

Abstract
Structure-based mutational analysis was employed to probe an unusual intramolecular interaction between partially buried glutamate residues adjacent to the active site of Escherichia coli glutaminyl-tRNA synthetase (GlnRS). Crystal structures of unliganded GlnRS and of the GlnRS-tRNAGln complex reveal that the Glu34 and Glu73 side-chain carboxylates contact each other only in the tRNA-bound state, and that the interaction is formed via mutual induced-fit transitions that occur en route to the ground-state Michaelis complex. Steady-state and transient kinetic analysis of mutant enzymes suggest that formation of this intermolecular contact is a key event that facilitates proper formation of the active site. Mutants at both positions destabilize the binding of substrate glutamine at the opposite side of the active-site cleft, while Glu73 appears to play an additional important role by promoting correct binding of the 3′-acceptor end of tRNA adjacent to both ATP and glutamine. The data suggest the existence of multiple structural pathways by which the binding of tRNA propagates conformational transitions leading to proper formation of the glutamine binding site. The single-turnover kinetic analysis also establishes that the Glu34 carboxylate does not play a direct enzymatic role as a catalytic base to help deprotonate the tRNA-A76 nucleophilic 2′-hydroxyl group. The elimination of this previously proposed mechanism, together with recent chemical modification experiments in the histidyl-tRNA synthetase system, emphasizes that substrate-assisted catalysis by the phosphate of the aminoacyl adenylate may be a common means by which all tRNA synthetases facilitate the aminnoacyl transfer step of the reaction.
Keywords: RNA-protein interaction, induced fit, aminoacyl-tRNA synthetase, enzyme catalysis
Aminoacyl tRNA-synthetases comprise an ancient class of enzymes that are crucial to maintaining fidelity in the genetic code. They catalyze specific aminoacyl-tRNA synthesis in a two-step reaction. First, energy from ATP hydrolysis is utilized to generate an activated aminoacyl adenylate intermediate possessing a mixed anhydride linkage (1). In a second step, the 2′ or 3′ ribose oxygen of the 3′-terminal A76 tRNA nucleotide attacks the carbonyl carbon atom of this linkage, forming aminoacyl-tRNA with release of AMP. The tRNA synthetases are divided into two classes possessing topologically distinct active-site domains (2). Other domains in the structure are primarily involved in discriminating against noncognate tRNAs, and diverge significantly among the enzymes.
Crystal structures of all twenty canonical tRNA synthetases, many determined in multiple liganded states including as complexes with tRNA, reveal the common presence of induced-fit conformational rearrangements (3). Interestingly, despite the common active-site Rossmann fold (class I) or mixed α/β domain (class II), the conformational changes upon ligand binding are not conserved. Instead, distinct rearrangements, often including both global repositioning of domains and local movements of surface loops and side-chains, appear to be idiosyncratic to each enzyme (4). The ubiquity of induced fit in tRNA synthetases reflects the common presence of this phenomenon in RNA-protein interactions generally (5). Catalytic roles of induced fit may include (i) exploiting intrinsic flexibility to lower the free-energy barrier for complex formation, and/or (ii) fostering specificity via incomplete triggering of rearrangements in noncognate complexes, leading to the improper juxtaposition of reactive moieties on bound substrates (6, 7).
E. coli glutaminyl-tRNA synthetase (GlnRS) is a monomeric class I enzyme that has served as a model system to address the detailed stereochemical pathway of catalysis and the molecular origins of specificity for the amino acid and tRNA (8). GlnRS is one of four class I tRNA synthetases that require binding of cognate tRNA to catalyze amino acid activation, suggesting that structural transitions associated with tRNA complex formation are crucial to formation of the correct active site conformation (9). Substantial other evidence also points to the importance of induced fit in mediating GlnRS substrate specificity. First, fluorescence experiments have yielded direct physical evidence for enzyme rearrangements upon tRNA binding (10, 11). Second, mutation of tRNAGln anticodon nucleotide U35 sharply reduces both the rate of the chemical steps for aminoacylation as well as the affinity for glutamine, demonstrating the existence of a structural pathway for long-range communication from the C-terminal β-barrel domains to the active site some 40 Å distant (12). Third, comparison of the crystal structures of unliganded GlnRS and the GlnRS-tRNAGln complex shows in detail how tRNA binding generates structural changes throughout the entire enzyme structure, particularly with respect to the positioning of key active site peptides that bind glutamine and ATP (4, 13).
When tRNA binds to GlnRS, the 110 amino acid acceptor-binding domain (ABD; residues 100-209), which is inserted between the two halves of the dinucleotide fold (DNF), makes a rigid body rotation of about 10° towards the active site (Figure 1). In addition to properly positioning a complementary surface for binding the hairpinned single-stranded tRNA 3′-acceptor end, rotation of the ABD also results in the reorganization of a DNF surface loop spanning amino acids 64-76 within the N-terminal half of the fold (depicted in purple in Figure 1). This surface loop is only loosely ordered in the unliganded enzyme. Its reorientation has several important consequences: (i) the side chain of Asp66 is repositioned to point into the substrate binding pocket, where it makes a direct salt bridge with the α-NH3+ group of the amino acid; (ii) the side-chain carboxylate of Glu73 makes a new contact with the carboxylate of Glu34. The new Glu34-Glu73 interaction, which presumably also involves uptake of a proton for charge neutralization between the carboxylates, apparently assists in reorganization of the active-site peptide spanning Pro32-Pro33-Glu34-Pro35, and facilitates formation of a new Asp66-Pro33 main-chain hydrogen-bond. Together, these active site motions also allow binding of the tRNA-A76 ribose sugar directly adjacent to the ATP α-phosphate, as required for catalysis (14) (Figures 2, 3).
