = g + e, where e is a normally distributed random variable with mean 0 and variance 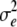 . The parameter T is the threshold value of x; the risk is b if x < T and 1 otherwise. With these definitions
. The parameter T is the threshold value of x; the risk is b if x < T and 1 otherwise. With these definitions 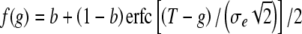 , where erfc is the complementary error function,
, where erfc is the complementary error function, 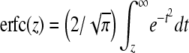 . For
. For  f is equivalent to a step function of g, considered by Lindsey (2005). If
f is equivalent to a step function of g, considered by Lindsey (2005). If  and 0 < T < 1, the threshold model is equivalent to the heterogeneous model analyzed by Risch (1990). For larger values of σe, f is a sigmoid function centered at g = T with a slope at T proportional to 1/σe.
and 0 < T < 1, the threshold model is equivalent to the heterogeneous model analyzed by Risch (1990). For larger values of σe, f is a sigmoid function centered at g = T with a slope at T proportional to 1/σe.
OBSERVABLE QUANTITIES
Prevalence and recurrence risk:
The data available for a complex disease are the prevalence K and the recurrence risks to relatives of relationship R, KR. The risk ratio is defined to be 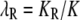 . In the notation used here,
. In the notation used here,
 |
(2) |
where G represents the multilocus genotype (i1, i2, i3) and the sum is over all possible genotypes.
From James (1971),
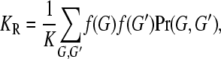 |
(3) |
where G′ is the genotype of a relative with relationship R. The joint probability of G and G′ is obtained from Equation 1 and the assumption of no linkage. For MZ twins, Equation 3 is equivalent to Equation 2 with f(G) replaced by 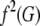 . Consequently, KM depends on the variance in risk:
. Consequently, KM depends on the variance in risk: 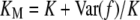 .
.
Here, I consider five classes of relatives: MZ twins (R = M: θ = 0, γ = 1), full siblings (R = S: 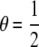 ,
, 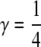 ), first-degree relatives (parent–offspring) (R = 1: θ = 1, γ = 0), second-degree relatives (grandparent–offspring, half siblings, aunt– or uncle–niece or nephew) (R = 2:
), first-degree relatives (parent–offspring) (R = 1: θ = 1, γ = 0), second-degree relatives (grandparent–offspring, half siblings, aunt– or uncle–niece or nephew) (R = 2: 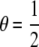 , γ = 0), and third-degree relatives (first cousins) (R = 3:
, γ = 0), and third-degree relatives (first cousins) (R = 3:  , γ = 0). Results are reported in terms of the prevalence, K, the concordance probability for MZ twins, KM, and the risk ratios for other relatives,
, γ = 0). Results are reported in terms of the prevalence, K, the concordance probability for MZ twins, KM, and the risk ratios for other relatives,  . Of particular interest are the values of λ1, λ2, and λ3. Risch (1990) showed that for the additive model,
. Of particular interest are the values of λ1, λ2, and λ3. Risch (1990) showed that for the additive model,  and for the multiplicative model
and for the multiplicative model 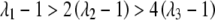 .
.
Population-attributable risk and odds ratios:
The population-attributable risk of locus j (PARj) of + at locus j is the scaled difference between the frequency in affected individuals and the frequency in the population,
 |
(4) |
where  is the frequency of + at locus j among affected individuals (i.e., cases) (Bengtsson and Thomson 1981). Other denominators are also used.
is the frequency of + at locus j among affected individuals (i.e., cases) (Bengtsson and Thomson 1981). Other denominators are also used.
The odds ratios for a locus are the ratios of average risks to individuals with known genotypes. Let f+/+, f+/−, and f−/− be the average risks to individuals with genotypes +/+, +/−, and −/− at a locus. The two odds ratios of interest are OR2 = f+/+/f−/− and OR1 = f+/−/f−/−.
