

Abstract
A combined electrodynamic ion funnel and ion trap coupled to an orthogonal acceleration (oa)-time-of-flight mass spectrometer (oa-TOF MS) was developed and characterized. The ion trap was incorporated through the use of added terminal electrodynamic ion funnel electrodes enabling control over the axial DC gradient in the trap section. The ion trap operates efficiently at a pressure of 1 Torr, and measurements indicate a maximum charge capacity of ~3×107 charges. An order of magnitude increase in sensitivity was observed in the analysis of low concentration peptides mixtures with oa-TOF MS in the trapping mode as compared to the continuous regime. A signal increase in the trapping mode was accompanied by reduction in the background chemical noise, due to more efficient desolvation of e.g., solvent related clusters. Controlling the ion trap ejection time was found to result in efficient removal of singly charged species and improving signal-to-noise ratio (S/N) for the multiply charged analytes.
Introduction
The orthogonal acceleration time-of-flight mass spectrometer (oa-TOF MS) is a powerful platform for fast on-line detection of biopolymers. Oa-TOF MS is characterized by unsurpassed speed of analysis, high sensitivity, high mass measurement accuracy and high mass resolving power1-5, and thus representing an attractive platform for high throughput analysis. Due to the inherently pulsed separation of ions in a TOF flight tube, the efficient coupling of a continuous ion source, such as electrospray ionization (ESI), to TOF MS is challenging. Following the pioneering work by Dodonov et al5, 6, ESI-oa-TOF MS has been widely applied7, 8 and successfully commercialized. However, the sensitivity of ESI-oa-TOF MS is limited by the instrument duty cycle, which depends on the mass-to-charge ratio (m/z) of the detected analytes and typically remains within 5-20%. Increasing the instrument duty cycle, e.g., by using higher pulsing frequency, reduces the detectable m/z range.
Various ion traps such as 3D, linear quadrupole, and ring electrode traps9-14 have been introduced between the ESI source and a TOF MS to effectively accumulate ions prior to analysis.15 When coupling an ion trap to a TOF instrument, three key characteristics should be considered: trapping efficiency, charge capacity of the trap and the speed of ion ejection from the trap. Increasingly being used are the linear ion traps for their capabilities that are similar to those of 3D traps, but with higher charge capacity.16 However, these traps are limited by lower operating pressures (usually within 10-5- 10-3 Torr). Since both trapping efficiency and collisional relaxation (i.e., ion cooling) are directly proportional to the number density of the collision gas, accumulation of ions at higher pressures potentially offers higher sensitivity. An ion trap that works at a pressure of several Torr is also particularly beneficial for use with gas phase ion separations, such as ion mobility spectrometry (IMS), coupled to TOF MS. Efficient implementation of higher charge capacity ion trap prior to IMS would increase overall sensitivity, otherwise constrained by the low IMS duty cycle associated with pulsed ion introduction. Though segmented quadrupoles were shown to operate at higher pressures,17 higher order multipoles provide greater radial confinement18, and given the dependence of the effective potential on pressure19, are better suited for trapping larger ion populations at higher pressures. Several groups have reported on efforts to improve the efficiency of ion ejection from traps, which operate at intermediate pressures of 10-5-10-3 Torr, by segmenting the quadrupole rods,17, 20 adding resistively-coated electrodes around the trap,13 using conical rods,21 or inserting slopped T-shaped rods between multipole rods.22
Particularly attractive for fast ion ejection is a “stacked-ring” assembly, which is similar to the high order multipole and has been thoroughly characterized both analytically and experimentally.13, 18 The “stacked-ring” ion trap is comprised of ring-shaped electrodes where 180° phase-shifted RF fields are applied to adjacent electrodes to create a radial confining field. Axial confinement is achieved by applying DC potentials to the trap terminus. To improve ion ejection, an axial DC field is generated by superimposing a pulsed gradient onto the RF field. The effective potential force in the “stacked-ring” assembly depends on the distance between adjacent electrodes and increases exponentially on approaching the electrode edges.18 At a sufficiently small gap between the electrodes, such a device can provide highly efficient RF-confinement. Another advantage is that generation of a pulsed gradient in an assembly of ring electrodes for fast ion ejection is also highly effective.
A modification of the “stacked-ring” assembly is the electrodynamic ion funnel,23 which is characterized by ring electrodes of progressively reduced inner diameters that serve to collimate a diffuse ion beam. Introduced and significantly enhanced by our laboratory for ion sampling from ESI sources,23-26 the electrodynamic ion funnel improves ion utilization compared to standard skimmer-based MS interfaces. Since it can efficiently operate at pressures of ~1-30 Torr,27 the ion funnel represents an attractive interface with higher pressure ionization sources.
Another important aspect of ion trap operation relates to its ability to reduce “chemical noise” while enhancing the analyte signal, which leads to an increase in signal-to-noise ratios (S/N). Improving S/N is critical for biological experiments with, for example, human blood plasma in which biological applications (e.g. disease biomarkers), are often present at very low concentrations among higher abundance analytes and chemical noise. Chemical noise can arise due to many factors, and in some cases it can be due to singly charged partially solvated ions that typically generate abundant signals at the low m/z range of a mass spectrum.28 The presence of such species is a result of a compromise typically made in the ESI interface between conditions that provide good desolvation but that also avoid analyte activation/dissociation. Although MS-based methods for separating multiply charged analytes from singly charged species have been developed to reduce chemical noise, these approaches are limited by either low duty cycle or pronounced losses of doubly charged species under the conditions used to suppress singly charged ions. 29, 30
In the present work, we have characterized an electrodynamic ion funnel trap at a pressure of ~1 Torr. The electrodynamic ion funnel was coupled to a high-performance oa-TOF MS, which was operated in both trapping and continuous modes for comparison. Analysis of peptide mixtures with oa-TOF MS in the trapping mode revealed reduction in the chemical noise as well as significant improvements in the S/N of the analyte species.
Experimental Setup
The experiments were performed using a prototype dual-stage reflectron oa-TOF mass spectrometer as shown schematically in Figure 1a. Ions from the electrospray source were transmitted through a 508 μm-i.d., 10 cm-long stainless steel capillary interface, resistively heated to 165°C, and into an electrodynamic ion funnel that worked at pressure of ~ 1 Torr. A schematic diagram of the ion funnel is depicted in Figure 1b. The 180° phase-shifted RF fields were applied to adjacent ring-electrodes at a peak-to-peak amplitude of 70 Vp-p and a frequency of 600 kHz. Ion transmission through the funnel was improved by superimposing a DC field onto the RF field applied to each electrode. In continuous mode, the DC gradient applied to the funnel was 20 V/cm.
Figure 1.