Figure 1.
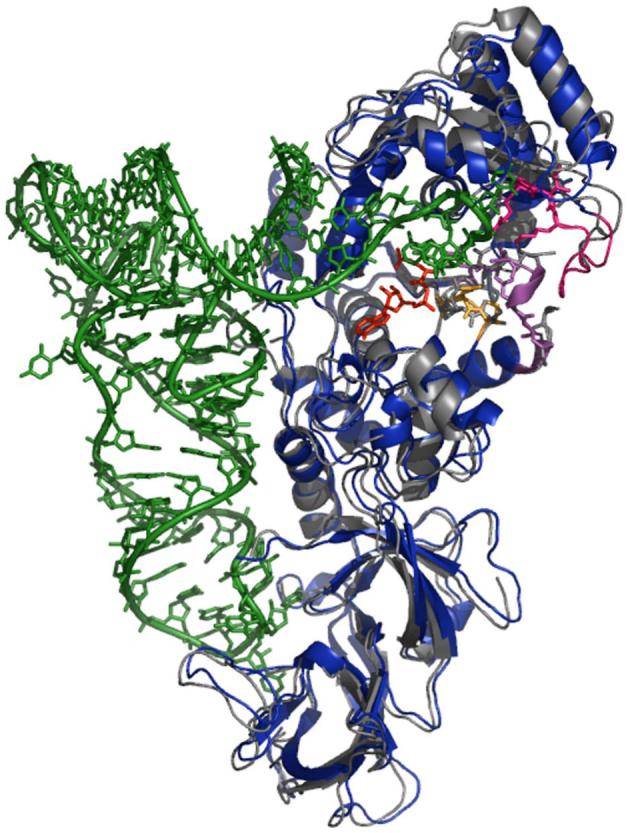
Superimposition of unliganded (gray) and tRNA-bound (blue) GlnRS structures based on protein backbone atoms within the dinucleotide fold, as defined previously (4). Peptide segments implicated in the induced-fit rearrangement studied here are colored as follows: amino acids 195-204 (pink), amino acids 63-76 (purple), amino acids 33-35 (gold). The QSI inhibitor (see text) is shown is red.
Figure 2.

Interactions in and adjacent to the GlnRS active site. The QSI inhibitor is shown in red and tRNA is depicted in green. Hydrogen bonds are shown as dotted gold lines. Peptides in GlnRS are colored as in Figure 1.
Figure 3.

Stereoview of interactions made by Glu34 in the GlnRS active site. Atoms are colored as follows: carbon, green; oxygen, red; nitrogen, blue; phosphoris, orange. Waters are shown as red spheres, and hydrogen bonds are shown by dashed black lines.
This examination of crystal structures strongly suggests that the unusual intramolecular Glu34-Glu73 contact formed upon tRNA binding is central to mediating active site assembly by induced fit. Moreover, another role for Glu34 has been proposed based on examination of the crystal structure of GlnRS bound to tRNAGln and the aminoacyl adenylate analog 5′-O-[N-(L-glutaminyl)sulphamoyl] adenosine (QSI) (15). In that study, it was suggested that the Glu34 carboxylate functions as a general base in the second step of the aminoacylation reaction, by increasing the nucleophilicity of the 2′-OH of tRNA A76 via a water-mediated interaction. An earlier alternative proposal, however, is that a nonbridging oxygen of the glutaminyl adenylate α-phosphate group functions as a general base for this reaction (14). The latter mechanism has been suggested to operate in the class II aspartyl (AspRS) and histidyl-tRNA synthetases (HisRS) as well (16, 17). Indeed, since this mechanism is substrate-mediated, it could function generally in the tRNA synthetase enzyme family.
Here we investigate the roles of Glu34 and Glu73 in catalysis by E. coli GlnRS by mutational analysis coupled to steady-state and pre-steady state kinetic measurements of glutaminyl-tRNA synthesis. By single-turnover kinetics, we find that the rate of the chemical steps for Gln-tRNAGln synthesis by the E34Q mutant is nearly identical to that of the wild-type enzyme, indicating that the carboxylate group of Glu34 does not have an obligate role in facilitating the second-step aminoacyl transfer. Surprisingly, rates of aminoacyl-tRNA synthesis in mutants of the more distal Glu73 residue are slower than rates measured for mutants at Glu34, suggesting a unique role for the former side-chain in promoting proper juxtaposition of the A76 ribose in the active site. Mutations at both positions weaken the binding affinity for glutamine at the opposite side of the active site, highlighting the interdependent nature of substrate binding and the extraordinary precision of architectural construction that is required to properly juxtapose all three reactive moieties for the overall two-step reaction.
EXPERIMENTAL PROCEDURES
Mutagenesis
Oligodeoxynucleotide primers for mutagenesis were purchased from IDT. Mutations were made using the Quikchange™ site directed mutagenesis (Stratagene) protocol using the GlnRS-6His (QRSH) plasmid as a template (18), and followed by transformation into XL-1 blue supercompetent cells (Stratagene). DNA sequencing of mutant genes was performed by the University of Iowa DNA sequencing facility. After sequence confirmation, mutant plasmids were transformed into PlysS one-shot supercompetent cells (Invitrogen) for expression.