Analytic results for the unconstrained multiplicative model:
For the unconstrained multiplicative model (Risch 1990), the overall risk to an individual is the product across loci: 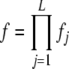 . Therefore, in the notation used here,
. Therefore, in the notation used here,
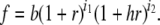 |
(5) |
The unconstrained multiplicative model is particularly simple to analyze because the average risk (K) and the risks to relatives of affected individuals are obtained by finding the contribution for each locus and then multiplying across loci (Risch 1990).
The average risk attributable to locus j is  and hence the prevalence is
and hence the prevalence is
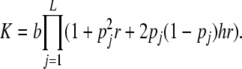 |
(6) |
Risch (1990) showed that 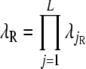 , where
, where
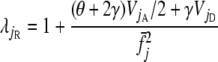 |
(7) |
and VjA and VjD are the additive and dominance components of the variance in risk attributable to locus j. Using the standard theory of quantitative genetics (Falconer 1981),
 |
(8) |
and
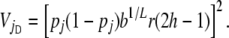 |
(9) |
The PAR for the multiplicative model depends only on pj:
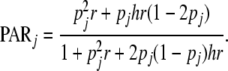 |
(10) |
If 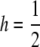 , this equation reduces to
, this equation reduces to 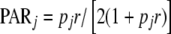 .
.
Obviously OR1 = 1 + r and OR2 = 1 + hr.
Numerical analysis:
To obtain numerical results I wrote a Mathematica program to evaluate the above expressions. It provides exact results but is very slow if L > 15. To analyze models with larger L, I wrote a stochastic simulation program in C that randomly generates pairs of genotypes in relatives by using Equation 1 with specified values of θ and γ, computes the risk for each genotype generated, and then averages over a large number of replicates. Results from the simulation program agree with those from the Mathematica program and with the analytic results for the multiplicative model. Both the Mathematica and C programs numerically determine the average allele frequency necessary to obtain a specified K. Copies of the Mathematica and C programs are available from the Slatkin lab web site.
I first consider the unconstrained multiplicative model and then show that the other models produce comparable results. If 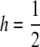 and pj = p, the analytic results for the unconstrained multiplicative model reduce to
and pj = p, the analytic results for the unconstrained multiplicative model reduce to
 |
(11) |
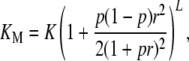 |
(12) |
and
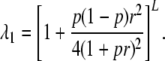 |
(13) |
There are only four parameters, p, b, r, and L. Typical values for the odds ratios found in recent genomewide association (GWA) studies are on the order of 2 for individuals homozygous for the SNP associated with the disease, so it is reasonable to set r to 1. There are then three equations satisfied by the remaining three parameters. These equations can be used to find parameter values consistent with low K, and relatively high KM, and λ1. For example, if b = 5.7 × 10−8, p = 0.1882, and L = 70, then K = 0.01, KM = 0.4, and λ1 = 6.48. Other results for this model are shown in Table 1 in the column for the unconstrained multiplicative model.
TABLE 1.
Comparison of results from simulations of the multiplicative, threshold, and constrained additive models with parameter values chosen so that the dependence of f on i2 with i1 = 0 is similar
| Multiplicative
|
||||
|---|---|---|---|---|
| Unconstrained | Constrained | Threshold | Additive | |
| p | 0.1883 | 0.1889 | 0.198 | 0.204 |
| KM | 0.33 | 0.16 | 0.13 | 0.29 |
| λ1 | 6.5 | 4.9 | 4.6 | 7.80 |
| λ2 | 3.2 | 2.3 | 2.3 | 3.3 |
| λ3 | 1.6 | 1.5 | 1.6 | 1.8 |
| PAR | 0.08 | 0.08 | 0.08 | 0.10 |
| OR1 | 1.5 | 1.48 | 1.46 | 1.57 |
| OR2 | 2.0 | 1.94 | 2.08 | 2.40 |
In all cases, K = 0.01. The parameter values for the four models illustrated in Figure 1 were used.