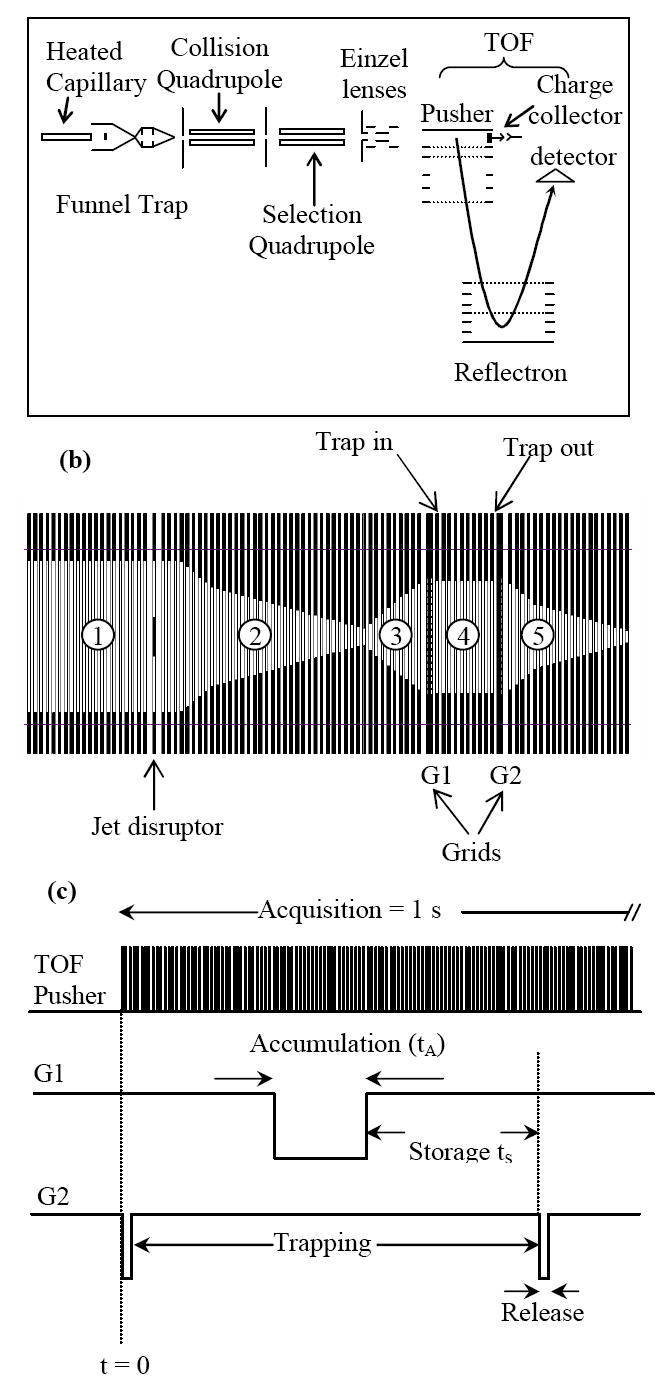
a) Outline of experiment setup. b) Schematic diagram of the ion funnel trap. The numbers refer to the different sections of the funnel (see text). c) The pulse sequence for ion accumulation, storage and ejection from the trap.
The funnel consists of 98 brass ring electrodes, each 0.5 mm thick and separated 0.5 mm apart with Teflon spacers. The ring electrodes are assembled onto four ceramic rods that ensure proper alignment. The inner diameters of the ring electrodes vary from 25.4 mm at the funnel entrance, 19.1 mm at the trap section (between grids G1 and G2 in Figure 1b), and 2.4 mm at the funnel exit plate. Section 1 of the ion funnel in Figure 1b, which accepts ions exiting the heated capillary, is composed of 24 ring electrodes, each having a 25.4-mm i.d. A jet disrupter made of a 6.5-mm brass disk is located ~ 20 mm downstream of the funnel entrance to reduce the gas load to the subsequent stages of differential pumping while maintaining high ion transmission.31 An independent DC voltage was applied to the jet disrupter.
The ions exiting section 1 were collimated into section 2, which is characterized by a converging geometry (Figure 1b). Section 3, which is characterized by diverging geometry, couples section 2 to the ion trap. In trapping mode, ions are accumulated in section 4, which is comprised of 10 ring electrodes, each having a 19.1-mm i.d. The trapping region (section 4) is separated from sections 3 and 5 by two grids fabricated from 95 %-transmission nickel mesh (InterNet Inc., Minneapolis, MN).
Pulsing voltages applied to the grids G1 and G2 were used to control ion populations that could be introduced into the trap, as well as to control the ion storage and extraction times, respectively. The DC gradient in the trapping region was varied independently from the rest of the ion funnel by adjusting potentials at the first (“Trap IN”) and last electrodes (“Trap OUT”) in section 4. In continuous mode, the potentials on grids G1 and G2 were optimized to ensure efficient ion transmission through the trapping region. The ions passing section 4 were re-collimated in the tapered portion of section 5 and then focused into a 15-cm long collisional quadrupole, operating at a pressure of ~6×10-3 Torr.
Following collisional relaxation and focusing, ions were transmitted through a 20-cm long selection quadrupole at a pressure of 1.5×10-5 Torr and focused by an Einzel lens assembly into a TOF extraction region. Both the collisional and selection quadrupoles were operated at an RF amplitude of 2500 Vp-p and an RF frequency of 2 MHz. The TOF chamber encompasses a stack of acceleration electrodes, a dual-stage ion mirror and a 40-mm-diameter bipolar detector (Burle ElectroOptics, Sturbridge, MA). The length of the TOF flight tube is 100 cm and the distance between the center of 40-mm long TOF extraction region and the detector axis is 75 mm. A typical full width at half maximum (fwhm) of signal peaks was 3-3.5 ns, yielding an optimum resolving power of 10,000 and a routine resolving power of 7000-8000. The TOF detector was impedance matched to a 2 GS/s 8-bit analog-to-digital converter AP200 (Acqiris, Geneva, Switzerland) that enabled routine mass measurement accuracy of ~ 5 ppm. Prior to ion introduction into the TOF acceleration stack, both continuous and pulsed ion currents were measured with a Faraday cup charge collector positioned on the interface axis immediately downstream of the TOF extraction region (Figure 1a). Ion current pulses were acquired using a fast current amplifier (Keithley Model 428, Cleveland, OH) coupled to a digital oscilloscope (Tektronix, Richardson, TX).