Enzyme and tRNA Preparation
His-tagged GlnRS mutants were expressed in BL21-DE3(pLysS) by induction with 1 mM IPTG at OD600 between 0.4 and 0.6. Cells were resuspended in a buffer containing 0.5 M NaCl, 50 mM HEPES (pH 7.2), 10 mM imidazole, and were disrupted by French press. 100 mM PMSF, 100 mM benzamidine, 50 μl of RQ1 DNase (Promega), and 15 mM MgCl2 were added to the lysate, and the mixture was stirred at room temperature for 20 min. Enzyme was purified on a 5 ml. NTA-agarose nickel affinity column (Qiagen) equilibrated in 10 mM imidazole, 20 mM HEPES (pH 7.2), 1 mM β-mercaptoethanol, and 0.5 M NaCl; the elution buffer was identical to the equilibration buffer except for the inclusion of 120 mM imidazole. His-tagged GlnRS was recovered at better than 99% purity as judged by SDS polyacrylamide gel electrophoresis, and was stored at high concentration at -20°C, as described for the native enzyme (21). E. coli tRNA2Gln and tRNA variants (each containing a catalytically neutral U1G mutation to promote efficient transcription initiation) were transcribed in high yield from a synthetic DNA template, as described (19). The template was constructed from complementary synthetic oligonucleotides containing a short central overlapping region, which were extended to form the full-length duplex by treatment with the Klenow fragment of E. coli DNA polymerase I. The DNA template incorporated 2′-O methyl sugars at the two 5′ nucleotides in the noncoding DNA strand, resulting in a high proportion of enzymatically active tRNA transcripts (19).
Aminoacylation Assay using 32P-labeled tRNA
tRNA was 32P-labelled at the 3′-terminal internucleotide linkage using the exchange reaction of tRNA nucleotidyltransferase, as described (12, 20, 21). To ensure maximal substrate activity, labeled and unlabeled tRNA were mixed to the appropriate final concentration and heated at 65°C for 3 min., followed by addition of MgCl2 to 7.5 mM and cooling to ambient temperature. All reactions were quenched in a buffer containing 0.1 % SDS, 0.15 M sodium acetate (pH 5.2). P1 nuclease digestions were performed by adding 1-5 uL of the reaction mixture to a microtitre well containing 3-5 μL of 0.1 mg/ml P1 nuclease (Fluka), 0.15 M sodium acetate (pH 5.2) and incubating 10 min. at ambient temperature. Aminoacylated tRNA (as 3′-aminoacylated A76) and unreacted substrate (as unmodified AMP) were separated by TLC and quantitated by phosphorimaging analysis. Steady-state assays were performed at 20 μM tRNAGln, 3-100 mM glutamine, and 30 mM ATP with the enzyme concentration for each mutant as follows: E34A (either 0.25 or 0.5 μM); E34Q (0.28 μM); E34D (0.25 μM); E73Q (2 μM). For E73A, a value for kcat/Km was estimated from initial velocities, because reactions were too slow to permit estimation of the individual kcat and Km parameters under conditions of sufficient substrate excess. Timecourses were also adjusted in order to ensure that the maximal product formation remained under 10% of the total acylable tRNA (50-70%).
Rapid Chemical Quench Kinetics
Single-turnover measurements of E34 mutants to determine kchem were performed using a rapid chemical quench flow apparatus (Kintek RQF-3) at saturating substrate concentrations (20 μM tRNAGln; 3-100 mM glutamine; 30 mM ATP) by the same protocol and under the same buffer conditions as the steady-state reactions. The temperature was maintained at 37° C for all experiments. Enzyme and substrates in two 20 μl sample loops were rapidly mixed into a single reaction loop of specified dimensions to control the time of the reaction. The mole ratio of GlnRS to tRNAGln was maintained at 4:1 for all experiments (80 μM GlnRS). Reactions were quenched in 50 μl of 400 mM sodium acetate (pH 5.2), 0.1 % SDS. 8-10 timepoints were collected for each kchem determination. Reaction rates were fit to a first-order exponential equation (Y = A *e-kchem*t) and plotted using Kaleidagraph™ software.
The concentration dependencies of the single-turnover reactions were determined by titrating the concentration of one substrate while maintaining saturating levels of the other two, following procedures recently described for the wild-type enzyme (18). To determine the glutamine Kd, reactions contained 60-100 μM GlnRS, 15-20 μM tRNAGln, 30 mM ATP, 13.75 μM MgCl2, 50 mM Tris (pH 7.5), 5 mM DTT, and 3 mM - 125 mM glutamine. For all experiments, the concentration of 3′ end-labeled tRNAGln was negligible compared to the unlabeled concentration. kchem values determined at each substrate concentration were fit to the hyperbolic (Y = S0* kchem/S0 + Kd) binding equation to derive the Kd under aminoacylation conditions. All single-turnover data under saturating conditions were acquired in triplicate and plotted using Kaleidagraph™.
RESULTS AND DISCUSSION
The recent availability of a crystal structure for unliganded GlnRS offers the first opportunity for a detailed comparison with the GlnRS-tRNAGln complex bound to ATP and amino acid substrates (4). Such an analysis is important for the insights obtainable into the process of mutual induced-fit: the conformational rearrangements in both macromolecular partners that occur between the formation of an initial “encounter complex”, and the ground-state Michaelis complex. Although a structure of unliganded tRNAGln is not available, its bound conformation very strongly suggests that hairpinning of the single-stranded 3′-acceptor terminus into the active site is a primary rearrangement that occurs upon binding (Figure 1; (13)). The tRNA hairpin is stabilized by complementary protein interactions with the extruded C74 base, by an intramolecular hydrogen bond between the exocyclic amino group of G73 and the phosphate of A72, and by ion-pair and hydrogen-bonding electrostatic contacts with the sugar-phosphate backbone at nucleotides 74-76. To make these contacts with tRNA, the entire acceptor-binding domain (ABD) of GlnRS is rotated by 10° toward the active site, propagating a series of conformational changes in enzyme surface loops that terminate in the amino acid and ATP binding sites (Figures 1, 2).
