

Abstract
Most quantitative trait loci (QTL) mapping experiments typically collect phenotypic data on multiple correlated complex traits. However, there is a lack of a comprehensive genomewide mapping strategy for correlated traits in the literature. We develop Bayesian multiple-QTL mapping methods for correlated continuous traits using two multivariate models: one that assumes the same genetic model for all traits, the traditional multivariate model, and the other known as the seemingly unrelated regression (SUR) model that allows different genetic models for different traits. We develop computationally efficient Markov chain Monte Carlo (MCMC) algorithms for performing joint analysis. We conduct extensive simulation studies to assess the performance of the proposed methods and to compare with the conventional single-trait model. Our methods have been implemented in the freely available package R/qtlbim (http://www.qtlbim.org), which greatly facilitates the general usage of the Bayesian methodology for unraveling the genetic architecture of complex traits.
COMPLEX traits involve effects of a multitude of genes in an interacting network. Mapping quantitative trait loci (QTL) means inferring the genetic architecture (number of genes, their positions, and their effects) underlying these complex traits. The QTL mapping problem has several salient features: first, the predictor variables in the regression (the genotypes of QTL) are not observed; second, it is really a model selection problem as there are typically thousands of loci to choose from; and third, the genomic loci on the same chromosome are correlated. Much has been done in this regard, especially in the univariate case (e.g., Lander and Botstein 1989; Jiang and Zeng 1997; Broman and Speed 2002). Bayesian methods have been very successful in the QTL mapping framework (Satagopan and Yandell 1996; Yi and Xu 2002; Yi et al. 2003, 2005, 2007; Yi 2004); see a recent review by Yi and Shriner (2008).
Most of these methods are applicable to mapping QTL for a single trait. However, in QTL experiments typically data on more than one trait are collected and, more often than not, they are correlated. It seems natural to jointly analyze these correlated traits. There are two distinct advantages for jointly analyzing correlated traits: including information from all traits can increase the power to detect QTL and the precision of the estimated QTL effects. Biologically, it is imperative to jointly analyze correlated traits to answer questions like pleiotropy (one gene influencing more than one trait) and/or close linkage (different QTL physically close to each other influencing the traits). Testing these hypotheses is key to understanding the underlying biochemical pathways causing complex traits, which is the ultimate goal of QTL mapping.
Several methods have been developed to jointly analyze multiple correlated traits. Some of them use a maximum-likelihood-based approach (Jiang and Zeng 1995; Jackson et al. 1999; Williams et al. 1999a,b; Vieira et al. 2000; Huang and Jiang 2003; Lund et al. 2003; Xu et al. 2005) or a least-squares approach (Knott and Haley 2000; Hackett et al. 2001). Most of these methods involve a single-QTL model or at most very few QTL. A problem with the likelihood-based approach is that with increasing complexity, due to the increase in the number of parameters to be estimated, the increase in degrees of freedom of the test statistic can restrain its practical use when the number of traits is large (Mangin et al. 1998). As a result, the advantage of joint analysis is lost over single-trait analysis. Another approach for joint analysis is to use a dimension reduction technique, namely, principal component analysis (PCA) or discriminant analysis (DA) or using canonical variables associated with the traits (Mangin et al. 1998; Mähler et al. 2002; Gilbert and Le Roy 2003, 2004), and then use the linear combination of traits to map QTL. The problem with this approach is that linear combinations of traits are not biologically interpretable and can cause spurious linkages (Mähler et al. 2002; Gilbert and Le Roy 2003). Gilbert and Le Roy (2003) compared the performance of PCA, DA, and the multivariate model in a full-sib family and half-sib families under different scenarios. Lange and Whittaker (2001) use a nonparametric generalized estimating equations approach to multivariate QTL mapping.
Meuwissen and Goddard (2004) used a Markov chain Monte Carlo (MCMC) algorithm to map QTL, using linkage disequilibrium and linkage information for multiple-traits data. Recently, Liu et al. (2007) developed a Bayesian approach to map QTL for a combination of normal and ordinal traits in a full-sib design based on the variance components approach. They used a reversible-jump (RJ) MCMC to estimate the unknown number of QTL. The problem with RJ-MCMC is that increased complexity drastically increases the computational burden, rendering it unsuitable for genomewide scans where typically thousands of positions are scanned for a putative QTL. Another major challenge is to ascertain convergence of the RJ sampler and obtain a rapidly converging sampler (Yi 2004). Yang and Xu (2007) extended the Bayesian shrinkage analysis with a fixed-interval approach (Wang et al. 2005), where a QTL is placed in each marker interval, to a moving-interval approach, where the position of a QTL can be searched in a range that covers many marker intervals for dynamic/longitudinal traits using a Legendre polynomial. Their method, however, focuses on the study of the growth trajectory of time-dependent or repeated-measures types of outcomes (called dynamic traits) and is very different from our approach.
All the multivariate methods mentioned here use the traditional multivariate regression model, which assumes the same genetic model for all traits. However, almost all correlated traits are actually affected to some extent by a different multilocus network. To capture this facet of multiple traits we use the so-called “seemingly unrelated regression” (SUR) model (Zellner 1962), which allows each trait to have a different set of QTL. Verzilli et al. (2005) implemented a Bayesian version of SUR using RJ-MCMC to jointly analyze multiple correlated traits with SNP data in a human population. They found it difficult “to deal with very many loci” and restricted attention to only 12 SNPs. Their method appears unsuitable to genomewide scans.
In the literature of joint analysis for QTL mapping, there is a lack of comprehensive genomewide strategies to map multiple pleiotropic and nonpleiotropic QTL. In this article, we extend the composite model space approach of Yi (2004) to jointly analyze multiple correlated continuous traits. Multiple traits are modeled using novel QTL SUR models that enable us to detect either the same or different QTL for different traits, facilitating the separation of pleiotropy and close linkage. The QTL SUR models include the traditional multivariate model and the single trait-by-trait model as special cases. We develop computationally efficient MCMC algorithms for performing joint analysis. Finally, we conduct extensive simulation studies to assess the performance of the proposed methods.
BAYESIAN MODELING OF MULTIPLE QTL FOR MULTIPLE TRAITS
QTL SUR models:
We focus our attention on experimental crosses derived from two inbred lines. Observed data in QTL mapping consist of phenotypic values of complex traits and molecular marker data. We extend the composite model space approach of Yi (2004) to jointly analyze multiple correlated continuous traits. We assume that the marker data include not only the marker genotypes but also the genomic positions of the markers. We approximate positions for all possible QTL using a partition of the entire genome into evenly spaced loci, including all observed markers and additional loci (called pseudomarkers) between flanking markers (Sen and Churchill 2001; Yi et al. 2005). Inserting pseudomarkers enables us to detect potential QTL within the marker intervals, but introduces a special statistical problem; i.e., QTL genotypes are unobserved. Before mapping QTL, we calculate the probabilities of genotypes at these preset loci given the observed marker data as priors of QTL genotypes in our Bayesian framework.