Although these parameters predict the desired results, there are two problems. First, the risk is >1 for individuals with a sufficient number of + alleles. In fact, with these parameters, the risk for an individual homozygous for + at all loci exceeds 1010. Second, assuming 70 unlinked loci with odds ratios of 2 makes the assumption of free recombination implausible. With only 26 chromosome pairs, 70 randomly chosen loci are not likely to be all freely recombining.
The effect of allowing f > 1 is minor because, under HWLE, individuals with risks >1 are present only in very low frequencies. In this case, Pr(f > 1) = 3.67 × 10−4. Table 1 shows that simply setting the risks for those individuals to 1—the constrained multiplicative model—makes only a small difference in p although a somewhat larger difference in the recurrence risks. The average risk, from which p is computed, depends on the first moment of f while the recurrence risks depend on the second moment: hence the greater sensitivity of the recurrence risks to disallowing unfeasibly large f. Other results for this model are shown in Table 1.
In general, linkage of pairs of loci increases recurrence risk. The reason is that linkage causes the probabilities of IBD at pairs of loci to differ from the product of the probabilities for each locus separately. For example, in full siblings the probability that loci on two randomly chosen chromosomes have both alleles IBD is not the square of the one-locus probabilities,  , but is slightly larger, (1 + (1 − 2c)2)/4, where c is the recombination rate between the loci (Lynch and Walsh 1998, p. 147). Consequently, the covariance in risk between full siblings is slightly elevated because multiplicative interactions between loci create a small additive × additive epistatic component of the variance. The effect will be minor unless loci are very closely linked or several loci are linked, in which case a simulation study would be required to determine the exact dependence of recurrence risks on the linkage map. Interestingly, the recurrence risk for parent–offspring pairs is unaffected by linkage because exactly one allele at every locus is necessarily IBD.
, but is slightly larger, (1 + (1 − 2c)2)/4, where c is the recombination rate between the loci (Lynch and Walsh 1998, p. 147). Consequently, the covariance in risk between full siblings is slightly elevated because multiplicative interactions between loci create a small additive × additive epistatic component of the variance. The effect will be minor unless loci are very closely linked or several loci are linked, in which case a simulation study would be required to determine the exact dependence of recurrence risks on the linkage map. Interestingly, the recurrence risk for parent–offspring pairs is unaffected by linkage because exactly one allele at every locus is necessarily IBD.
We can understand why we are getting these results from the two multiplicative models by considering further the relationship between recurrence risk and the distribution of risk. In general, large KM is associated with large λ1. Recall that 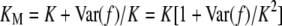 . If KM ≫ K, then Var(f)/K2 ≫ 1. For example, if K = 0.01 and KM = 0.3, then Var(f)/K2 = 29. Because f > 0, that condition can be satisfied only if most of the population has very low risk and a few individuals have much higher risks. For example, if f is beta distributed, i.e.,
. If KM ≫ K, then Var(f)/K2 ≫ 1. For example, if K = 0.01 and KM = 0.3, then Var(f)/K2 = 29. Because f > 0, that condition can be satisfied only if most of the population has very low risk and a few individuals have much higher risks. For example, if f is beta distributed, i.e., 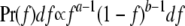 , then a = 0.34 and b = 3.31 for Var(f)/K2 = 29. The assumption of multiplicative interactions, with or without the constraint that f ≤ 1, is only one way to obtain a risk function with such a large coefficient of variation of risk. Any function for which the risk increases steeply in the narrow range of genotypes present in relatively high frequency will have the same qualitative properties. To illustrate, consider the threshold model with parameters chosen to approximate the constrained multiplicative model. The solid line in Figure 1 shows the risk under the multiplicative model as a function of i2 with i1 = 0 for the parameters given above. The dashed line in Figure 1 shows the dependence of f on i2 for the threshold model with b = 5.7 × 10−8, p = 0.198, T = 22, σe = 1.5, and L = 70. The simulation results for this model are similar to those from both multiplicative models (Table 1). Even the additive model produces similar results provided that it is constrained so that values <0 are set to 0 and values >1 are set to 1 (Table 1). These results demonstrate that it is not the multiplicative interaction among loci but the steep increase in risk that creates the pattern of low prevalence and high recurrence risk.