The comparisons between the unliganded and tRNA-bound GlnRS structures suggest that new interactions between the surface loop spanning amino acids 64-76, and an active-site peptide at residues Pro32-Pro35, may be of particular importance for promoting assembly of the catalytic site. These two enzyme segments form a complementary interface at the edge of the active site that includes numerous van der Waals and electrostatic contacts. In addition to a new hydrogen bond between main-chain amides of Asp66 and Pro33, the most notable interaction between these peptides is an unusual contact between the partially buried side-chain carboxylates of Glu73 and Glu34. In the tRNA-bound structure, Glu34 is positioned 3.7 Å from the 2′-OH group of the A76 ribose sugar - the nucleophile for the second aminoacyl transfer step in the mechanism - and is located in a key β-strand-loop-α-helix motif containing the class I HIGH signature motif that provides a platform for ATP binding. In addition to its contact with Glu34, Glu73 also makes several new intramolecular interactions with Asn69, which in turn mediates electrostatic contacts of Lys102 and Arg104 with the tRNA phosphates (Figures 2, 3). Thus, the surface loop spanning amino acids 64-76, which is partially disordered in the unliganded structure, appears to provide the key connection between tRNA 3′-end positioning, and the reconfiguring of the ATP binding site.
The approach of the partially buried Glu34 and Glu73 carboxylates within hydrogen-bonding distance of each other also suggests that the pKa of one of these moieties is sufficiently elevated to render likely the acquistion of a proton at conditions of physiological pH. To test the importance of this interaction in mediating induced fit, we constructed and purified the following GlnRS mutant enzymes: E34Q, E34D, E34A, E73Q, and E73A. All of the mutant enzymes were highly overproduced and contained a (His)6 C-terminal poly-histidine tag for rapid purification by nickel-ion affinity chromatography. The C-terminal tag is without effect on the kinetic parameters of the wild-type enzyme (18), and allows clear separation of the mutant from the much lower levels of endogenous native GlnRS produced from the chromosomal copy of the gene. Proper folding of the mutants is highly likely given the abundant overexpression and partitioning entirely into the soluble cell fraction in each case.
Steady-state kinetics
Kinetic reactions were performed using a highly sensitive assay in which the 3′-terminal internucleotide linkage of the tRNA is labelled with 32P (18, 20, 21). After the reaction, the tRNA is cleaved with either P1 or S1 nuclease, and the aminoacylated A76 nucleotide is separated from unreacted substrate by thin-layer chromatography. All reactions monitored the two-step conversion of ATP, glutamine, and tRNAGln to form Gln-tRNAGln with release of AMP and pyrophosphate. The steady-state parameters were determined with respect to glutamine, under conditions in which both ATP and tRNAGln were saturating (see Methods).
The three mutants at the Glu34 position exhibited similar kinetic behavior in the steady-state. Each of the mutant enzymes was reduced by 103-104 fold in kcat/Km, with increases of 124 to 165-fold in the Km for glutamine, and decreases of 21 to 88-fold in kcat for the two-step aminoacylation reaction (Table 1). Given the proximity of Glu34 to the active site, and the structural and chemical diversity of the introduced side-chains, the strong similarity in kinetic parameters among the mutants (particularly at the level of kcat) was unexpected. The significantly deleterious effects on the Km for glutamine, even for the conservative E34Q mutant, suggest that the chemical identity of the amino acid at position 34 is of importance to mediating active-site assembly. This is because Glu34 is located some 10-12 Å from the binding site of the substrate glutamine side-chain, at the opposite side of the active-site cleft (Figure 2).
Table I.
Kinetic analysis of wild-type and mutant GlnRS enzymes
| Wild Type† | E34A | E34Q | E34D | E73Q | E73A | |
|---|---|---|---|---|---|---|
| kcat (s-1) | 3.2 ± 0.5 | 0.14 +/- 0.06 (23)# | 0.065 +/- 0.026 (46) | 0.034 +/- 0.026 (90) | 0.004 +/- 0.001 (800) | n.d.* |
| Km (mM) | 0.26 ± 0.04 | 46.3 +/- 23 (165) | 34.9 +/- 4.26 (120) | 45 +/- 10 (160) | 22.3 +/- 6.3 (80) | n.d.* |
| kcat / Km (M-1s-1) | 1*104 | 3 (3300) | 1.8 (5600) | 0.76 (1.3*104) | 0.19 (5.2*104) | 0.004 (2.5*106) |
| kchem (s-1) | 28 ± 2 | > 0.3 (< 93) | 24 (--) | 0.96 +/- 0.26 (28) | 0.021 +/- 0.01 (1300) | 0.0028 +/- 0.0012 (1* 104) |
| Kd [Gln] (mM) | 1.1 ± 0.04 | > 200 (< 200) | 20 (20) | 45 +/- 12 (45) | 16 +/- 2.9 (16) | 70 +/- 0.03 (70) |
Values for wild-type GlnRS are taken from ref. 12.
The steady-state parameters for E73A were not measurable within the experimental timeframe because of the deacylation of the Gln-tRNAGln product that occurs after dissociation from the enzyme.
Numbers in parentheses designate the fold change from wild-type
It was also found that the E73Q and E73A mutants are more significantly affected in their steady-state behavior than are the mutants at position 34 (Table 1). E73Q is decreased by nearly 105-fold in kcat/Km, with the major effect at the level of kcat. The much stronger kcat effect for E73Q as compared with E34Q was not anticipated, given that Glu73 is more remote from the position at which the reaction chemistry occurs in the active site (Figure 2). E73A is decreased by a further 50-fold in kcat/Km as compared with E73Q; reactions of this mutant were too slow to permit estimation of the individual kcat and Km parameters at conditions of reasonable substrate excess. The E73A enzyme is decreased by 3 × 106-fold in kcat/Km as compared with wild-type GlnRS, indicating a severe disruption in catalytic efficiency from the single substitution. The Km of E73Q for glutamine is elevated by nearly 200-fold, an effect similar to that observed for the mutants at position 34. This observation further substantiates an important role for the Glu34-Glu73 interaction in mediating proper formation of the glutamine binding pocket at the opposite side of the active site cleft.