The actual number of detectable QTL for each trait in a particular experiment is unknown, but usually not too large. We employ a composite model space approach (Yi 2004; Yi et al. 2005) and consider at most L possible loci. The upper bound L is larger than the number of detectable QTL with high probability for a given data set and can be set on the basis of the initial analyses using conventional mapping methods on each trait (Yi 2004; Yi et al. 2005). Conditioning on the genotypes at these L loci for all individuals, the phenotypic values yti for individual i on trait t can be expressed as a linear regression,
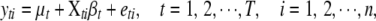 |
(1) |
where T and n represent the numbers of traits and individuals, respectively, the subscripts t and i represent the tth trait and the ith individual, respectively, μt is the overall mean for trait t, Xti is the row vector of the main-effect predictors of L loci, determined from the genotypes by using a particular genetic model [we use the Cockerham genetic model, although other genetic models are possible (Kao and Zeng 2002; Zeng et al. 2005)], βt is the vector of all main effects for L loci of trait t, and the vector of residual errors across traits, ei, is independent and normal with mean 0 and covariance matrix  ; i.e.,
; i.e., 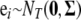 . Thus, the residual errors are independent among individuals, but are correlated among traits within individuals. The above equations can be rewritten as
. Thus, the residual errors are independent among individuals, but are correlated among traits within individuals. The above equations can be rewritten as
 |
(2) |
where 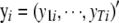 ,
,  ,
,  , and
, and  . This model can include a large number of effects, many of which are irrelevant to modeling the phenotype and should be excluded from the model. We use an unobserved vector of indictor variables
. This model can include a large number of effects, many of which are irrelevant to modeling the phenotype and should be excluded from the model. We use an unobserved vector of indictor variables 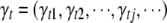 to indicate which effects
to indicate which effects 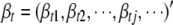 are included in (
are included in (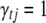 ) or excluded from (
) or excluded from ( ) the model for the tth trait. We denote the genomic positions of L loci for trait t by the vector
) the model for the tth trait. We denote the genomic positions of L loci for trait t by the vector 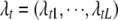 . The vector
. The vector 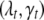 thus determines the genetic architecture of the tth trait, i.e., the actual number of QTL, their positions, and the activity of the associated genetic effects. Our goal is to infer the posterior distribution of
thus determines the genetic architecture of the tth trait, i.e., the actual number of QTL, their positions, and the activity of the associated genetic effects. Our goal is to infer the posterior distribution of 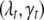 and estimate the associated genetic effects.
and estimate the associated genetic effects.
Model (1) or (2) uses trait-specific effect predictors 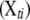 , positions
, positions 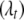 , and indicator variables
, and indicator variables 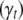 , allowing each trait to have a different set of QTL or a different genetic model. Therefore, models for different traits seem unrelated, but actually are related through correlated residual errors (or observed phenotypes) or the genotypes of linked QTL. Hereafter, we refer the above model as the QTL SUR model. We consider two different SUR models. In the first model as described above, different traits can have different sets of L loci
, allowing each trait to have a different set of QTL or a different genetic model. Therefore, models for different traits seem unrelated, but actually are related through correlated residual errors (or observed phenotypes) or the genotypes of linked QTL. Hereafter, we refer the above model as the QTL SUR model. We consider two different SUR models. In the first model as described above, different traits can have different sets of L loci 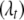 and thus different indicator variables
and thus different indicator variables 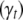 and predictors
and predictors 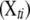 . The second SUR model uses the same set of L loci, i.e.,
. The second SUR model uses the same set of L loci, i.e., 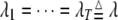 and thus
and thus 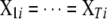 , but different indicator variables for different traits. We denote these two SUR models by SUR modeling with different loci used for all traits (SURd) and SUR modeling with the same loci used for all traits (SURs). Note that both QTL SUR models include two existing models as special cases, the univariate single-trait approach (STA) where the residual errors are unrelated, i.e.,
, but different indicator variables for different traits. We denote these two SUR models by SUR modeling with different loci used for all traits (SURd) and SUR modeling with the same loci used for all traits (SURs). Note that both QTL SUR models include two existing models as special cases, the univariate single-trait approach (STA) where the residual errors are unrelated, i.e., 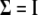 , and the traditional multivariate (TMV) model where all traits have the same set of loci and the same indicator variables, i.e.,
, and the traditional multivariate (TMV) model where all traits have the same set of loci and the same indicator variables, i.e., 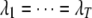 ,
, 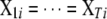 , and
, and 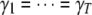 .
.
Prior distributions:
To complete Bayesian modeling of QTL SUR, we need to specify prior distributions for all unknowns. We describe the prior distributions for the model SURd in detail (appendix a), which can be easily adapted to the models SURs and TMV. For SURd, unknowns include the positions  , indicator variables
, indicator variables 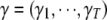 , main effects
, main effects 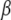 , overall mean
, overall mean  , residual covariance matrix
, residual covariance matrix 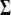 , and genotypes
, and genotypes 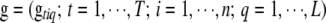 , where
, where 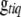 is the genotype of individual i for trait t at locus q.
is the genotype of individual i for trait t at locus q.
As described in the previous section, the prior on 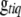 is the probability of the genotype given the observed marker data. For computational reasons, we directly work on the inverse matrix
is the probability of the genotype given the observed marker data. For computational reasons, we directly work on the inverse matrix 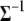 instead of
instead of 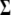 (see the next section and appendix b). The prior for
(see the next section and appendix b). The prior for 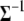 can be taken to be the commonly used noninformative prior; i.e.,
can be taken to be the commonly used noninformative prior; i.e., 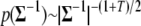 (see Gelman et al. 2004). We assume that the unknowns
(see Gelman et al. 2004). We assume that the unknowns 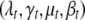 are independent among the traits. For each trait, the priors on
are independent among the traits. For each trait, the priors on  can be specified as in Yi et al. (2005, 2007), which we describe in appendix a.
can be specified as in Yi et al. (2005, 2007), which we describe in appendix a.
MARKOV CHAIN MONTE CARLO ALGORITHM
We fit the models using the MCMC algorithm, applied to the joint posterior density of all the unknowns 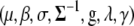 . The joint posterior distribution can be expressed as
. The joint posterior distribution can be expressed as
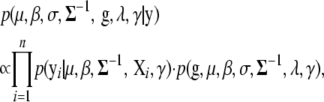 |
(3) |
where the likelihood 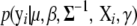 is defined by model (2), and the prior
is defined by model (2), and the prior 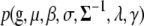 is described in the last section and appendix a, and the augmentation with hyperparameters σ presents the prior variances for the effects
is described in the last section and appendix a, and the augmentation with hyperparameters σ presents the prior variances for the effects 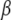 (Yi et al. 2007; see appendix a). For notational convenience, we suppress the dependence on the observed marker data here and afterward.
(Yi et al. 2007; see appendix a). For notational convenience, we suppress the dependence on the observed marker data here and afterward.
The joint posterior distribution can be simulated using the Gibbs sampler and Metropolis algorithm, alternately updating each unknown conditional on all other parameters and the observed data. We show all the conditional distributions in appendix b. Conditional updates of 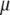 ,
, 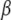 ,
, 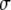 , and
, and 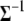 are the same for the models SURd, SURs, and TMV. However, conditional updates of g,
are the same for the models SURd, SURs, and TMV. However, conditional updates of g, 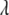 , and
, and  are illustrated only for the SURd model, which can be easily adapted to the SURs and TMV models (see appendix b). Below, we describe our algorithm, with more details on steps for unknowns where the method involves explicit extension for multiple correlated traits.
are illustrated only for the SURd model, which can be easily adapted to the SURs and TMV models (see appendix b). Below, we describe our algorithm, with more details on steps for unknowns where the method involves explicit extension for multiple correlated traits.
A commonly used updating scheme for the overall means and the coefficients is performed by updating jointly 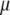 and
and 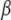 for all traits (see Smith and Kohn 2000; Griffiths 2001; Verzilli et al. 2005). This scheme requires large matrix operations at each simulation iteration, resulting in prohibitive computational burden for genomewide multiple-QTL analysis. We have developed a pure Gibbs sampler to update one parameter at a time: for each t and j, we sample
for all traits (see Smith and Kohn 2000; Griffiths 2001; Verzilli et al. 2005). This scheme requires large matrix operations at each simulation iteration, resulting in prohibitive computational burden for genomewide multiple-QTL analysis. We have developed a pure Gibbs sampler to update one parameter at a time: for each t and j, we sample 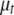 and
and  from their conditional posterior distributions, respectively, which are normal distributions (see Equations B1 and B2 in appendix b). This one-at-a-time algorithm never requires matrix operations and is computationally very efficient. Note that if
from their conditional posterior distributions, respectively, which are normal distributions (see Equations B1 and B2 in appendix b). This one-at-a-time algorithm never requires matrix operations and is computationally very efficient. Note that if 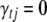 , we do not need to sample
, we do not need to sample  .
.