, then a = 0.34 and b = 3.31 for Var(f)/K2 = 29. The assumption of multiplicative interactions, with or without the constraint that f ≤ 1, is only one way to obtain a risk function with such a large coefficient of variation of risk. Any function for which the risk increases steeply in the narrow range of genotypes present in relatively high frequency will have the same qualitative properties. To illustrate, consider the threshold model with parameters chosen to approximate the constrained multiplicative model. The solid line in Figure 1 shows the risk under the multiplicative model as a function of i2 with i1 = 0 for the parameters given above. The dashed line in Figure 1 shows the dependence of f on i2 for the threshold model with b = 5.7 × 10−8, p = 0.198, T = 22, σe = 1.5, and L = 70. The simulation results for this model are similar to those from both multiplicative models (Table 1). Even the additive model produces similar results provided that it is constrained so that values <0 are set to 0 and values >1 are set to 1 (Table 1). These results demonstrate that it is not the multiplicative interaction among loci but the steep increase in risk that creates the pattern of low prevalence and high recurrence risk.
Figure 1.—

Graphs of risk functions  for the models described in the text plotted against i2, the number of heterozygous loci. For the two multiplicative models, b = 5.7 × 10−8, r = 1; for the threshold model, b = 5.7 × 10−8, T = 22, and σe = 2.5; for the constrained additive model,
for the models described in the text plotted against i2, the number of heterozygous loci. For the two multiplicative models, b = 5.7 × 10−8, r = 1; for the threshold model, b = 5.7 × 10−8, T = 22, and σe = 2.5; for the constrained additive model,  and
and 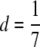 .
.
Additional simulation results confirm this conclusion. The patterns are easiest to see in the threshold model because the background risk (b), the range of genotypes for which risk increases (T), and the steepness of increase (1/σe) can be varied independently. In the multiplicative model, the overall shape of the risk function depends on combinations of the parameters. For example, the value of i2 for which f = 0.5 (corresponding to T in the threshold model) is 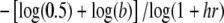 .
.
The background risk, b, makes little difference in the results as long as it is substantially smaller than the average risk, K. Table 2 shows some typical results for a series of cases in which K was constrained to 0.01. This lack of sensitivity to changes in b confirms that the behavior of the risk function only as risk starts to increase determines patterns of recurrence risk and other measurable quantities.
TABLE 2.
Effect of varying b, the background risk, in the threshold model
| b | p | PAR | OR1 | OR2 | KM | λ1 |
|---|---|---|---|---|---|---|
| 10−6 | 0.199 | 0.08 | 1.46 | 2.09 | 0.13 | 4.6 |
| 10−5 | 0.199 | 0.08 | 1.46 | 2.08 | 0.13 | 4.5 |
| 10−4 | 0.198 | 0.08 | 1.45 | 2.08 | 0.13 | 4.4 |
| 10−3 | 0.197 | 0.08 | 1.41 | 1.98 | 0.12 | 4.0 |
In all cases, L = 70, T = 22, σe = 2.5, h = 0.5, and p is adjusted so that K = 0.01. All results are based on the averages of 106 replicates of the simulation program described in the text.
If the model is fixed and L is allowed to vary, again holding K constant, the main effect is to increase p, with a smaller effect on KM, λ1, and OR2 as shown in Figure 2. The results are similar for other combinations of parameters. Assuming a smaller number of loci does not affect the qualitative conclusions and it reduces any effects of linkage.