Single-turnover kinetics
The interpretation of steady-state kinetic data in structural terms is necessarily limited, because the Michaelis parameters kcat and Km cannot be directly interpreted in terms of individual rate and equilibrium constants. For wild-type GlnRS, we have previously shown that the rate of the composite two-step aminoacylation reaction (kchem) exceeds kcat by nearly 10-fold, while the glutamine Kd of about 1 mM is some five-fold higher than the steady-state Km (12). Pre-steady state measurements also revealed burst kinetics for the wild-type enzyme, demonstrating that the product release step is rate-limiting. Because kchem and Kd are microscopic parameters that pinpoint particular sites on the crystal structure (the site of the chemical transformations and that of glutamine binding, respectively), their evaluation for the mutant enzymes allows a much more definitive assessment of the effects of the amino acid substitutions. By contrast, the precise meanings of kcat and Km likely vary among the mutants depending on the rate-limiting step in each particular case, and may often represent complex combinations of a variety of microscopic constants.
The rate of the single-turnover reaction (kchem) was measured under conditions of four-fold molar excess of enzyme over tRNA, using a rapid chemical quench instrument for reactions too rapid to be sampled by hand (12, 18). Control experiments established that 20 μM tRNAGln and 30 mM ATP provided substrate saturation for both wild-type and mutant GlnRS enzymes, while 3-100 mM glutamine was required for amino acid saturation (depending on the particular mutant). Further controls established that the order of substrate premixing did not affect reaction rates. kchem was derived from single-exponential fits of production formation with time, and encompasses all reaction steps up to and including formation of Gln-tRNAGln on the enzyme. However, for wild-type GlnRS, the second-order rate constant estimated for the association of enzyme with tRNA is approximately 108 M-1s-1, at or near the diffusion control limit (18). Assuming that this association rate is not greatly altered for the mutants, and given also the very high enzyme and substrate concentrations used in the single-turnover experiments, it is then virtually certain that kchem directly reflects the rate of the chemical steps. For all mutants, we also examined the dependence of kchem on the concentration of glutamine. Hyperbolic plots of kchem vs. glutamine concentration then allow determination of the Kd for glutamine. Titrations of glutamine showed single-exponential kinetics over the full concentration range tested for each mutant, indicating that glutamine is in rapid equilibrium with the enzyme, and that the Kd so derived is equivalent to the thermodynamic binding constant (12).
The improved insight obtained from transient kinetic measurements is evident in the comparisons of kinetic parameters among the wild-type and Glu34 mutant enzymes. Thus, while kcat for the two-step aminoacylation reaction is similar among E34Q, E34D and E34A, the rate of the chemical steps varies significantly. Most striking is the observation that the nearly isosteric E34Q mutant aminoacylates tRNAGln with an efficiency that is nearly identical to that of the wild-type enzyme (Table 1; Figure 4). Hence, a negatively charged moiety at this position, adjacent to the A76 ribose sugar (Figure 3), is not important to facilitating the bond-breaking and bond-making events (see further discussion below). By contrast, the E34D mutant, which preserves a negatively charged group, is diminished by 25-fold in kchem as compared with wild-type and E34Q-GlnRS (Table 1). Thus, the withdrawal of the side-chain Glu 34 carboxylate by a spacing corresponding to a single methylene group - that is; about 1.5 Å - produces a clear functionally consequential effect. It may be reasonable to presume that the water-mediated interaction of the Glu34 carboxylate with the reactive A76 ribose is preserved in E34Q, but is destabilized or eliminated in E34D (Figure 3). If this is the case, then the 25-fold decrease in kchem observed for E34D could arise from the removal of a key determinant that contributes to precise positioning of the A76 ribose with respect to the reactive moieties of glutamine and ATP.
Figure 4.

a. kchem dependence on glutamine concentration for the E34Q GlnRS mutant, fit to a hyperbolic binding equation to determine the kchem(max) and Kd. The kinetic constant kchem is very close to that of the wild-type reaction, however, the Kd (Gln) is 20-fold higher The inset shows a TLC plate demonstrating the separation of the substrate (AMP; center of TLC plate) and product (aa-AMP; top of TLC plate). Reactions were spotted at the bottom of the plate. The control reaction before rapid mixing is in the left-hand lane.
b. Replot of the dependence of kchem on glutamine concentration for the E34A GlnRS mutant. This plot shows a linear fit to binding data indicating that saturation was not achieved even at concentrations of up to 150 mM glutamine. Hence, Table 1 indicates only a lower bound estimate for the glutamine Kd in this case.
The behavior of the E34 GlnRS mutants with respect to glutamine affinity is also of interest. We find that the binding affinities of E34Q and E34D are weakened by 20-fold and 45-fold, respectively. Thus, both mutants appear to propagate a long-range structural change, or cause a defect in induced-fit assembly of the active site, that results in the destabilization of glutamine binding at the opposite side of the cleft. The effect of E34Q on glutamine binding, but not on catalytic efficiency, suggests that there are different pathways by which mutations at this position exert their effects. One possibility is that, while E34Q may not disrupt tRNA A76 ribose binding, it does destabilize the local conformation of its active-site peptide - which may, in turn, propagate a structural change through the ATP binding site (Figures 2, 3). Glu34 is embedded in a highly unusual tetrapeptide comprising the sequence Pro32-Pro33-Glu34-Pro35, which is located at the C-terminal end of the first β-strand of the active-site Rossman fold, preceding a loop that bridges to the ATP-binding class I signature sequence His40-Ile41-Gly42-His43. Any destabilization in this portion of the structure might be transmitted in turn to the glutamine binding site, resulting in a weakening of glutamine affinity. In contrast, the strong effects of E34D and E34A on kchem suggest direct destabilization of A76 ribose binding, and possibly propagation of destabilizing structural changes to the glutamine binding site through this portion of the tRNA. The E34A substitution is so destabilizing that saturation of glutamine binding is not detectable, so that only lower limits on kchem and Kd could be estimated for this mutant.