The variance parameters 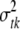 are updated one at a time: for each t and k, the conditional posterior distribution of
are updated one at a time: for each t and k, the conditional posterior distribution of 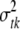 is a scaled inverse
is a scaled inverse 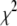 -distribution and can be directly sampled (see Equation B3 in appendix b). For computational convenience, we work on the inverse matrix
-distribution and can be directly sampled (see Equation B3 in appendix b). For computational convenience, we work on the inverse matrix 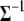 instead of
instead of 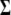 (see appendix b). The conditional posterior distribution of
(see appendix b). The conditional posterior distribution of 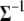 is a standard Wishart distribution, and thus both the Gibbs sampler and the Metropolis algorithm can be applied to update
is a standard Wishart distribution, and thus both the Gibbs sampler and the Metropolis algorithm can be applied to update 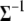 (see Equations B4 and B5).
(see Equations B4 and B5).
The genotypes are usually updated one at a time from the conditional posterior distributions. If locus q is included in the model and the genotype 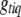 is not observed, the conditional posterior distribution of
is not observed, the conditional posterior distribution of 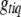 is a simple multinomial (or binomial) distribution and thus can be sampled directly (see Equation B6); otherwise, we do not need to sample
is a simple multinomial (or binomial) distribution and thus can be sampled directly (see Equation B6); otherwise, we do not need to sample  . The positions
. The positions 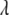 are also updated one at a time. As above, we need to update only those loci that are included in the current model. The conditional posterior distribution of (
are also updated one at a time. As above, we need to update only those loci that are included in the current model. The conditional posterior distribution of (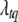 ,
, 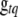 ) is not a standard distribution, and thus a Metropolis algorithm is needed to update (
) is not a standard distribution, and thus a Metropolis algorithm is needed to update (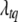 ,
,  ) (see Equations B7 and B8 in appendix b).
) (see Equations B7 and B8 in appendix b).
The indicator variables  are also updated one at a time. The binary indicator variables
are also updated one at a time. The binary indicator variables 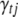 for the SUR models have independently binomial conditional posterior distributions (see Equations B9 and B12 in appendix b). At each iteration, therefore, the Gibbs sampler can be used to generate each indicator from its conditional posterior. However, for the QTL SUR models, using the Gibbs samplers is computationally demanding because the SUR models contain T times the number of indicators as a single-trait model and most of the indicators are zero. To speed up the algorithm we extend the Metropolis–Hastings (MH) algorithm proposed by Yi et al. (2007) to the QTL SUR models in a natural way (see Equation B11). This MH algorithm can be easily adapted to the TMV model.
for the SUR models have independently binomial conditional posterior distributions (see Equations B9 and B12 in appendix b). At each iteration, therefore, the Gibbs sampler can be used to generate each indicator from its conditional posterior. However, for the QTL SUR models, using the Gibbs samplers is computationally demanding because the SUR models contain T times the number of indicators as a single-trait model and most of the indicators are zero. To speed up the algorithm we extend the Metropolis–Hastings (MH) algorithm proposed by Yi et al. (2007) to the QTL SUR models in a natural way (see Equation B11). This MH algorithm can be easily adapted to the TMV model.
SUMMARIZING AND INTERPRETING THE POSTERIOR SAMPLES
Assessing the convergence and mixing behavior of any MCMC algorithm is somewhat difficult to ascertain and it is intensified for a high-dimensional problem. Several methods have been developed so far; many are implemented in R/coda (Plummer et al. 2004), an R package providing an object-based infrastructure for analyzing output of MCMC simulations and performing convergence diagnostics.
The posterior samples generated by the above MCMC algorithm contain all available information about the unknowns in the QTL SUR and thus the genetic architecture of the multiple traits. The vector (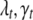 ) determines the number of QTL, their positions, and the main effects of QTL, for the tth trait and hence identifies its genetic architecture. The posterior inclusion probability for each locus is estimated as its frequency in the posterior samples. The larger the effect size is for a locus, the more frequently the locus is sampled. Taking the prior probability into consideration, we use Bayes factors (BF) to show evidence for inclusion against exclusion of a locus. Bayes factors are calculated on the basis of the idea of model averaging. The Bayes factor of the jth locus for the tth trait can be represented as the ratio of the posterior to prior odds of selecting that particular locus. Model averaging accounts for model uncertainty and hence provides more robust inference compared to a single “best” model approach (Raftery et al. 1997; Ball 2001; Sillanpää and Corander 2002).
) determines the number of QTL, their positions, and the main effects of QTL, for the tth trait and hence identifies its genetic architecture. The posterior inclusion probability for each locus is estimated as its frequency in the posterior samples. The larger the effect size is for a locus, the more frequently the locus is sampled. Taking the prior probability into consideration, we use Bayes factors (BF) to show evidence for inclusion against exclusion of a locus. Bayes factors are calculated on the basis of the idea of model averaging. The Bayes factor of the jth locus for the tth trait can be represented as the ratio of the posterior to prior odds of selecting that particular locus. Model averaging accounts for model uncertainty and hence provides more robust inference compared to a single “best” model approach (Raftery et al. 1997; Ball 2001; Sillanpää and Corander 2002).
Since the information about correlation between multiple traits is taken into account, the proposed QTL SUR model is expected to increase the probability of detecting QTL, especially weak-effect QTL. More importantly, the QTL SUR model allows for a statistically rigorous procedure to test a number of biologically important questions involving multiple traits, such as pleiotropy and pleiotropy vs. close linkage. To test if the jth locus is a pleiotropic QTL we considered all models that include the jth locus for all traits (i.e., all models with 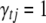 for all t) and compute the joint posterior inclusion probabilities. By jointly considering the positions λ and the indicators
for all t) and compute the joint posterior inclusion probabilities. By jointly considering the positions λ and the indicators  , one can distinguish pleiotropy and close linkage.
, one can distinguish pleiotropy and close linkage.
IMPLEMENTATION IN R/QTLBIM
The proposed methods have been implemented in R/qtlbim (Yandell et al. 2007), which is a freely available R library. The previous version of R/qtlbim performs only single-trait analysis. R/qtlbim is built on top of the widely used R/qtl (Broman et al. 2003) and provides an extensible, interactive environment for Bayesian analysis of multiple interacting QTL in experimental crosses. The MCMC algorithm is written in C and the graphics and data manipulation are performed in R.
R/qtlbim provides tools to monitor mixing behavior and convergence of the simulated Markov chain, either by examining trace plots of the sample values of scalar quantities of interest, such as the numbers of QTL and main effects, or by using formal diagnostic methods provided in the package R/coda. R/qtlbim provides extensive informative graphical and numerical summaries of the MCMC output to infer and interpret the genetic architecture of complex traits (Yandell et al. 2007).
SIMULATION STUDIES
Design and method:
With an increased complexity and sophistication of a proposed method, it is very important to compare its performance with existing methods in an objective way. To achieve this end, we conduct extensive simulation studies to compare the proposed methods for joint analysis of multiple traits among themselves and also with a single trait-by-trait analysis. Any simulation experiment is necessarily incomplete and does not represent real QTL experiments. Nevertheless, we try to simulate a relatively “realistic” QTL model and evaluate the performance with different sample sizes and correlation structures.
We consider a backcross population with sample sizes of 100, 200, and 500 to represent very small, small, and large sample sizes. Two continuous traits (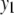 and
and 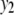 ) are considered for simplicity. We simulate a genome with 19 chromosomes, each of length 100 cM with 11 equally spaced markers (markers placed 10 cM apart) on each chromosome. Ten percent of the genotypes of these markers were assumed to be randomly missing in all cases. For each of the three sample sizes, we consider two correlation structures, namely, low and high with
) are considered for simplicity. We simulate a genome with 19 chromosomes, each of length 100 cM with 11 equally spaced markers (markers placed 10 cM apart) on each chromosome. Ten percent of the genotypes of these markers were assumed to be randomly missing in all cases. For each of the three sample sizes, we consider two correlation structures, namely, low and high with 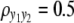 and
and  . Therefore, we have six cases with three samples sizes and two correlation structures. For each of these six cases, we simulate six QTL (Q1–Q6) that control the phenotypes: Q1 and Q2 (Q3 and Q4) are nonpleitropic QTL, influencing only the trait
. Therefore, we have six cases with three samples sizes and two correlation structures. For each of these six cases, we simulate six QTL (Q1–Q6) that control the phenotypes: Q1 and Q2 (Q3 and Q4) are nonpleitropic QTL, influencing only the trait  (
(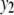 ) with moderate-sized and weak effects, respectively;
) with moderate-sized and weak effects, respectively; 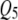 is a moderate-sized pleiotropic QTL affecting both
is a moderate-sized pleiotropic QTL affecting both  and
and 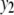 ; while
; while 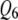 is a weak pleiotropic QTL affecting both
is a weak pleiotropic QTL affecting both 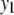 and
and 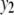 . Table 1 presents the simulated positions of six QTL, their effect values, and their heritabilities (proportion of the phenotypic variation explained by a QTL).