The pulsing sequence for ion trapping is schematically depicted in Figure 1c. By toggling one of the TOF MS control bits (Run/Stop) high at the beginning of each spectrum acquisition, a waveform generator (Hewlett Packard, Palo Alto, CA) was triggered to release a burst of trigger pulses. The repetition rate and the number of burst pulses determined the trapping and acquisition times, respectively. Each trigger pulse activated a delay generator (Stanford Research Systems, San Jose, CA), which in turn determined the pulse widths and time delay in pulsing grids G1 and G2. The output TTL signals from the delay generator were then fed into two independent high-voltage switches (Behlke, Kronberg, Germany) that provided pulsed voltages for the two pulsing grids. For the experiments reported herein, grid G1 was not pulsed and ESI-generated ions entered the trap continuously.
All samples were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO) and used without further purification. The samples were prepared in 50% aqueous methanol acidified with 1% acetic acid and infused into the instrument at a flow rate of 0.4 μl/min.
Results and Discussion
The ion funnel was initially optimized by adjusting RF and DC fields in the trap region for higher sensitivity. Figure 2 shows the average monoisotopic intensity of triply-charged neuroten sin as a function of the RF amplitude at a constant RF frequency of 600 kHz. Although an optimum was found for the RF amplitude in the trapping mode, no significant signal variation was observed over a wide range of RF amplitudes in the continuous mode. Therefore, 55 Vp-p was used as the optimal RF amplitude for the results reported here. The optimum RF amplitude was also found to be consistent with relationships reported recently.32 The relationship for high m/z limit (m/z)high as a function of the RF frequency f and the radial DC electric field component En can be estimated as follows:
Figure 2.
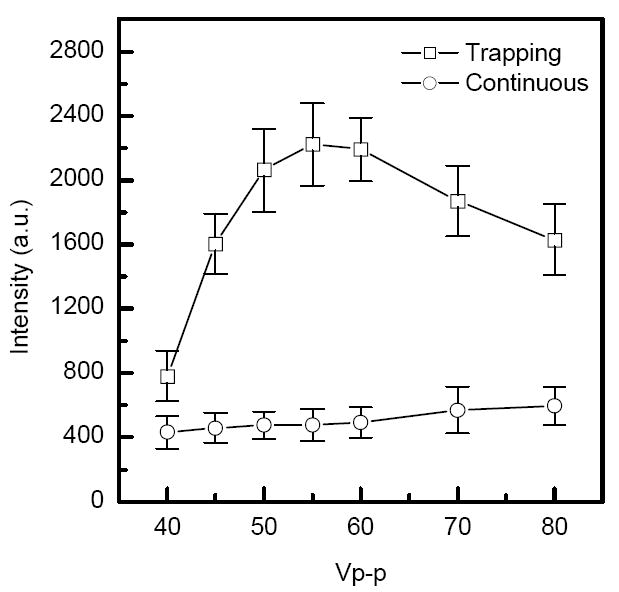
Comparison of monoisotopic [M+H3]3+ signal intensities from ESI of a 5 nM neurotensin solution in the continuous and trapping modes. Intensity is plotted as a function of the funnel RF amplitude at constant RF frequency of 600 kHz. Similar results were obtained for angiotensin I [Ang I+H2]2+ and fibrinopeptide A [Fib A+H2]2+ ions. Accumulation time = 100 ms, extraction time = 45 μs.
| (1) |
Here, e is the elementary charge, mu = 1.6605* 10-27 kg is the atomic mass unit, ω = 2πf is the angular frequency, h0 ≈ 1 mm is the distance corresponding to the onset of ion losses on the surface of the ion funnel ring electrodes, and δ is related to the distance between the ring electrodes, d=1 mm, as δ = d/π . Assuming that the trapped ion ensemble is limited to (m/z)high ≈ 2000 amu, using f = 600 KHz and the electric field characteristic for the DC trapping conditions in our experiments, En = 20 V/cm, one can obtain from eq.(1) the RF voltage VRF ≈ 30V, or 60 Vp-p, which is consistent with the experimentally observed RF amplitude (Figure 2). In the continuous mode both DC trapping and space charge components of En are reduced, which explains the different VRF behavior in Figure 2.
We have also found that trapping efficiency strongly depends on the axial DC gradient. Figure 3 shows the dependence of reserpine monoisotopic peak intensity on the extraction time at two different DC gradients in the trap region. Using DC gradient of 20 V/cm (which is similar to that employed in the rest of the ion funnel) led to poor ion accumulation efficiency. Reduction of the DC gradient from 20 to 4 V/cm resulted in a more than two-order-of-magnitude improvement in sensitivity and an ion extraction time of 100 μs. Fast removal of ions from the trap is important for efficient coupling of the ion trap to oa-TOF MS. Evaluation of a number of DC gradients revealed a general trade-off: lower DC gradients give rise to more efficient ion accumulation, but slower ion ejection. The drastic decrease in ion accumulation efficiency with an increase in the ion trap DC field is related to axial compression of the ion cloud and associated space charge effects. Due to the cylindrical geometry of the trap, the DC trapping field has a radial component that tends to eject ions in radial direction where they experience higher RF oscillations and lost to the electrodes. When the axial electric field is sufficiently low (4 V/cm), the accumulated ion cloud extends axially, thus increasing the trap capacity and its efficiency. Additional evidence from SIMION simulations is provided in the Supporting Information.
Figure 3.
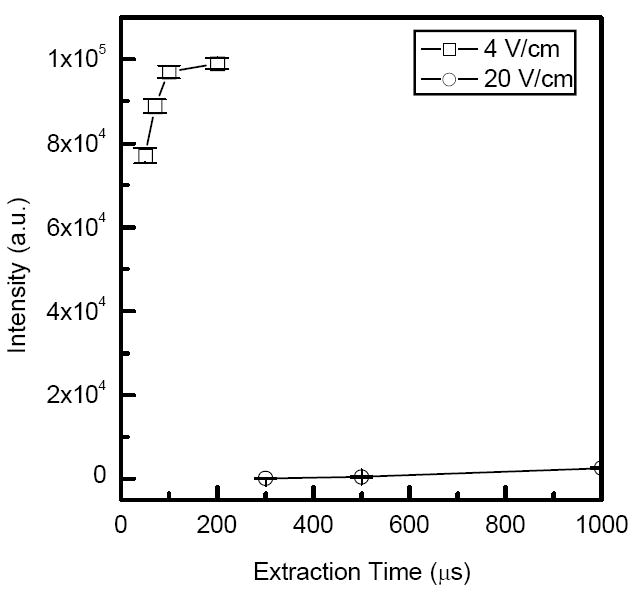
Monoisotopic [M+H]+ peak intensity from ESI of a 1 μM reserpine solution as a function of the extraction time for two different DC gradients in the trap region.