A strong burst of product formation observed under pre-steady state conditions is characteristic of the wild-type GlnRS aminoacylation reaction (12). This indicates that the physical step comprising release of aminoacylated tRNA and AMP is the rate-limiting step in the reaction sequence. Since the binding and induced-fit transition steps are very fast, the fact that product release is rate-limiting may also be deduced from the relative magnitudes of kcat and kchem, which differ by 10-fold in the native enzyme (Table 1). While the identity of the rate-limiting step is difficult to deduce in E34A due to the lack of glutamine saturation, both E34Q and E34D resemble the wild-type enzyme with respect to the fact that kcat is much slower than kchem, indicating that product release is rate-limiting for these mutants as well. Indeed, for E34Q, kchem exceeds kcat by nearly 400-fold. Thus, under conditions of substrate saturation, the effect of the mutation in lowering kcat is manifested almost entirely at the product release step. Possibly, the removal of the Glu34 negative charge directly adjacent to the position where the negatively charged AMP is formed helps to stabilize the GlnRS:Gln-tRNAGln:AMP ternary complex from dissociating. The retention of the negative charge in E34D might account for why the relative magnitude of kchem and kcat for this mutant is very similar to that observed in wild-type GlnRS (Table 1).
A distinct role in induced-fit for Glu73
Characterization of the E73Q and E73A GlnRS mutants by both steady-state and transient kinetics revealed that alterations at this position cause more severe catalytic defects than do the mutations introduced at position Glu34 (Table 1). At the steady-state level, kcat/Km with respect to glutamine is reduced by 105-fold for E73Q (with a larger effect on kcat) and by 3 × 106-fold for E73A. The activity of E73A is so attenuated that determination of the individual Michaelis parameters was not possible. Single-turnover kinetics showed that the effects on glutamine binding due to E73Q and E73A are similar to those produced by the mutations at position Glu34 - with the exception of E34A, all the mutants at either position bind glutamine from 20 to 70-fold more weakly than wild-type GlnRS. By contrast, the distinct feature of the E73Q and E73A mutants is the sharply decreased rate of aminoacylation. E73Q and E73A are reduced in kchem by 1400-fold and 105-fold, respectively, compared to wild-type GlnRS. This represents a further weakening of 50-350 fold as compared with the already significantly weakened E34D mutation (Table 1). Comparison of kcat and kchem shows that product release remains the rate-limiting step in E73Q.
A particularly striking comparison appears between the respective kchem values determined for E34Q and E73Q. Since these two side-chains directly interact (Figures 2, 3), the mutation of either to glutamine produces essentially the same isosteric, singly negative-charged interaction between them. Despite this, the E73Q mutation results in over 103-fold loss in the rate of aminoacylation, while the E34Q enzyme is virtually unimpaired in catalytic rate as compared with native GlnRS. A rationale for this distinction may be provided by the fact that Glu73 appears to play a key role in properly orienting the hairpinned 3′ acceptor end of tRNA. When tRNA binds, the rotation of the acceptor-binding domain and concomitant rearrangement of surface loops to produce the Glu73-Glu34 contact also generates a complementary protein interface for binding of the C74-C75-A76 nucleotides. In addition to its contact to Glu34, the Glu73 carboxylate also contacts the polypeptide backbone at Asn69, helping to stabilize the internal conformation of the surface loop spanning these residues. The side-chain of Asn69, in turn, makes a direct hydrogen bond with the ribose sugar at C75. Further, the oxygen atom of the Asn69 side-chain amide also accepts hydrogen bonds from the side-chains of both Lys102 and Arg104, both of which form salt bridges with tRNA phosphates from the 3′-hairpin (Figure 2). Mutations at Glu73, even the conservative E73Q substitution, thus have potential to disrupt stable tRNA 3′-end binding into its precisely required position adjacent to the glutamine and ATP substrates.
Taken together, the structure-based mutational analysis presented here suggests that the rearrangements in the enzyme promoted by tRNA binding may have an important focal point in the Glu34-Glu73 interaction adjacent to the active site. The unusual, partially buried direct contact between these two carboxylates is apparently positioned at a key architectural junction, where it can mediate the rearrangements arising from the global rotation of the acceptor binding domain, which ultimately converge on the active site to provide stable, properly juxtaposed binding sites for the three substrates. The binding energy liberated by initial tRNA complex formation presumably liberates the free energy required to drive these changes. How the conformational rearrangements may differ upon noncognate tRNA binding is a clear outstanding question for future studies. While this work has focused on transitions that probably occur late in the induced-fit process, the key enzyme structure elements that drive discrimination against noncognate tRNAs are perhaps more likely to be located nearer to tRNA identity elements in the acceptor stem and anticodon loop. Mutational analysis coupled to rapid kinetics has revealed that anticodon loop contacts are transmitted to the active site, but the precise pathway(s) of transmission remain obscure. How such pathways converge to facilitate the active site assembly - including the important Glu73-Glu34 contact elucidated here - is an important question for future research.