. Table 1 presents the simulated positions of six QTL, their effect values, and their heritabilities (proportion of the phenotypic variation explained by a QTL).
TABLE 1.
True positions of six QTL, their effects, and heritabilities
| Q1 | Q2 | Q3 | Q4 | Q5 | Q6 | |
|---|---|---|---|---|---|---|
| Chromosome | 1 | 1 | 2 | 2 | 3 | 4 |
| Position (cM) | 22 | 55 | 22 | 65 | 65 | 45 |
| y1 | 0.8 | 0.6 | 0 | 0 | 0.8 | 0.6 |
| y2 | 0 | 0 | −0.8 | −0.6 | 0.8 | 0.6 |
| y1 (%) | 8.8 | 4.9 | 0 | 0 | 8.8 | 4.9 |
| y2 (%) | 0 | 0 | 9.3 | 5.2 | 9.3 | 5.2 |
For each of the six cases, we generate 100 replicated data sets, resulting in 600 total data sets. For each of these 600 data sets we perform analysis using four methods, namely, the STA, joint analysis using a TMV model, joint analysis using a SURd model, and joint analysis using a SURs model. For all analyses, pseudomarkers were placed every 1 cM across the entire genome, resulting in a total of 1919 possible QTL positions. The prior expected number of main-effect QTL was set at l0 = 4, and the upper bound on the number of QTL was then L = 10 (= 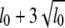 , also see Yi et al. 2005). To check posterior sensitivity to these prespecified values, we analyzed the data with several other values of l0 and L and obtained essentially identical results. We ran the MCMC algorithm for 12 × 104 times after discarding the first 1000 iterations as burn-in. The chain was thinned by considering one in every 40 samples, rendering 3000 samples from the joint posterior distribution. The saved posterior samples were used to make inference about the genetic architecture.
, also see Yi et al. 2005). To check posterior sensitivity to these prespecified values, we analyzed the data with several other values of l0 and L and obtained essentially identical results. We ran the MCMC algorithm for 12 × 104 times after discarding the first 1000 iterations as burn-in. The chain was thinned by considering one in every 40 samples, rendering 3000 samples from the joint posterior distribution. The saved posterior samples were used to make inference about the genetic architecture.
To illustrate the advantages of using a more complex method of analysis it is important to have an objective and reproducible plan of evaluation. However, in the model selection framework of multiple QTL mapping this assessment becomes a little more complicated as one has to account for model uncertainty (Burnham and Anderson 2002). The model selection uncertainty can lead to underestimation about the quantities of interest, which could be quite large as shown by Miller (1984) in the regression context. One could use the Jeffreys relative scaling of Bayes factors to assess strength of evidence, but the behavior of Bayes factors in complex situations like multiple-QTL mapping is unknown. Nonetheless, to assess the performance of different methods we adopt a simple approach. For all six cases we simulate 100 null (no-QTL) data sets and compute the genomewide maximum 2 logeBF (twice the natural logarithm of Bayes factors) for each trait. The 95th percentile of the max 2 logeBF empirical distribution is considered as the threshold value above which a QTL would be deemed “significant.” At each replication, the number of correctly identified QTL and the number of incorrectly identified or extraneous QTL are recorded. A peak in the 2 logBF profile is considered a QTL if it crosses the significance threshold. It is deemed correct if it is within 10 cM (Broman and Speed 2002) of a true QTL. If there is more than one peak within 10 cM of the true QTL, only one is considered correct.
Results:
Table 2 represents the average correct and extraneous (incorrect) QTL detections for the six situations and for all four methods for 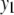 and
and 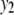 , respectively. It can be seen that TMV detects the highest number of correct as well as the highest number of extraneous QTL. All the multivariate methods detect the higher number of correct QTL compared to the univariate procedure (especially in high correlation cases). The performance of both the QTL SUR models is very close.
, respectively. It can be seen that TMV detects the highest number of correct as well as the highest number of extraneous QTL. All the multivariate methods detect the higher number of correct QTL compared to the univariate procedure (especially in high correlation cases). The performance of both the QTL SUR models is very close.
TABLE 2.
Average correct and incorrect QTL detected for traits y1 (first row) and y2 (second row)
| Correct
|
Extraneous
|
|||||||
|---|---|---|---|---|---|---|---|---|
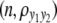 |
STA | TMV | SURs | SURd | STA | TMV | SURs | SURd |
| (100, 0.5) | 0.65 | 0.8 | 0.67 | 0.64 | 0.7 | 1.34 | 0.45 | 0.65 |
| 0.74 | 0.78 | 0.64 | 0.81 | 0.39 | 1.36 | 0.26 | 0.59 | |
| (100, 0.8) | 0.34 | 1.01 | 1.02 | 0.97 | 0.24 | 1.85 | 0.75 | 0.54 |
| 0.78 | 1.07 | 1.3 | 1.21 | 0.71 | 1.72 | 0.84 | 0.78 | |
| (200, 0.5) | 1.69 | 2.13 | 2.12 | 1.78 | 1.06 | 2.53 | 0.78 | 1.02 |
| 1.76 | 2.2 | 2.16 | 1.67 | 0.63 | 2.55 | 0.78 | 0.69 | |
| (200, 0.8) | 1.51 | 2.6 | 2.56 | 2.24 | 0.63 | 2.92 | 0.73 | 0.72 |
| 1.75 | 2.61 | 2.66 | 2.4 | 0.96 | 2.96 | 0.84 | 0.8 | |
| (500, 0.5) | 3.54 | 3.72 | 3.76 | 3.66 | 1.01 | 3.1 | 0.83 | 1.22 |
| 3.59 | 3.79 | 3.75 | 3.56 | 0.76 | 3.07 | 0.67 | 0.88 | |
| (500, 0.8) | 3.55 | 3.81 | 3.78 | 3.67 | 1.1 | 3.14 | 1.03 | 1.01 |
| 3.64 | 3.8 | 3.75 | 3.62 | 1.18 | 3.11 | 0.6 | 0.84 | |
Figures 1–3 display the 2 logeBF profile for chromosomes 1–4 for the three sample sizes (n = 100, 200, 500), respectively, in the four frameworks, namely, SURs, SURd, TMV, and STA. Chromosomes 5–19 had negligible QTL samples (not shown). As can be seen in Figure 1A, both SUR procedures barely detected the moderate effects 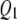 and
and 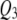 in the low correlation case, but strongly detected the same QTL in the high correlation case (Figure 1B); STA could barely detect them in either case; TMV incorrectly detected
in the low correlation case, but strongly detected the same QTL in the high correlation case (Figure 1B); STA could barely detect them in either case; TMV incorrectly detected  and
and 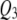 for both traits. Figure 2, A and B, shows SURd performed reasonably well in detecting all six QTL for both high and low levels of correlation between traits; SURs performed similarly but detected
for both traits. Figure 2, A and B, shows SURd performed reasonably well in detecting all six QTL for both high and low levels of correlation between traits; SURs performed similarly but detected 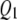 for both traits incorrectly; however, STA failed to detect the weak effects
for both traits incorrectly; however, STA failed to detect the weak effects 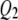 (
( ) in the high (low) correlation cases; TMV identified all six QTL for each trait but only four QTL were true for each trait. Finally, Figure 3, A and B, shows STA, SURs, and SURd could correctly identify all six QTL clearing the threshold for both correlation situations comfortably; TMV, however, strongly detected all six QTL for each trait, as in Figure 2, A and B.