Figure 2.—

Dependence of p, OR2, KM, and λ1 on L, the number of loci, while other parameters are held constant. In all cases, the threshold model was used with 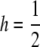 , b = 10−5, T = 22, and σe = 2.5. For each set of parameter values p was chosen so K = 0.01. All results are based on 106 replicates of the simulation program described in the text.
, b = 10−5, T = 22, and σe = 2.5. For each set of parameter values p was chosen so K = 0.01. All results are based on 106 replicates of the simulation program described in the text.
Increasing T while holding L and the other parameters fixed has the opposite effect to increasing L: p increases with increasing T, although KM and λ1 are somewhat more sensitive to changes in T (Figure 3).
Figure 3.—

Dependence of p, OR2, KM, and λ1 on T, the number of loci, while other parameters are held constant. In all cases, the threshold model was used with 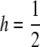 , b = 10−5, L = 70, and σe = 2.5. For each set of parameter values p was chosen so K = 0.01. All results are based on 106 replicates of the simulation program described in the text.
, b = 10−5, L = 70, and σe = 2.5. For each set of parameter values p was chosen so K = 0.01. All results are based on 106 replicates of the simulation program described in the text.
Changes in σe have almost no effect on p, but the recurrence risks all increase as σe becomes smaller, thus confirming the importance of the steepness of the risk function for recurrence risks. Some results are shown in Table 3.
TABLE 3.
Effect of varying σe in the threshold model
| σe | p | PAR | OR1 | OR2 | KM | λ1 |
|---|---|---|---|---|---|---|
| 1.0 | 0.22 | 0.11 | 1.6 | 2.5 | 0.49 | 10.7 |
| 1.5 | 0.22 | 0.10 | 1.6 | 2.4 | 0.32 | 8.2 |
| 2.5 | 0.20 | 0.08 | 1.5 | 2.1 | 0.13 | 4.6 |
| 3.5 | 0.18 | 0.06 | 1.4 | 1.9 | 0.06 | 2.7 |
| 4.5 | 0.15 | 0.04 | 1.3 | 1.7 | 0.03 | 2.0 |
In all cases, L = 70, T = 22, h = 0.5, b = 10−5, and p is adjusted so that K = 0.01. All results are based on the averages of 106 replicates of the simulation program described in the text.
The results presented so far assume p is the same at every locus. If p varies among loci, the results are surprisingly similar. An example is shown in Table 4. The parameter values are the same as for the threshold model in Table 1 and Figure 1. The value of p that yielded K = 0.01 was used as the mean of a beta distribution with a specified coefficient of variation (CV) to generate a set of pj. That set of pj was tested to determine whether 0.09 < K < 0.11 in the simulation program, and the process continued until a set of pj satisfying that condition was obtained. Then the simulation program computed the other quantities of interest. Results in Table 4 are based on averages of 106 replicates for each of five independent sets of pi. The realized coefficients of variation in the five sets are 0.78, 0.72, 0.77, 0.71, and 0.72.
TABLE 4.
Effect of allowing for variation in allele frequencies among loci
| CV | KM | λS | λ1 | λ2 | λ3 |
|---|---|---|---|---|---|
| 0 | 0.13 | 4.6 | 4.6 | 2.3 | 1.6 |
| 0.5 | 0.12 | 4.4 | 4.4 | 2.3 | 1.5 |
| 0.75 | 0.11 | 4.1 | 4.1 | 2.2 | 1.5 |
In all cases, the threshold model with L = 70, 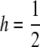 , b = 10−5, and T = 22 was used. All results for the cases with CV > 0 are averages of five sets of pj generated as described in the text, with 106 replicates for each set.
, b = 10−5, and T = 22 was used. All results for the cases with CV > 0 are averages of five sets of pj generated as described in the text, with 106 replicates for each set.