Mechanism of the aminoacyl transfer step in tRNA synthetases
Two proposals exist for the mechanism of the second step in aminoacylation - the transfer of the amino acid from the adenylate intermediate to the 2′-OH or 3′-OH group of the 3′-terminal A76 nucleotide of tRNA. It was first suggested (for the E. coli GlnRS-tRNAGln complex) that the phosphate of glutaminyl adenylate serves as the general base to abstract the proton from the 2′-OH of A76, as the oxygen nucleophile attacks the carbon of the mixed anhydride intermediate (14). Given the common presence of the aminoacyl adenylate intermediate, such a substrate-assisted mechanism has the advantage of potential generality among all tRNA synthetases. Indeed, because of the very low degree of sequence similarity among the enzymes, the direct use of a substrate moiety in promoting the approach to the transition state is the only way that a common mechanism could exist. It was also suggested that such a substrate-mediated mechanism could be an evolutionary remnant of a primordial tRNA synthetase that might have been partially or entirely composed of RNA (14).
This proposal for substrate-assisted catalysis was later criticized, however, on the grounds that the pKa for the phosphate in glutaminyl adenylate is approximately 1.5 - 2.0, similar to that of a phosphodiester in DNA (15). Hence, it was thought that efficient proton abstraction by this phosphate at physiological pH would be impossible. An alternative mechanism was then suggested: the nearby buried carboxylate of Glu34 could function indirectly as the general base (15). One oxygen atom of this moiety interacts with several nearby water molecules, one of which is located within hydrogen-bonding distance of the tRNAGln A76 2′-OH group (Figure 3). The Glu34 carboxylate was thus proposed to generate hydroxide ion by deprotonating water, allowing the new hydroxide in turn to abstract the proton from A76 2′-OH.
As suggested above, the proposal that Glu34 might play the role of general base is problematic from a biological point of view, because the residue is not conserved among tRNA synthetases. Even the closely related glutamyl-tRNA synthetases (GluRS), from which a distinct GlnRS is thought to have emerged late in evolutionary time (22), do not conserve an acidic group at this position in the active-site fold. Further, the high pKa of water disfavors ionization at physiological pH in the absence of an adjacent divalent metal ion or other strongly polarizing group, which is lacking in the GlnRS active site. The properties of the E34Q mutant reported here now confirm that Glu34 does not function as the general base. As described above (Table 1), the single-turnover rate for the two-step aminoacylation reaction catalyzed by E34Q is nearly identical to that of wild-type GlnRS. This measurement of kchem shows that no mechanistic step up to and including the chemical transformation on the enzyme is significantly slowed in the mutant. The very high activity of E34Q, together with the unsuitability of the glutamine amide for proton abstraction, thus demonstrate that the proposed mechanism involving direct participation by Glu34 cannot be correct (15).
Reconsideration of the substrate-assisted mechanism suggests that the low pKa of the adenylate phosphate group need not necessarily disqualify this group from consideration as the general base. This is because the phosphate of the AMP product protonates at pH 6.5 - 7.0. Thus, as the second step of the reaction begins and the charge distribution changes in the moieties poised for catalysis, the pKa of the adenylate phosphate begins to rise. This renders it much more likely to accept the proton than previously recognized (8, 14). A direct role for the phosphate is consistent with greatly decreased rates of tRNA aminoacylation by PheRS and MetRS, when phosphorothioate ATP analogs are substituted for ATP (23, 24). More recently, mutagenesis studies in tyrosyl-tRNA synthetase (TyrRS) and in HisRS, including examination of stereospecific phosphorothioate-modified histidyl adenylate analogs in the latter enzyme, each also concluded that the substrate-assisted mechanism operates in these tRNA synthetases (17, 25). Further, these experiments suggested that the transition state for aminoacyl transfer is reached via a concerted mechanism: bond formation between the tRNA nucleophile and the carbon of the mixed anhydride occurs concomitantly with breakage of the scissile acyl phosphate bond. Such a concerted mechanism is consistent with the original substrate-assisted mechanism proposed for GlnRS (14). We are not aware of other proposals for general base catalysis of the second step in tRNA aminoacylation by particular enzyme groups, and it appears that the substrate-assisted mechanism has gained general acceptance in the field.
Figure 5.
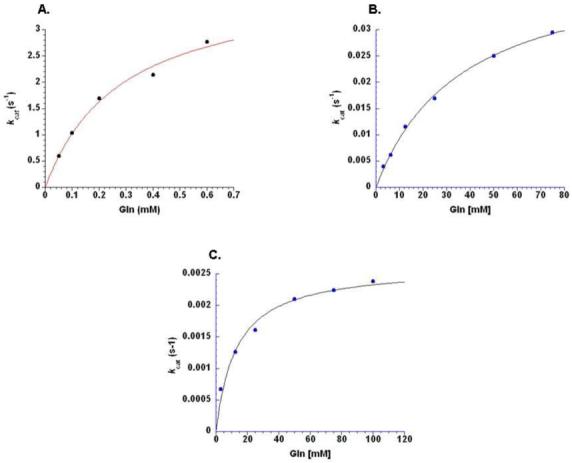
Velocity versus glutamine substrate concentration plots for steady-state aminoacylation by wild-type GlnRS (upper left), GlnRS E34Q (upper right), and GlnRS E73Q (bottom). The ordinates are normalized to the enzyme concentration used in the experiments for each mutant, and thus reflect kcat determinations at the subsaturating concentrations (see Methods). Vo versus S plots were fit to the Michaelis-Menten equation to derive the steady-state kinetic parameters summarized in Table 1.