) in the high (low) correlation cases; TMV identified all six QTL for each trait but only four QTL were true for each trait. Finally, Figure 3, A and B, shows STA, SURs, and SURd could correctly identify all six QTL clearing the threshold for both correlation situations comfortably; TMV, however, strongly detected all six QTL for each trait, as in Figure 2, A and B.
Figure 1.—


(A) 2 logBF profile for n = 100 and 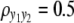 for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; the shaded dotted lines denotes the 95% threshold for y1 for the null model and the solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers. (B) 2 logBF profile for n = 100 and
for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; the shaded dotted lines denotes the 95% threshold for y1 for the null model and the solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers. (B) 2 logBF profile for n = 100 and 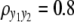 for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
Figure 2.—


(A) 2 logBF profile for n = 200 and 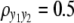 for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; the shaded dotted line denotes the 95% threshold for y1 for the null model and the solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers. (B) 2 logBF profile for n = 200 and
for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; the shaded dotted line denotes the 95% threshold for y1 for the null model and the solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers. (B) 2 logBF profile for n = 200 and 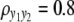 for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
Figure 3.—


(A) 2 logBF profile for n = 500 and  for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers. (B) 2 logBF profile for n = 500 and
for all four methods. Shaded curves represent 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers. (B) 2 logBF profile for n = 500 and  for all four methods. Shaded curves represent the 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
for all four methods. Shaded curves represent the 2 logBF profile for y1 and solid curves that for y2; shaded dotted lines denote the 95% threshold for y1 for the null model and solid dotted lines denote the same for y2. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
Figures 4–6 display the posterior probability profiles for the three sample sizes for testing pleiotropy (a certain locus is simultaneously included in the model for both traits) in the TMV, SURs, and SURd frameworks. We follow the same procedure to measure the threshold values for pleiotropic posterior probabilities. As can be seen in Figure 4, TMV incorrectly detected 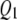 and
and 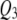 as pleiotropic QTL in the low correlation case; but in the high correlation case it could only feebly detect the true moderate pleiotropic QTL (
as pleiotropic QTL in the low correlation case; but in the high correlation case it could only feebly detect the true moderate pleiotropic QTL (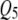 ) in addition to the incorrectly detected ones; SURs detected
) in addition to the incorrectly detected ones; SURs detected 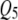 correctly and
correctly and 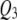 incorrectly in both correlation structures; SURd incorrectly detected
incorrectly in both correlation structures; SURd incorrectly detected 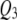 in the low correlation case, but correctly detected both pleiotropic QTL (
in the low correlation case, but correctly detected both pleiotropic QTL (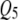 and
and 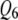 ) in the high correlation case. In Figures 4 and 6, TMV incorrectly detected all 6 QTL as pleiotropic QTL in both correlation structures. In Figures 5 and 6, SURs detected both pleiotropic QTL correctly but also detected some extraneous nonpleiotropic QTL for both correlation structures. SURd, however, detected both pleiotropic QTL correctly without any incorrect detection in the small and large sample size situations for both correlation structures.
) in the high correlation case. In Figures 4 and 6, TMV incorrectly detected all 6 QTL as pleiotropic QTL in both correlation structures. In Figures 5 and 6, SURs detected both pleiotropic QTL correctly but also detected some extraneous nonpleiotropic QTL for both correlation structures. SURd, however, detected both pleiotropic QTL correctly without any incorrect detection in the small and large sample size situations for both correlation structures.
Figure 4.—

Profile of posterior inclusion probabilities for the test of pleiotropy for n = 100. The dotted line represents the 95% threshold for the null model. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
Figure 5.—

Profile of posterior inclusion probabilities for the test of pleiotropy for n = 200. The dotted line represents the 95% threshold for the null model. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
Figure 6.—

Profile of posterior inclusion probabilities for the test of pleiotropy for n = 500. The dotted line represents the 95% threshold for the null model. On the x-axis, large tick marks represent chromosomes and small tick marks represent markers.
The average times taken to conduct each MCMC for all six cases and four methods are presented in Table 3. TMV was the fastest in all cases followed by SURs, STA, and SURd. However, the maximum difference between the fastest and the slowest was only 1.62 min (1 min 37 sec). So computationally complexity does not really pose a great threat.
TABLE 3.
Average MCMC time (in minutes) for four methods
 |
STA | TMV | SURs | SURd |
|---|---|---|---|---|
| (100, 0.5) | 1.17 | 0.96 | 1.10 | 1.18 |
| (100, 0.8) | 1.18 | 0.98 | 1.09 | 1.16 |
| (200, 0.5) | 2.47 | 1.99 | 2.23 | 2.52 |
| (200, 0.8) | 2.48 | 2.06 | 2.22 | 2.45 |
| (500, 0.5) | 6.94 | 6.14 | 6.51 | 7.76 |
| (500, 0.8) | 6.92 | 6.11 | 6.45 | 7.51 |
In conclusion, it is evident and expected that the multivariate procedures outperform STA in the small sample size and high correlation situations. However, one should not use the traditional multivariate model to detect nonpleiotropic QTL as there was astounding evidence of it being prone to erroneous detection. Both the SUR models performed well, but SURs provided slightly false evidence for a QTL influencing y1 (say) for y2. If one wants to detect only pleiotropic QTL, a traditional multivariate model can be used, but, in any other situations, a SURd procedure is recommended in light of a marginal increase in computational time.
DISCUSSION
Our goal in this article was to develop a comprehensive genomewide QTL mapping technique for multiple traits and assess its performance with existing single-trait analysis. When a QTL mapping experiment is conducted, an experimenter rarely measures only a single trait. However, even in the presence of data on more than one trait, there has been a lack of joint analysis of all traits primarily due to the lack of a comprehensive multivariate multiple-QTL mapping technique. From the simulation experiments we have observed that for relatively highly correlated traits the performance of multivariate methods is better compared to single-trait analysis in terms of QTL identification.
We have proposed two separate models for the joint analysis of multiple traits, namely, the seemingly unrelated regression and the traditional multivariate model. The advantage of the SUR model is that it permits all traits to have separate genetic models, much like an independent trait-by-trait analysis but including the correlation structure between the traits, thereby making it more powerful and precise. The traditional multivariate model, however, assumes the same genetic model for all traits. In the situation that we considered in the simulation experiment, we saw poor performance of the traditional multivariate model in terms of accuracy and extraneous detections. The traditional multivariate model is appropriate in the extreme sense when all detectable QTL are pleiotropic (influencing all traits simultaneously). Rarely, knowledge of this magnitude about a complex trait is known a priori. In general, we recommend using a SUR model.
We investigated two different QTL SUR models, namely, SURs and SURd. The performance of both these QTL SUR models has been good. SURs can favor, though very slightly, a QTL of no effect on one trait but having large effect on another trait. In these situations SURd is recommended, since it consistently inferred the correct underlying genetic architecture in simulations. However, the current sampling scheme for updating the genotypes of pleiotropic QTL based on SURd may be suboptimal (as indicated by one of the reviewers), because we always treat the genotypes for different traits separately. In the case where inferring genotypes is difficult we would advocate the use of SURs or replace the genotypes by their conditional expectation in our QTL SUR models (i.e., similar to Haley–Knott regression in QTL analysis). We also can improve the step of updating the genotypes of pleiotropic QTL by using a joint sampling method.
We have adopted the composite model space approach (Yi 2004) and extended it to the multivariate case. The advantage of this approach is that it provides a very efficient way to walk through the space of models, spending more time at “good” models. The key idea behind this approach is to reduce a variable dimensional problem (number of unknown QTL) to a fixed dimensional space and impose a constraint on the maximum number of QTL that can be detected. Our MCMC algorithm has smart strategies to improve efficiency and conduct genomewide scans quickly. For example, we developed a novel one-at-a-time Gibbs sampler to sample regression coefficients that allows us to avoid inverting matrices, saving a lot of precious computational time. In high dimensional problems, inverting extremely large matrices for typically >100,000 iterations can be very computationally taxing and prohibits the use of a multivariate algorithm (as seen in the implementation of Verzilli et al. 2005). We also use the inverse of the variance–covariance matrix for the same reason. We have used informative hierarchical priors for the regression coefficients that typically reflect most QTL mapping situations.