The ion current was measured at the collisional quadrupole and the charge collector (Figure 1a) in both trapping and continuous modes. Comparison of the observed ion current signals on these two elements provides insight into transmission efficiency through the quadrupole interface and electrostatic ion optics. In addition, an estimate of trapping efficiency can be made based on comparison of ion signals at the collisional quadrupole in continuous and trapping modes. Figure 4a shows the ion current measured at the collisional quadrupole rods obtained from ESI of a 1 μM solution of reserpine. Ion current pulses were acquired at different accumulation times in the ion trap. Note the maximum amplitude of the ion current pulse (28 nA at 100 ms accumulation time) exceeded that of the continuous beam (216 pA) by more than two orders of magnitude. Figure 4b shows both pulsed and continuous ion currents at the charge collector obtained with the same solution. The traces in Figure 4b have a full width at half maximum (fwhm) of ~ 1-2 ms as compared to ~200 μs for the traces shown in Figure 4a, reflecting diffusion broadening of the ion current pulse in the collisional quadrupole. The reason for the lower ion currents at the charge collector is explained below.
Figure 4.
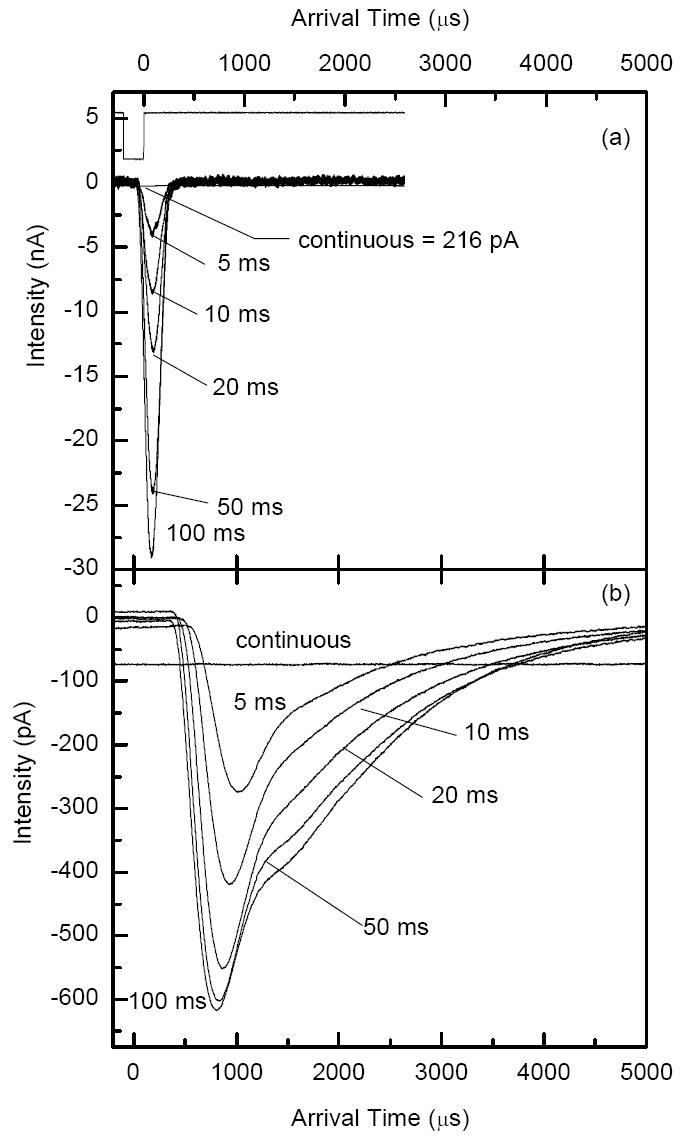
Current pulse measurements for ESI of a 1 μM reserpine solution at various accumulation times. a) Current at the collisional quadrupole. b) Current at the charge collector. Extraction time was 200 μs. Note that the intensity unit is nA for panel a and pA for panel b.
The number of charges released from the ion trap was calculated from the areas under the traces in Figure 4a and is plotted as solid squares in Figure 5a. The number of charges increases as the accumulation time increases. While the total number of charges reaches ~3×107, the linear range for the ion trap, extends to only ~1×107 charges. The trapping efficiency (depicted in Figure 5a as open squares) was calculated as the ratio of the charge impinging on the collisional quadrupole rods after a single accumulation event to the charge delivered to the same quadrupole by the continuous beam over the same accumulation period (see Figure 4a). The trapping efficiency reached 70-80% at shorter accumulation times (< 10 ms), and then decreased to 20-30% on approaching the charge capacity of the trap (for accumulation time > 50 ms). The ion losses at the charge collector were due to reduction of ion transmission from the collision quadrupole to the charge collector. Figure 5b shows the transmission efficiency of the quadrupole and focusing ion optics interface in detecting pulsed ion currents at the charge collector. Transmission efficiency was determined as the ratio of the pulsed ion current (expressed as number of charges) at the charge collector to the pulsed ion current at the collisional quadrupole rods in Figure 5a, and is plotted in Figure 5b as a function of the number of charges exiting the ion trap. Note that the pulsed ion current transmission decreased as the total number of ions transmitted through the collisional quadrupole increased. This trend is indicative of increased radial expansion of the ion packet due to the increased space charge effects and the associated ion losses on the conductance limit orifices and in the elements of the electrostatic Einzel lens. In comparison, total ion transmission efficiency of the continuous beam from the ion funnel to the charge collector was ~ 35%, which also included ion losses due to the low m/z cut off (<200 m/z) in the collisional quadrupole. Further improvements in transmission of dense ion packets through the quadrupole interface are feasible through more efficient ion focusing at higher residual gas pressures. However, in proteomic experiments, rigorous control over ion populations accumulated in the ion trap can be accomplished using automated gain control33, to be described in subsequent publication.
Figure 5.