ABBREVIATIONS
- GlnRS
glutaminyl-tRNA synthetase
- GluRS
glutamyl-tRNA synthetase
- ABD
acceptor-binding domain
- DNF
dinucleotide fold
- AspRS
aspartyl-tRNA synthetase
- HisRS
histidyl-tRNA synthetase
- IPTG
isopropyl-thio galactoside
- TLC
thin-layer chromatography
- PMSF
phenyl methyl sulfonyl fluoride
Footnotes
Supported by NIH grant GM63713 (to J.J.P.)
REFERENCES
- 1.Ibba M, Söll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequences motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 3.Francklyn C, Perona JJ, Puetz J, Hou Y-M. Aminoacyl-tRNA synthetases: versatile players in the changing theater of translation. RNA. 2002;8:1363–1372. doi: 10.1017/s1355838202021180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherlin LD, Perona JJ. tRNA-dependent active-site assembly in a class I aminoacyl-tRNA synthetase. Structure. 2003;11:591–603. doi: 10.1016/s0969-2126(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 5.Williamson JR. Induced fit in RNA-protein recognition. Nature Struct. Biol. 2000;7:834–837. doi: 10.1038/79575. [DOI] [PubMed] [Google Scholar]
- 6.Ribas de Pouplana L, Auld DS, Kim S, Schimmel P. A mechanism for reducing entropic cost of induced fit in protein-RNA recognition. Biochemistry. 1996;35:8095–8102. doi: 10.1021/bi960256a. [DOI] [PubMed] [Google Scholar]
- 7.Post CB, Ray WJ., Jr. Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry. 1995;34:15881–15885. doi: 10.1021/bi00049a001. [DOI] [PubMed] [Google Scholar]
- 8.Perona JJ. Glutaminyl-tRNA synthetases. In: Ibba M, Francklyn C, Cusack S, editors. The Aminoacyl-tRNA Synthetases. Landes Bioscience/Eurekah.com; 2004. pp. 73–88. [Google Scholar]
- 9.Ibba M, Becker HD, Stathopoulos C, Tumbula DL, Söll D. The adaptor hypothesis revisited. Trends in Biochem. Sci. 2000;25:311–316. doi: 10.1016/s0968-0004(00)01600-5. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharyya T, Roy S. A fluorescence spectroscopic study of substrate-induced conformational changes in glutaminyl-tRNA synthetase. Biochemistry. 1993;32:9268–9273. doi: 10.1021/bi00087a002. [DOI] [PubMed] [Google Scholar]
- 11.Mandal AK, Bhattacharyya A, Bhattacharyya S, Bhattacharyya T, Roy S. A cognate tRNA specific conformational change in glutaminyl-tRNA synthetase and its implication for specificity. Protein Science. 1998;7:1046–1051. doi: 10.1002/pro.5560070422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uter NT, Perona JJ. Long-range intramolecular signalling in a tRNA synthetase complex revealed by pre-steady state kinetics. Proc. Natl. Acad. Sci. USA. 2004;101:14396–14401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rould MA, Perona JJ, Söll D, Steitz TA. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNAGln and ATP at 2.8 Å resolution. Science. 1989;246:1135–1142. doi: 10.1126/science.2479982. [DOI] [PubMed] [Google Scholar]
- 14.Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by E. coli glutaminyl-tRNA synthetase. Biochemistry. 1993;32:8758–8771. doi: 10.1021/bi00085a006. [DOI] [PubMed] [Google Scholar]
- 15.Rath VL, Silvian LF, Beijer B, Sproat BS, Steitz TA. How glutaminyl-tRNA synthetase selects glutamine. Structure. 1998;6:439–449. doi: 10.1016/s0969-2126(98)00046-x. [DOI] [PubMed] [Google Scholar]
- 16.Eiler S, Dock-Bregeon AC, Moulinier L, Thierry J-C, Moras D. Synthesis of Asp-tRNAAsp in E. coli - a snapshot of the second step. EMBO J. 1999;18:6532–6541. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guth E, Connolly SH, Bovee M, Francklyn CS. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry. 2005;44:3785–3794. doi: 10.1021/bi047923h. [DOI] [PubMed] [Google Scholar]
- 18.Uter NT, Gruic-Sovulj I, Perona JJ. Amino acid-dependent transfer RNA affinity in a class I aminoacyl-tRNA synthetase. J. Biol. Chem. 2005;280:23966–23977. doi: 10.1074/jbc.M414259200. [DOI] [PubMed] [Google Scholar]
- 19.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, LaRiviere F, Uhlenbeck OC, Scaringe S. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc. Natl. Acad. Sci. USA. 2002;99:5965–5970. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bullock TL, Uter NT, Nissan TA, Perona JJ. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J. Mol. Biol. 2003;328:395–408. doi: 10.1016/s0022-2836(03)00305-x. [DOI] [PubMed] [Google Scholar]
- 22.Woese CR, Olsen GJ, Ibba M, Söll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connolly BA, Von der Haar F, Eckstein F. Structure of the metal.nucleotide complex in the yeast phenylalanyl transfer ribonucleic acid synthetase reaction as determined with diastereomeric phosphorothioate analogs of ATP. J. Biol. Chem. 1980;255:11301–11307. [PubMed] [Google Scholar]
- 24.Smith LT, Cohn M. Reactions of thio analogues of adenosine 5′-triphosphate catalyzed by methionyl-tRNA synthetase from Escherichia coli and metal dependence of stereospecificity. Biochemistry. 1982;21:1530–1534. doi: 10.1021/bi00536a010. [DOI] [PubMed] [Google Scholar]
- 25.Xin Y, Li W, First EA. Stabilization of the transition state for the transfer of tyrosine to tRNATyr by tyrosyl-tRNA synthetase. J. Mol. Biol. 2000;303:299–310. doi: 10.1006/jmbi.2000.4126. [DOI] [PubMed] [Google Scholar]