We have developed SUR models for QTL that act in a strictly additive manner. However, it is important to mention that this might not be a good assumption especially in light of the growing number of QTL studies providing evidence in favor of interactions between QTL. Our method can extend to include gene–gene and gene–environment interactions in a natural way. In the presence of such interactions, the search space for possible QTL increases dramatically. We plan to investigate the performance of epistatic SUR methods in the future. We also plan to extend the multivariate framework to a mixture of continuous, binary, and ordinal traits.
Acknowledgments
We thank the reviewers for their helpful comments on the previous version of this manuscript. This work was supported by the following National Institutes of Health grants: R01 GM069430 (N.Y. and B.Y.), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) 5803701 (B.Y.), NIDDK 66369-01 (B.Y.), and National Institute of General Medical Sciences/R01 PA-02-110 (B.Y.).
APPENDIX A: PRIOR DISTRIBUTIONS
The independent priors across traits are straightforward extensions of Yi et al. (2005, 2007). We describe the priors on 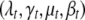 for each trait, highlighting the distinctions pertinent to multiple correlated traits.
for each trait, highlighting the distinctions pertinent to multiple correlated traits.
The prior distribution on QTL locations is uniformly distributed over the preset loci across the genome (Yi et al. 2005). Two constraints can be incorporated into the prior on QTL locations to reduce the model space: the first restricts the spacing among multiple linked QTL and the second restricts the number of detectable QTL on each chromosome (see Yi et al. 2007).
For the vector of indicators 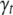 , we could use an independence prior,
, we could use an independence prior, 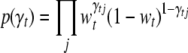 , with wt being the prior inclusion probability of each effect for the tth trait. A useful reduction can be achieved by setting
, with wt being the prior inclusion probability of each effect for the tth trait. A useful reduction can be achieved by setting  . To specify wt, we first determine the prior expected numbers of main-effect QTL and then solve for wt from the expressions of the prior expected numbers (see Yi et al. 2005). The prior expected number of main-effect QTL could be set to the number of QTL detected by traditional mapping methods.
. To specify wt, we first determine the prior expected numbers of main-effect QTL and then solve for wt from the expressions of the prior expected numbers (see Yi et al. 2005). The prior expected number of main-effect QTL could be set to the number of QTL detected by traditional mapping methods.
The prior for the overall mean 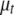 is chosen to be normally distributed with mean and variance being sample mean and variance of the tth trait, respectively. For the genetic effects
is chosen to be normally distributed with mean and variance being sample mean and variance of the tth trait, respectively. For the genetic effects 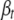 , we extend the prior of Yi et al. (2007) that assumes that different types of effects (e.g., additive effects or dominance effects) follow different prior distributions. For type k, effects
, we extend the prior of Yi et al. (2007) that assumes that different types of effects (e.g., additive effects or dominance effects) follow different prior distributions. For type k, effects 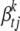 have the prior,
have the prior,  , where
, where 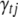 is the indicator variable for
is the indicator variable for 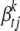 , and
, and 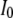 is a point mass at 0. Under this prior, when
is a point mass at 0. Under this prior, when 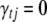 ,
, 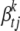 is assigned to be 0 and thus is actually removed from the model; when
is assigned to be 0 and thus is actually removed from the model; when 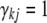 ,
,  follows a normal distribution
follows a normal distribution  . The variance
. The variance 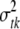 is treated as a random variable with an inverse-χ2 hyperprior distribution; i.e.,
is treated as a random variable with an inverse-χ2 hyperprior distribution; i.e., 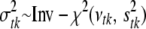 . The degrees of freedom νtk control the skewness of the prior for
. The degrees of freedom νtk control the skewness of the prior for 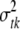 , with larger values recommended (here νtk = 6) to tightly center the prior around
, with larger values recommended (here νtk = 6) to tightly center the prior around 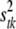 (see Yi et al. 2007). The scale parameter
(see Yi et al. 2007). The scale parameter 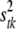 controls the prior proportion of phenotypic variance explained by
controls the prior proportion of phenotypic variance explained by 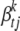 . We set
. We set 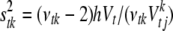 , leading to the proportion of phenotypic variance explained by
, leading to the proportion of phenotypic variance explained by 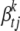 being h, where Vt is the phenotypic variance of trait t, and
being h, where Vt is the phenotypic variance of trait t, and 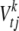 is the sample variance for the column of X associated with effect
is the sample variance for the column of X associated with effect 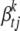 . Expected effect heritability, h, can be set small (say 0.05–0.2) to reflect prior knowledge about genetic architecture.
. Expected effect heritability, h, can be set small (say 0.05–0.2) to reflect prior knowledge about genetic architecture.
APPENDIX B: CONDITIONAL POSTERIOR DISTRIBUTIONS
We here derive conditional posterior distribution for each unknown from the joint posterior distribution (3). Denote all the unknowns by 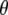 ; i.e.,
; i.e., 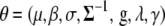 . We first present the conditional posterior distributions for the model SURd and then make some changes to the models SURs and TMV if necessary.
. We first present the conditional posterior distributions for the model SURd and then make some changes to the models SURs and TMV if necessary.
Conditional posterior distribution of each μt:
The conditional posterior distribution for the overall mean of the tth trait,  , can be shown to be
, can be shown to be
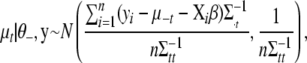 |
(B1) |
where 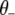 represents all elements of
represents all elements of 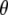 except
except 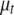 ,
,  is the vector
is the vector 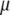 with the tth element
with the tth element 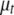 replaced by 0,
replaced by 0, 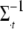 is the tth column of
is the tth column of 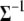 , and
, and 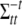 is the (t, t) element of
is the (t, t) element of 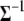 . Since the conditional posterior is a standard distribution, a Gibbs sampler can be easily performed.
. Since the conditional posterior is a standard distribution, a Gibbs sampler can be easily performed.
Conditional posterior distribution of each βtj:
If the jth effect of the tth trait, 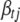 , is included in the model, the conditional posterior distribution of
, is included in the model, the conditional posterior distribution of 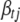 can be shown to be
can be shown to be
 |
(B2) |
where 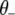 represents all elements of
represents all elements of 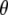 except
except 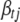 ,
, 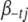 is the vector
is the vector 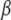 with the element
with the element 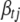 replaced by 0,
replaced by 0, 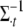 and
and 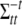 are defined as in (A1), and
are defined as in (A1), and 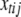 is the main-effect contrast for the jth effect for the tth trait and the ith individual. Note that
is the main-effect contrast for the jth effect for the tth trait and the ith individual. Note that 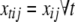 for SURs and TMV.
for SURs and TMV.
Conditional posterior distribution of each σtk2:
For each type of genetic effects (additive and dominance), the conditional posterior distribution of 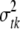 is an inverse-
is an inverse-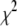 distribution,
distribution,
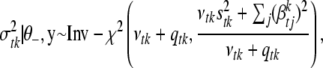 |
(B3) |
where 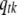 is the number of nonzero effects in
is the number of nonzero effects in  , and other parameters are defined earlier.
, and other parameters are defined earlier.
Conditional posterior distribution of Σ−1:
Keeping the computationally efficient goal in mind, it should be noted that generating 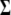 would involve computing its inverse to draw samples from (B1) and (B2) in each iteration. So, it is not only convenient to work with
would involve computing its inverse to draw samples from (B1) and (B2) in each iteration. So, it is not only convenient to work with 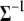 but computationally efficient as well. The conditional posterior distribution for
but computationally efficient as well. The conditional posterior distribution for 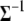 can be calculated
can be calculated
 |
(B4) |
where 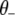 represents all elements of
represents all elements of 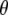 except
except 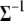 , and
, and 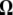 is a T × T matrix of residuals where the (t, t′)th element of
is a T × T matrix of residuals where the (t, t′)th element of 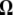 ,
, 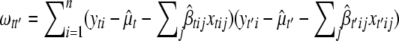 . Since the posterior of
. Since the posterior of 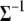 follows a standard Wishart distribution, a Gibbs sampler can be used to generate samples. An alternative Metropolis algorithm could also be used to generate samples where a newly generated iterate
follows a standard Wishart distribution, a Gibbs sampler can be used to generate samples. An alternative Metropolis algorithm could also be used to generate samples where a newly generated iterate  is accepted over an old value
is accepted over an old value 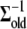 with probability
with probability
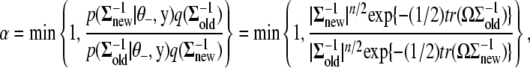 |
(B5) |
where q(.) is the proposal density that is assumed to be the same as its prior. We have implemented both the Gibbs sampler and the Metropolis algorithm for updating 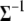 and in either case we get similar results.
and in either case we get similar results.