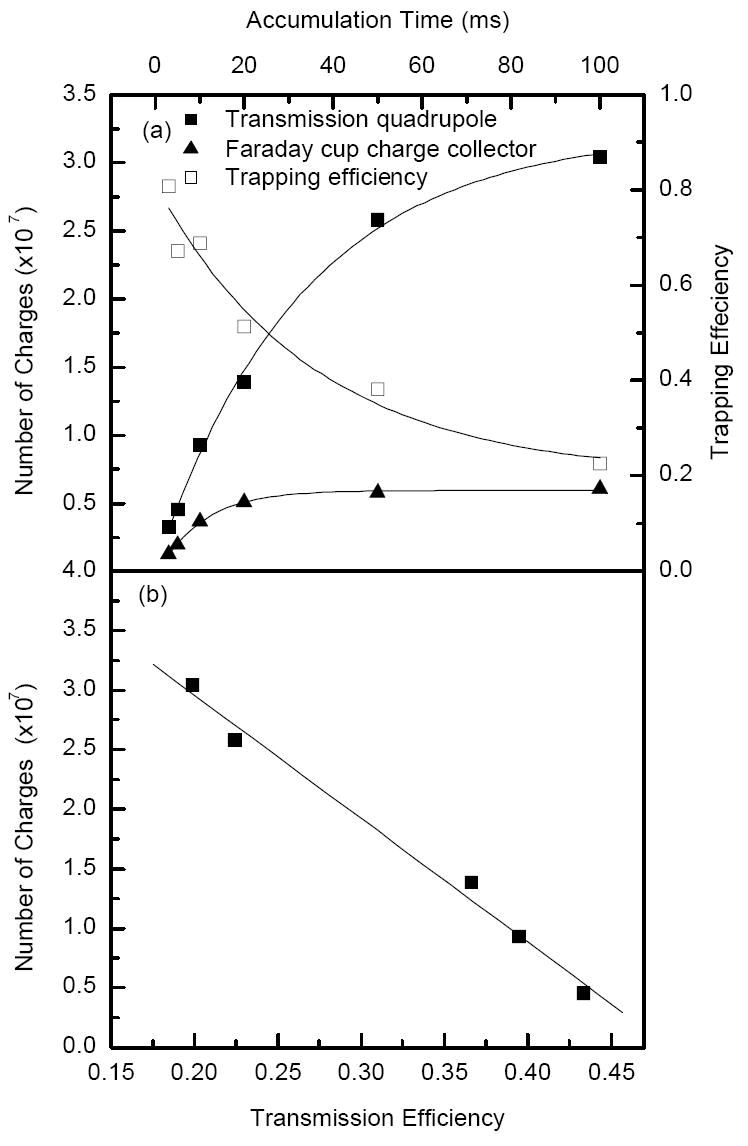
a) Number of charges detected at the collisional quadrupole (solid squares) and charge collector (solid triangles) as calculated from the areas under the traces shown in Figure 4. b) Transmission efficiency through the quadrupole interface as calculated from the ratio of the number of charges at the collisional quadrupole and charge collector in Figure 5 a.
The linear dynamic range of the ion trap was studied using reserpine solutions at concentrations ranging from 10 nM to 1 μM. Figure 6 shows the intensity of monoisotopic peak of reserpine normalized to the number of ion trap releases per 1 second TOF acquisition as a function of accumulations time at different concentrations. As indicated in Figure 6, the ion trap has a linear response at accumulation time ≤ 20 ms independent of sample concentration. The nonlinearities at longer accumulation time are due to limitation in ion trap charge capacity and increased transmission losses at higher ion densities.
Figure 6.
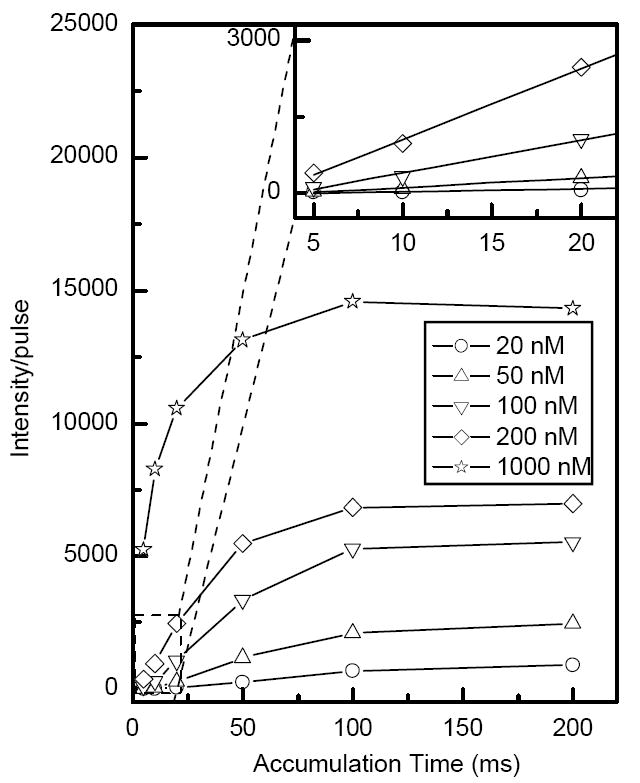
Monoisotopic [M+H]+ peak intensity for ESI of a resperine solution normalized to the number of trap releases per 1 second acquisition time as a function of accumulation time at different concentrations. The inset shows the linear dynamic range of the trap.
The improved data quality resulting from the use of the ion trap becomes more evident at low concentrations. Figure 7a shows a portion of a mass spectrum for 10 nM mixture of bradykinin and fibrinopeptide A. For the same TOF acquisition time of 1 second, the intensities of doubly-charged bradykinin and fibrinopeptide A ions in the trap mode are more than an order of magnitude higher than those in the continuous (no trapping) mode. In the trapping mode, the mass spectrum corresponds to 20 trap releases per 1s (or a sum of 200 TOF pulses) while in the continuous mode the mass spectrum is obtained as a sum of 9000 TOF pulses. Figure 7b-c shows the ratios of intensities of doubly-charged bradykinin and fibrinopeptide A ions in the trapping and continuous modes as a function of the accumulation time at different analyte concentrations. The trends shown in Figure 7b-c indicate that the intensities of doubly-charged bradykinin and fibrinopeptide A exceeded those from the continuous beam by a factor of 13-20-fold at a concentration of 10 nM. When the ion population reaches trap capacity, no further increase in sensitivity is expected in the trapping mode. Furthermore, an increase in accumulation time results in lower duty cycle (and signal) as fewer ion packets are introduced to the TOF MS per unit time, as illustrated in Figure 7b-c at longer accumulation times.
Figure 7.