Overall the effect of increasing CV is to reduce recurrence risks slightly. The reason is simple. Allowing variation in pj reduces the extent of variation in i1 and i2 in the population. Recurrence risks reflect the shape of the risk function within a narrow range of genotypes. Hence, reducing the range of genotypes somewhat reduces recurrence risks.
The results so far assume 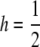 . Varying h has a strong effect on p: lower h requires larger p to obtain the same K, as shown in Figure 4. Once again, whether p is large or small depends on the details of the risk model. Surprisingly, the recurrence risks are slightly smaller for intermediate h, probably because assuming the same p at all loci constrains the variation in genotypic values more for h = 0 or 1 than for intermediate values of h.
. Varying h has a strong effect on p: lower h requires larger p to obtain the same K, as shown in Figure 4. Once again, whether p is large or small depends on the details of the risk model. Surprisingly, the recurrence risks are slightly smaller for intermediate h, probably because assuming the same p at all loci constrains the variation in genotypic values more for h = 0 or 1 than for intermediate values of h.
Figure 4.—

Dependence of p, KM, and λ1 on h, the degree of dominance, while other parameters are held constant. In all cases, the threshold model was used with L = 70, b = 10−5, T = 22, and σe = 2.5. For each set of parameter values p was chosen so K = 0.01. All results are based on 106 replicates of the simulation program described in the text.
Testing for epistasis:
Several GWA studies have tested for interactions among SNPs found in their studies and have concluded that there is no evidence for deviations from the multiplicative model. The most extensive testing was done by Maller et al. (2006) in a study of three loci containing five SNPs affecting the risk of age-related macular degeneration (AMD). Together these five SNPs account for about half of the recurrence risk to siblings. Maller et al. found no significant deviations from the multiplicative model. The question is whether significant deviations would have been found if the threshold model were the true model. To answer this question, I simulated a threshold model with L = 20, b = 10−5, T = 10, σe = 1.5, and pj generated from a beta distribution with CV = 0.75, as described above. The average frequency needed to obtain K = 0.01 is p = 0.259. Then the first five loci for which pj > 0.1 were assumed to be recognized as causative. One thousand cases and 1000 controls were randomly sampled from the simulated population and tested for a significant deviation from the multiplicative model. The sample sizes in the Maller et al. study were 1238 cases and 934 controls. Eight of 100 replicates of this experiment deviated from the multiplicative model at the 5% level. In a similar experiment with the multiplicative model, 5 of 100 replicates deviated at the 5% level.
Population-attributable risk:
The PAR of an allele is proportional to the difference in allele frequency in cases and controls. It is a convenient description of an allele's effect because the difference in allele frequencies can be related to standard population-genetics theory of selection (Lynch and Walsh 1998). The constant of proportionality differs among studies. Here I use the scaling suggested by Bengtsson and Thomson (1981, Equation 4). When p varies among loci, the simulation program described above computes PAR for each locus. Figure 5a shows scatter plots of PARj against pj for the unconstrained multiplicative, threshold, and constrained additive models shown in Figure 1, with the CV in pj being 0.75. The results for the unconstrained multiplicative model follow the prediction of Equation 10. The comparable threshold and additive models produce similar results, as shown.
Figure 5.—

Allele frequency vs. population-attributable risk (PAR). (a) Results from simulations of the three models (unconstrained multiplicative, threshold, and constrained additive) illustrated in Figure 1. In all cases, K = 0.01, CV = 0.75, and results are based on a single set of pj generated as described in the text and 106 replicates for each model. PAR is the population-attributable risk calculated from Equation 4 in the text. (b) Scatter plot of data in Table 5.
To determine whether there is any similarity between these results and available information from GWA studies, I compiled frequencies of SNPs significantly associated with type II diabetes in three recent GWA studies (Scott et al. 2007; Sladek et al. 2007; Zeggini et al. 2007). The results are summarized in Table 5 and the scatter plot of PAR vs. p is shown in Figure 5b. Except for one SNP (rs9300039), values of PAR are not far from a single line, suggesting that the assumption of equal effects across loci, at least those detected in these studies, may be roughly valid.