Conditional posterior distribution of each gtiq:
If locus q for trait t is included in the model and the genotype  of individual i is not observed, the conditional posterior distribution of
of individual i is not observed, the conditional posterior distribution of 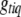 is
is
 |
(B6) |
where 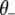 represents all elements of
represents all elements of 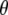 except
except 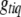 ,
, 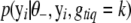 is the likelihood for individual i calculated by model (2), and
is the likelihood for individual i calculated by model (2), and 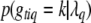 is the prior probability of
is the prior probability of 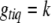 . This posterior is a simple multinomial distribution and thus can be sampled directly. If locus q is excluded from the model or
. This posterior is a simple multinomial distribution and thus can be sampled directly. If locus q is excluded from the model or 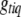 is observed (e.g., for fully observed markers), we do not need to sample
is observed (e.g., for fully observed markers), we do not need to sample 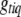 .
.
Conditional posterior distribution of λ:
If locus q for trait t is included in the model, the joint conditional posterior distribution of the position 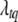 and the genotypes
and the genotypes 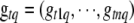 is
is
 |
(B7) |
where  represents all elements of
represents all elements of 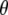 except
except 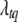 and gtq,
and gtq,  is the likelihood calculated by model (2),
is the likelihood calculated by model (2), 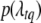 is the prior of
is the prior of 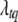 , and
, and 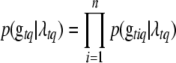 is the prior probability of
is the prior probability of 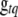 .
.
This posterior is not a standard distribution, and thus a Metropolis algorithm is needed to update 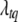 and gtq jointly. We first propose a new position
and gtq jointly. We first propose a new position 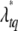 from a proposal distribution
from a proposal distribution  and then generate new genotypes,
and then generate new genotypes, 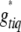 , at this new position for all individuals from the conditional posterior (B6). The proposals for
, at this new position for all individuals from the conditional posterior (B6). The proposals for  and
and 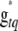 are then accepted simultaneously with probability
are then accepted simultaneously with probability
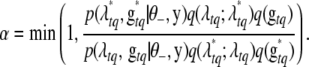 |
(B8) |
The proposal distribution for the new position 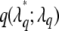 is usually constructed as uniformly distributed over 2d most flanking loci of
is usually constructed as uniformly distributed over 2d most flanking loci of 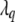 , with d being a predetermined tuning integer. In our implementation, we take d = 2 and incorporate the preset constraints on QTL positions into our algorithm.
, with d being a predetermined tuning integer. In our implementation, we take d = 2 and incorporate the preset constraints on QTL positions into our algorithm.
Conditional posterior distribution of each γtj:
The conditional posterior distribution of γtj can be expressed as
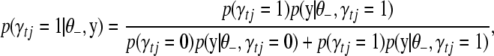 |
(B9) |
where 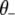 represents all elements of
represents all elements of 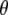 except
except 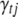 and
and 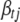 ,
, 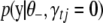 is calculated using model (2) with
is calculated using model (2) with 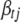 replaced by 0, and
replaced by 0, and  does not depend on
does not depend on 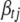 and can be calculated using the identity of simple conditional probability
and can be calculated using the identity of simple conditional probability
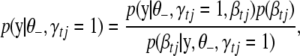 |
(B10) |
where  is the phenotype likelihood calculated using model (2),
is the phenotype likelihood calculated using model (2), 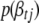 is the prior distribution of
is the prior distribution of  , and
, and  is the conditional posterior distribution of
is the conditional posterior distribution of 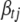 calculated by (B2). Notationally, the right side of (B10) depends on
calculated by (B2). Notationally, the right side of (B10) depends on 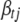 , but from the definition of
, but from the definition of 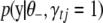 , we know it cannot depend on
, we know it cannot depend on 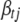 in a real sense. That is, the factors that depend on
in a real sense. That is, the factors that depend on 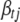 in the numerator and the denominator must cancel. Thus, we can compute (B10) by inserting any value of
in the numerator and the denominator must cancel. Thus, we can compute (B10) by inserting any value of  into the expression. A convenient, stable choice is the conditional posterior mean of
into the expression. A convenient, stable choice is the conditional posterior mean of 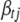 (Gelman et al. 2004; Yi et al. 2007).
(Gelman et al. 2004; Yi et al. 2007).
To calculate the conditional posterior probability (B9), we may need the values of parameters associated with 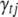 . If
. If 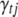 is currently 0 and the involved QTL(s) is (are) not currently in the model, we first sample new QTL position(s) from their corresponding priors as needed, new genotypes for all individuals, and the prior variance of
is currently 0 and the involved QTL(s) is (are) not currently in the model, we first sample new QTL position(s) from their corresponding priors as needed, new genotypes for all individuals, and the prior variance of 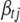 if this parameter is currently out of the model. If the current value of
if this parameter is currently out of the model. If the current value of  is 1, the associated unknowns were already generated at the preceding iteration.
is 1, the associated unknowns were already generated at the preceding iteration.
The Gibbs sampler can be used to generate each indicator 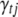 from its conditional posterior (B9). However, for the QTL SUR models, using the Gibbs samplers is computationally demanding because the SUR models contain T times the number of indicators as a single-trait model and most of the indicators are zero. To speed up the algorithm we extend the Metropolis–Hastings algorithm proposed by Yi et al. (2007) to the QTL SUR models. As with the Gibbs sampler, the MH scheme proceeds to update all indicator variables. Denote the current value of
from its conditional posterior (B9). However, for the QTL SUR models, using the Gibbs samplers is computationally demanding because the SUR models contain T times the number of indicators as a single-trait model and most of the indicators are zero. To speed up the algorithm we extend the Metropolis–Hastings algorithm proposed by Yi et al. (2007) to the QTL SUR models. As with the Gibbs sampler, the MH scheme proceeds to update all indicator variables. Denote the current value of 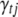 by C (= 0 or 1). The MH algorithm proposes a new value P (= 0 or 1) for
by C (= 0 or 1). The MH algorithm proposes a new value P (= 0 or 1) for 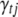 from the prior probability p(γtj = C). If P = C, the MH acceptance probability is 1, and thus γtj remains at C and there is no need to compute any values. Otherwise, we update γtj from the current value C to the proposal 1 − C with acceptance probability
from the prior probability p(γtj = C). If P = C, the MH acceptance probability is 1, and thus γtj remains at C and there is no need to compute any values. Otherwise, we update γtj from the current value C to the proposal 1 − C with acceptance probability
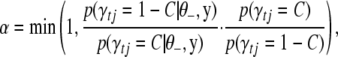 |
(B11) |
where  and
and 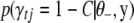 are calculated in (B9).
are calculated in (B9).