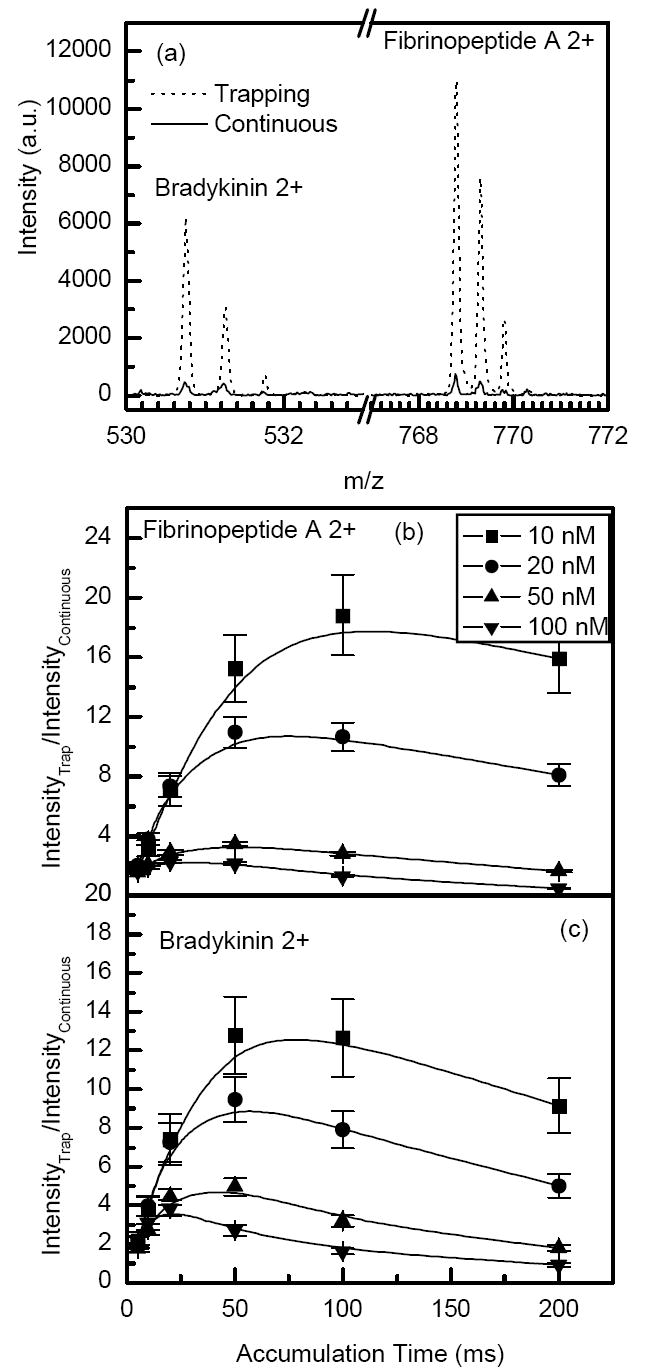
a) Portions of the mass spectra for ESI of a 10 nM mixture of bradykinin and fibrinopeptide A solution in the continuous and trapping mode (100 ms accumulations, 45 μs extraction time). Lower panels show the ratio of ion signals in the trapping and continuous modes for doubly-charged b) fibrinopeptide A and c) bradykinin as a function of accumulation time at different concentrations. The mass spectrum acquisition time was 1 second in all cases.
Sensitivity improvement in the trapping mode is also related to the more effective ion desolvation and the resulting reduction of chemical noise. The S/N’s and noise levels for the data acquired with a 10 nM mixture of bradykinin and fibrinopeptide A are listed in Table 1. Although Figure 7c indicates a ~13-fold signal enhancement for bradykinin in the trapping mode as compared to that obtained in the continuous regime, the actual S/N gain was ~35 due to a 3-fold lower chemical noise. The reduction in noise reported in Table 1 is most likely related to the gentle (relatively slow) declustering of solvated ions during their extended accumulation in the trap. A desolvation mechanism in the ion trap is expected to involve water/solvent cluster heating as a result of collisions with neutral gas assisted by the RF field. The RF heating of ions is also promoted by the DC electric field that drives ions to locations with increased effective potential resulting in more efficient activation.
Table 1.
Signal-to-noise ratio (S/N) and noise level for bradykinin and fibrinopeptide A.
| Bradykinin 2+530.75 amu | Fibrinopeptide A 2+768.85 amu | |||
|---|---|---|---|---|
| S/N | noisea | S/N | noise | |
| continuous | 15.4 | 32.1 | 49.8 | 14.3 |
| Trappingb | 534.9 | 11.8 | 988.8 | 13.5 |
| Ratio | 34.6 | 0.4 | 19.9 | 0.9 |
The noise is in units of mass spectrum intensity (arbitrary units) and is calculated as an average intensity in an m/z segment located 1 Da to the left from the analyte peak.
The accumulation time is 100 ms.
The enhancements in S/N is thus attributed to a combination of an increase in the number of transmitted ions to the TOF detector due to ion accumulation, to more efficient desolvation of the analyte ions, and to removal of chemical noise ion peaks following the desolvation of smaller ions in the ion trap. Since most of the solvent ions are in the low m/z range, a more pronounced background noise reduction is observed in the lower m/z range. The S/N enhancement due to the reduction in the background chemical noise plays an important role in improving the instrument limit-of-detection (LOD). Figure 8a-b shows portions of the mass spectra obtained for a 0.1 nM solution of neurotensin in both continuous and trapping modes. The solution was infused at a flow rate of 60 nL/min and both mass spectra were acquired over a 3 s period, corresponding to 300 zmol of analyte introduced. The mass spectrum in Figure 8a reveals pronounced chemical noise peaks at every m/z, which is common for TOF MS operating in the continuous mode. Figure 8b shows a mass spectrum in which the background noise is suppressed and the S/N is 37-fold enhanced. Although it is possible to reduce such solvent related chemical noise by greater activation (e.g. heating) in the ESI interface, this measure can also lead to dissociation of analyte species, and thus decreased sensitivity.
Figure 8.
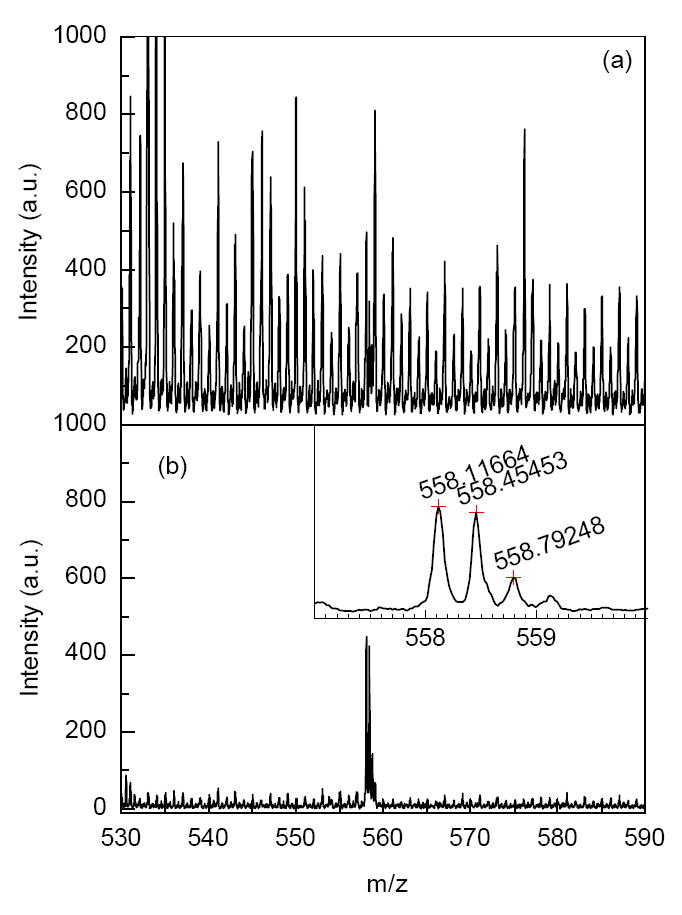
Improvement in the signal-to-noise ratio (S/N) of [M+H3]3+ for ESI of a 0.1 nM of neurotensin solution. a) Continuous regime. b) Trapping mode. Accumulation time = 100 ms, extraction time = 70 μs. In both the continuous and trapping modes the sample infusion rate was 60 nl/min and TOF acquisition time was 3 seconds.