DISCUSSION AND CONCLUSIONS
The conclusion from the analysis presented here is that the genetic architecture of complex inherited diseases with relatively high heritabilities is constrained in such a way that there has to be a large variance in risk among genotypes present in a population. A large variance in risk can be achieved under a variety of models of gene interaction, including the exchangeable multiplicative, threshold, and additive models examined here. The observations that have supported the use of the multiplicative model—namely the pattern of decrease of risk to first-, second-, and third-degree relatives and the failure to reject the multiplicative model for SNPs identified in GWA studies—are consistent with other models as well, provided that they result in the right pattern of variation in risk. The assumption of Hardy–Weinberg frequencies and linkage equilibrium implies that the distribution among individuals of the number of causative alleles is narrow. The risk function only for genotypes that are present in appreciable frequencies affects observable quantities.
An alternative and more positive way to view these results is that, because other models of gene interaction create patterns that are hardly distinguishable from the multiplicative model even under idealized conditions, that model can be used for many practical purposes. That is true, but the utility the multiplicative model for some purposes does not mean that its assumptions are true in general. Conclusions from population genetic models that assume multiplicative interactions across loci have to be checked for robustness to ensure they are still valid under more general models of the kind analyzed here.
The models analyzed in this article assumed equal effects on risk of a possibly unrealistic number of unlinked loci. These models are not intended to represent the architecture of any particular complex inherited disease but instead allow exploration of the consequences of what is assumed about many diseases, namely that they are affected by numerous loci that independently increase risk. More realistic models that are consistent with observations have to have the same overall property that the variance in risk has to be relatively large.
The results presented here have some bearing on the question of whether alleles that cause inherited diseases tend to be common or rare. Population genetics theory has been used to argue both for and against the generalization that complex diseases are caused by common alleles (Pritchard 2001; Reich and Lander 2001; Pritchard and Cox 2002). Even if the number of loci (L) that can carry causative alleles is relatively large, the number that has to interact to produce substantially elevated risk (T in the threshold model) may be small or large. Recurrence risk data do not strongly constrain T. If T is relatively small, then causative allele frequencies have to be in low frequency. Otherwise, average risk would be too high. If most causative alleles are common, T has to be much larger, implying that the combined effects of many loci are required for risk to be elevated.
Acknowledgments
I thank N. B. Freimer and G. J. Thomson for helpful discussions of this topic, A. Albrechtsen for writing the program in R that did the statistical test for epistasis, and R. R. Hudson and the referees for helpful comments on an earlier version of this manuscript. This research was supported in part by grant R01-GM40282 from the National Institutes of Health.
References
- Amundadottir, L. T., S. Thorvaldsson, D. F. Gudbjartsson, P. Sulem, K. Kristjansson et al., 2004. Cancer as a complex phenotype: pattern of cancer distribution within and beyond the nuclear family. PLoS Med. 1 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., and M. Turelli, 2004. Effects of genetic drift on variance components under a general model of epistasis. Evolution 58 2111–2132. [DOI] [PubMed] [Google Scholar]
- Bengtsson, B. O., and G. Thomson, 1981. Measuring the strength of associations between HLA antigens and diseases. Tissue Antigens 18 356–363. [DOI] [PubMed] [Google Scholar]
- Cannings, C., 1974. The latent roots of certain Markov chains arising in genetics: a new approach, 1. Haploid models. Adv. Appl. Probab. 6 260–290. [Google Scholar]
- Curnow, R. N., and C. Smith, 1975. Multifactorial models for familial diseases in man. J. R. Stat. Soc. Ser. A Stat. Soc. 138 131–169. [Google Scholar]
- Edwards, J. H., 1960. The simulation of Mendelism. Acta Genet. Stat. Med. 10 63–70. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., 1981. Introduction to Quantitative Genetics. Longman, New York.