The conditional posterior of  for the traditional multivariate model is a little tricky. Since the indicator variable of a particular effect is the same for all traits, the conditional posterior distribution of
for the traditional multivariate model is a little tricky. Since the indicator variable of a particular effect is the same for all traits, the conditional posterior distribution of 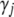 can be expressed as
can be expressed as
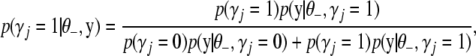 |
(B12) |
where  is the indicator variable for the jth effects for all traits,
is the indicator variable for the jth effects for all traits, 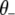 represents all elements of
represents all elements of  except
except 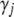 and
and 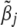 ,
, 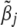 denotes the vector of the jth effects for all traits, and
denotes the vector of the jth effects for all traits, and 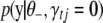 is calculated using model (2) with
is calculated using model (2) with  replaced by 0. The integration in (B10) should be with respect to joint distribution of all genetic effects for the traits in question. Proceeding similarly as above we can get
replaced by 0. The integration in (B10) should be with respect to joint distribution of all genetic effects for the traits in question. Proceeding similarly as above we can get
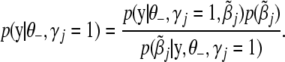 |
(B13) |
As before, a choice of 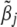 could be the posterior mean of the joint posterior distribution of
could be the posterior mean of the joint posterior distribution of 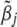 calculated below,
calculated below,
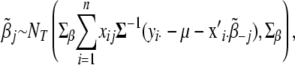 |
(B14) |
where 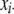 is the vector of main-effect contrast(s) for the ith individual for all loci,
is the vector of main-effect contrast(s) for the ith individual for all loci, 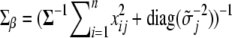 ,
, 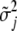 is the vector of the variances of the jth genetic effect for all traits, and
is the vector of the variances of the jth genetic effect for all traits, and 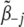 is the vector of coefficients with
is the vector of coefficients with 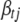 (t = 1, · · · T) replaced as 0.
(t = 1, · · · T) replaced as 0.
References
- Ball, R. D., 2001. Bayesian methods for quantitative trait loci mapping based on model selection: approximate analysis using the Bayesian information criterion. Genetics 159 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. B 64(4): 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., H. Wu, Ś. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19 889–890. [DOI] [PubMed] [Google Scholar]
- Burnham, K. P., and D. R. Anderson, 2002. Model Selection and Multi-Model Inference. Springer-Verlag, New York.
- Gelman, A., J. Carlin, H. Stern and D. Rubin, 2004. Bayesian Data Analysis. Chapman & Hall/CRC, London.
- Gilbert, H., and P. Le Roy, 2003. Comparison of three multitrait methods for QTL detection. Genet. Sel. Evol. 35 281–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, H., and P. Le Roy, 2004. Power of three multitrait methods for QTL detection in crossbreed populations. Genet Sel. Evol. 36 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, W. E., 2001. Bayesian Inference in the Seemingly Unrelated Regressions Model (Working Series Paper 793). Department of Economics, University of Melbourne, Melbourne, Australia.
- Hackett, C. A., R. C. Meyer and W. T. B. Thomas, 2001. Multi-trait QTL mapping in barley using multivariate regression. Genet. Res. Camb. 77 95–106. [DOI] [PubMed] [Google Scholar]
- Huang, J., and Y. Jiang, 2003. Genetic linkage analysis of a dichotomous trait incorporating a tightly linked quantitative trait in affected sib pairs. Am. J. Hum. Genet. 72 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, A. U., A. Fornés, A. Galecki, R. A. Miller and D. T. Burke, 1999. Multiple-trait quantitative trait loci analysis using a large mouse sibship. Genetics 151 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C., and Z.-B. Zeng, 1995. Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140 1111–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C., and Z.-B. Zeng, 1997. Mapping quantitative trait loci with dominant and missing markers in various crosses from two inbred lines. Genetica 101 47–58. [DOI] [PubMed] [Google Scholar]
- Kao, C.-H., and Z.-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott, S. A., and C. S. Haley, 2000. Multitrait least squares for quantitative trait loci detection. Genetics 156 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, C., and J. C. Whittaker, 2001. Mapping quantitative trait loci using generalized estimating equations. Genetics 159 1325–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J., Y. Liu, X. Liu and H.-W. Deng, 2007. Bayesian mapping of quantitative trait loci for multiple complex traits with the use of variance components. Am. J. Hum. Genet. 81 304–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, M. S., P. Sørensen, B. Guldbrandtsen and D. A. Sorensen, 2003. Multitrait fine mapping of quantitative trait loci using combined linkage disequilibria and linkage analysis. Genetics 163 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mähler, M., C. Most, S. Schmidtke, J. P. Sundberg, R. Li et al., 2002. Genetics of colitis susceptibility in IL-10-deficient mice: backcross versus F2 results contrasted by principal component analysis. Genomics 80 274–282. [DOI] [PubMed] [Google Scholar]
- Mangin, B., P. Thoquet and N. Grimsley, 1998. Pleiotropic QTL analysis. Biometrics 54 88–99. [Google Scholar]
- Meuwissen, T. H., and M. E. Goddard, 2004. Mapping multiple QTL using linkage disequilibrium and linkage analysis information and multitrait data. Genet. Sel. Evol. 36 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, A. J., 1984. Selection of subsets of regression variables. J. R. Stat. Soc. A 147 389–425. [Google Scholar]
- Plummer, M., N. Best, K. Cowles and K. Vines, 2004. Output Analysis and Diagnostics for MCMC, v. 0.9–5. Institute of Mathematical Statistics, Beachwood, OH.
- Raftery, A. E., D. Madigan and J. A. Hoeting, 1997. Bayesian model averaging for linear regression models. J. Am. Stat. Assoc. 92 179–191. [Google Scholar]
- Satagopan, J. M., and B. S. Yandell, 1996. Estimating the number of quantitative trait loci via model determination. Special Contributed Paper Session on Genetic Analysis of Quantitative Traits and Complex Diseases. Biometric Section, Joint Statistical Meeting, Chicago.
- Sen, Ś., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and J. Corander, 2002. Model choice in gene mapping: what and why. Trends Genet. 18 301–307. [DOI] [PubMed] [Google Scholar]
- Smith, M., and R. Kohn, 2000. Nonparametric seemingly unrelated regression. J. Econometrics 98 257–281. [Google Scholar]
- Verzilli, C. J., N. Stallard and J. C. Whittaker, 2005. Bayesian modeling of multivariate quantitative traits using seemingly unrelated regression. Genetic Epidemiol. 28 313–325. [DOI] [PubMed] [Google Scholar]
- Vieira, C., E. G. Pasyukova, Z.-B. Zeng, J. B. Hackett, R. F. Lyman et al., 2000. Genotype-environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Y. C. M. Zhang, X. Li, G. L. Masinde, S. Mohan et al., 2005. Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J. T., H. Begleiter, B. Porjesz, H. J. Edenberg, T. Foroud et al., 1999. a Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. II. Alcoholism and event-related potentials. Am. J. Hum. Genet. 65 1148–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J. T., P. Van Eerdewegh, L. Almasy and J. Blangero, 1999. b Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. I. Likelihood formulation and simulation results. Am. J. Hum. Genet. 65 1134–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C., Z. Li and S. Xu, 2005. Joint mapping of quantitative trait loci for multiple binary characters. Genetics 169 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, R., and S. Xu, 2007. Bayesian shrinkage analysis of quantitative trait loci for dynamic traits. Genetics 176 1169–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell, B. S., T. Mehta, S. Banerjee, D. Shriner, R. Venkataraman et al., 2007. R/qtlbim: QTL with Bayesian interval mapping in experimental crosses. Bioinformatics 23(5): 641–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., 2004. A unified Markov chain Monte Carlo framework for mapping multiple quantitative trait loci. Genetics 167 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and D. Shriner, 2008. Advances in Bayesian multiple QTL mapping in experimental designs. Heredity 100 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and S. Xu, 2002. Mapping quantitative trait loci with epistatic effects. Genet. Res. Camb. 79 185–198. [DOI] [PubMed] [Google Scholar]
- Yi, N., S. Xu and D. B. Allison, 2003. Bayesian model choice and search strategies for mapping interacting quantitative trait loci. Genetics 165 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., B. S. Yandell, G. A. Churchill, D. B. Allison, E. J. Eisen et al., 2005. Bayesian model selection for genomewide epistatic quantitative trait loci analysis. Genetics 170 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., D. Shriner, S. Banerjee, T. Mehta, D. Pomp et al., 2007. An efficient Bayesian model selection approach for interacting QTL models with many effects. Genetics 176 1865–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellner, A., 1962. An efficient method of estimating seemingly unrelated regressions and tests for aggregation bias. J. Am. Stat. Assoc. 57 348–368. [Google Scholar]
- Zeng, Z.-B., T. Wang and W. Zou, 2005. Modeling quantitative trait loci and interpretation of models. Genetics 169 1711–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]