Due to relatively high pressure and the applied DC gradient, a trapped ion cloud exhibits mobility separation during ejection from the trap. As a result, multiply charged ions are separated from their singly charged counterparts in the ejection step. Since mobility is directly related to charge state and inversely proportional to the collisional cross section,34 multiply charged ions tend to exit the ion trap faster as illustrated in Figure 9a-b. The mixture of peptides was ejected from the ion trap using two different gate opening events: 100 μs (Figure 9a) and 40 μs (Figure 9b). Limiting the extraction time to 40 μs led to discrimination against singly charged species because of their longer drift time through the gate. This effect is exemplified with the singly charged peak at m/z 387.2. The intensity of this peak decreased by two orders of magnitude when the extraction time was shortened from 100 μs (Figure 9a) to 40 μs (Figure 9b). Similar effect was also observed for other singly charged species shown in Figure 9a-b.
Figure 9.
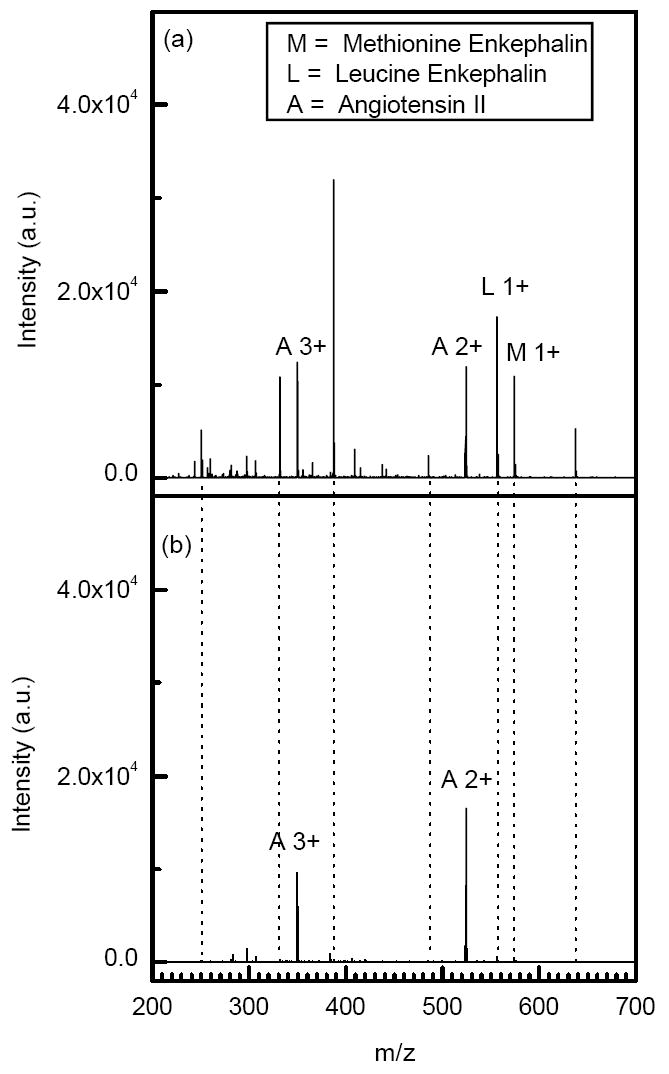
Portions of the mass spectra from ESI of a 28 ng/ml solution of peptides (methionine enkephalin, leucine enkephalin and angiotensin II) at different ejection times. Trapping conditions: a) accumulation time = 50 ms, ejection time = 100 μs and trap DC gradient = 4V/cm. c) accumulation time = 50 ms, ejection time = 40 μs and trap DC gradient = 4V/cm.
The controlled separation of multiply charged species from singly charged ones by changing the extraction time could be used to improve S/N in proteomic experiments with multiply charged tryptic peptides. Gating the ion trap for a period of 40 μs not only substantially decreased the noise level and suppressed the singly charged species, but also improved the S/N of multiply charged species by more than an order of magnitude. This approach is advantageous over other methods in which 50-85% of multiply charged species can be lost to suppress singly charged species.30
Conclusions
We developed and characterized an ion trap operating at pressures that enable seamless interfacing to atmospheric pressure ionization sources. The trap operating pressure can also be further increased 27 for e.g. more efficient coupling to mobility separations. For example, in the current arrangement, the ion trap is characterized by an extraction time of 40 μs for multiply charged ions and 100 μs for singly charged species. We found that an increase in pressure slowed the ion cloud release from the ion trap as a result of enhanced viscous effects. To address the increased drag effects, we implemented further modifications in the trap design that resulted in significant improved IMS-TOF performance (to be reported in the subsequent publication).
In the current study, the performance of TOF MS was examined in both trapping and continuous modes. In continuous mode, TOF MS provides a high pulsing rate of ~10 kHz, and given sufficient ion current, each successive TOF pulse can deliver ions to the detector. In the trapping mode, only 100 to 1000 ion packets are delivered to the TOF detector over the same acquisition period. However, packets of ions accumulated in the trap are characterized by higher charge density than those in the continuous mode. Therefore, to more fully realize the advantages of ion accumulation, the TOF acquisition system should provide a linear detection response to multiple ions within a single TOF digitizer bin, which implies benefit from the implementation of a fast multi-bit ADC-based data acquisition system.
Lastly, the improved S/N in the trapping mode resulted from a combination of several factors that contributed to an increase in signal intensity and a decrease in the chemical noise level. Ion accumulation in the trap appeared to be particularly advantageous at very low analyte concentrations. The ion packets exiting the trap are characterized by higher ion densities and, therefore, result in higher S/N. In addition, the ion trap facilitates more efficient desolvation of ions resulting in substantial reduction in background noise and further S/N improvement.
Supplementary Material
Acknowledgments
Portions of this research were supported by the U.S. Department of Energy (DOE) Office of Biological and Environmental Research, the National Institute of Allergy and Infectious Diseases (NIH/DHHS) through interagency agreement Y1-AI-4894-01 and the National Cancer Institute (NCI) through grant number 1R21 CA126191-01. Experimental work was performed in the Environmental Molecular Sciences Laboratory, a DOE national scientific user facility at the Pacific Northwest National Laboratory (PNNL) in Richland, Washington. PNNL is operated by Battelle for the DOE under Contract No. DE-AC05-76RLO 1830.