- Farrall, M., and S. Holder, 1992. Familial recurrence-pattern analysis of cleft lip with or without cleft palate. Am. J. Hum. Genet. 50 270–277. [PMC free article] [PubMed] [Google Scholar]
- James, J. W., 1971. Frequency in relatives for an all-or-none trait. Ann. Hum. Genet. 35 47–49. [DOI] [PubMed] [Google Scholar]
- Lindsey, J. W., 2005. Familial recurrence rates and genetic models of multiple sclerosis. Am. J. Med. Genet. A 135A 53–58. [DOI] [PubMed] [Google Scholar]
- Liu, W., and B. Weir, 2005. Genotypic probabilities for pairs of inbred relatives. Philos. Trans. R. Soc. B Biol. Sci. 360 1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Maller, J., S. George, S. Purcell, J. Fagerness, D. Altshuler et al., 2006. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat. Genet. 38 1055–1059. [DOI] [PubMed] [Google Scholar]
- Morton, N. E., S. Yee, R. C. Elston and R. Lew, 1970. Discontinuity and quasi-continuity: alternative hypotheses of multifactorial inheritance. Clin. Genet. 1 81–94. [Google Scholar]
- Oksenberg, J. R., and L. F. Barcellos, 2005. Multiple sclerosis genetics: leaving no stone unturned. Genes Immun. 6 375–387. [DOI] [PubMed] [Google Scholar]
- Peng, B., and M. Kimmel, 2007. Simulations provide support for the common disease-common variant hypothesis. Genetics 175 763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permutt, M. A., J. Wasson and N. Cox, 2005. Genetic epidemiology of diabetes. J. Clin. Invest. 115 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., 2001. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 69 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and N. J. Cox, 2002. The allelic architecture of human disease genes: common disease - common variant… or not? Hum. Mol. Genet. 11 2417–2423. [DOI] [PubMed] [Google Scholar]
- Reich, D. E., and E. S. Lander, 2001. On the allelic spectrum of human disease. Trends Genet. 17 502–510. [DOI] [PubMed] [Google Scholar]
- Risch, N., 1990. Linkage strategies for genetically complex traits: I. Multilocus models. Am. J. Hum. Genet. 46 222–228. [PMC free article] [PubMed] [Google Scholar]
- Romano-Zelekha, O., R. Hirsh, L. Blieden, M. S. Green and T. Shohat, 2001. The risk for congenital heart defects in offspring of individuals with congenital heart defects. Clin. Genet. 59 325–329. [DOI] [PubMed] [Google Scholar]
- Schliekelman, P., and M. Slatkin, 2002. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am. J. Hum. Genet. 71 1369–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, L. J., K. L. Mohlke, L. L. Bonnycastle, C. J. Willer, Y. Li et al., 2007. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek, R., G. Rocheleau, J. Rung, C. Dina, L. Shen et al., 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445 881–885. [DOI] [PubMed] [Google Scholar]
- Smith, C., 1971. Discriminating between different modes of inheritance in genetic disease. Clin. Genet. 2 303–314. [DOI] [PubMed] [Google Scholar]
- Smoller, J. W., and C. T. Finn, 2003. Family, twin, and adoption studies of bipolar disorder. Am. J. Med. Genet. C Semin. Med. Genet. 123C 48–58. [DOI] [PubMed] [Google Scholar]
- Sullivan, P. F., K. S. Kendler and M. C. Neale, 2003. Schizophrenia as a complex trait—evidence from a meta-analysis of twin studies. Arch. Gen. Psych. 60 1187–1192. [DOI] [PubMed] [Google Scholar]
- Szatmari, P., A. D. Paterson, L. Zwaigenbaum, W. Roberts, J. Brian et al., 2007. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini, E., M. N. Weedon, C. M. Lindgren, T. M. Frayling, K. S. Elliott et al., 2007. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]