References
- 1.Verentchikov AN, Ens W, Standing KG. Anal Chem. 1994;66:126–133. doi: 10.1021/ac00073a022. [DOI] [PubMed] [Google Scholar]
- 2.Morris HR, Paxton T, Dell A, Langhorne J, Berg M, Bordoli RS, Hoyes J, Bateman RH. Rapid Commun Mass Spectrom. 1996;10:889–896. doi: 10.1002/(SICI)1097-0231(19960610)10:8<889::AID-RCM615>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Guilhaus M, Selby D, Mlynski V. Mass Spectrom Rev. 2000;19:65–107. doi: 10.1002/(SICI)1098-2787(2000)19:2<65::AID-MAS1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 4.Wolff JC, Eckers C, Sage AB, Giles K, Bateman R. Anal Chem. 2001;73:2605–2612. doi: 10.1021/ac001419a. [DOI] [PubMed] [Google Scholar]
- 5.Dodonov AF, Kozlovski VI, Soulimenkov IV, Raznikov VV. Eur J Mass Spectrom. 2000;6:481–490. [Google Scholar]
- 6.Mirgorodskaya OA, Shevchenko AA, Chernushevich IV, Dodonov AF, Miroshnikov AI. Anal Chem. 1994;66:99–107. [Google Scholar]
- 7.Chernushevich IV, Ens W, Standing KG. Anal Chem. 1999:452A–461A. doi: 10.1021/ac990524l. [DOI] [PubMed] [Google Scholar]
- 8.Chernushevich IV, Thomson B. 6,507,019 US Patent.
- 9.Michael SM, Chien BM, Lubman DM. Anal Chem. 1993;65:2614–2620. [Google Scholar]
- 10.Purves RW, Li L. J Am Soc Mass Spectrom. 1997;8:1085–1093. [Google Scholar]
- 11.Doroshenko VM, Cotter RJ. Journal of Mass Spectrometry. 1998;33:305–318. doi: 10.1002/(SICI)1096-9888(199804)33:4<305::AID-JMS635>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 12.Campbell JM, Collings BA, Douglas DJ. Rapid Commun Mass Spectrom. 1998;12:1463–1474. doi: 10.1002/(SICI)1097-0231(19980815)12:15<1003::AID-RCM275>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 13.Luca A, Schlemmer S, Čermák I, Gerlich D. Rev Sci Instrum. 2001;72:2900–2908. [Google Scholar]
- 14.Collings BA, Campbell JM, Mao D, Douglas DJ. Rapid Commun Mass Spectrom. 2001;15:1777–1795. doi: 10.1002/rcm.440. [DOI] [PubMed] [Google Scholar]
- 15.Dresch T, Gulcicek EE, Whitehouse C. 6,020,586 US Patent.
- 16.Loboda A, Krutchinsky A, Loboda O, McNabb J, Spicer V, Ens W, Standing K. Eur J Mass Spectrom. 2000;6:531–536. [Google Scholar]
- 17.Dodonov A, Kozlovsky V, Loboda A, Raznikov V, Sulimenkov I, Tolmachev A, Wollnik AKH. Rapid Commun Mass Spectrom. 1997;11:1649–1656. doi: 10.1002/(SICI)1097-0231(19971015)11:15<1649::AID-RCM67>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 18.Gerlich D. In: State-Selected and State-to-State Ion-Molecule Reaction Dynamics. Part 1. Experiment. Ng C-Y, Baer M, editors. Vol. 82. Wiley; New York: 1992. pp. 1–176. [Google Scholar]
- 19.Tolmachev AV, Chernushevich IV, Dodonov AF, Standing KG. Nucl Instrum Methods Phys Res, Sect B. 1997;124:112–119. [Google Scholar]
- 20.Belov ME, Nikolaev EN, Harkewicz R, Masselon CD, Alving K, Smith RD. Int J Mass Spectrom. 2001;208:205–225. [Google Scholar]
- 21.Mansoori BA, Dyer EW, Lock CM, Bateman K, Boyd RK, Thomson BA. J Am Soc Mass Spectrom. 1998;9:775–788. [Google Scholar]
- 22.Taban IM, McDonnell LA, Rompp A, Cerjak I, Heeren RMA. Int J Mass Spectrom. 2005;244:135–143. [Google Scholar]
- 23.Shaffer SA, Tang K, Anderson GA, Prior DC, Udseth HR, Smith RD. Rapid Commun Mass Spectrom. 1997;11:1813–1817. [Google Scholar]
- 24.Kim T, Tolmachev AV, Harkewicz R, Prior DC, Anderson GA, Udseth HR, Smith RD, Bailey TH, Rakov S, Futrell JH. Anal Chem. 2000;72:2247–2255. doi: 10.1021/ac991412x. [DOI] [PubMed] [Google Scholar]
- 25.Belov ME, Gorshkov MV, Udseth HR, Anderson GA, Smith RD. Anal Chem. 2000;72:2271–2279. doi: 10.1021/ac991360b. [DOI] [PubMed] [Google Scholar]
- 26.Tolmachev AV, Kim T, Udseth HR, Smith RD, Bailey TH, Futrell JH. Int J Mass Spectrom Ion Processes. 2001;203:31–47. [Google Scholar]
- 27.Ibrahim Y, Tang K, Tolmachev AV, Shvartsburg AA, Smith RD. J Am Soc Mass Spectrom. 2006;17:1299–1305. doi: 10.1016/j.jasms.2006.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cech NB, Enke CG. Mass Spectrom Rev. 2001;20:362–387. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 29.Thomson BA, Chernushevich IV. Rapid Commun Mass Spectrom. 1998;12:1323–1329. [Google Scholar]
- 30.Chernushevich IV, Fell LM, Bloomfield N, Metalnikov PS, Loboda AV. Rapid Commun Mass Spectrom. 2003;17:1416–1424. doi: 10.1002/rcm.1074. [DOI] [PubMed] [Google Scholar]
- 31.Kim T, Tang K, Udseth HR, Smith RD. Anal Chem. 2001;73:4162–4170. doi: 10.1021/ac010174e. [DOI] [PubMed] [Google Scholar]
- 32.Page JS, Tolmachev AV, Tang K, Smith RD. J Am Soc Mass Spectrom. 2006;17:586–592. doi: 10.1016/j.jasms.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz JC, Zhou X-G, Bier ME. 5,572,022 US Patent.
- 34.McDaniel EW, Mason EA. The Mobility and Diffusion of Ions in Gases. John Wiley & Sons; New York: 1973. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.