

Abstract
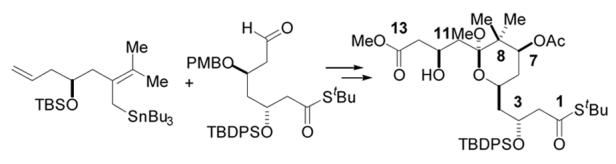
The synthesis of a C1-C13 A-ring subunit of bryostatin 1 is detailed. The key features of the approach include the convergent fragment assembly with a highly stereoselective construction of the C7-C8 bond indicated above.
Pettit and co-workers reported in 1982 the isolation and structural identification of bryostatin 1 (1) from the bryozoan Bugula neritina1. Seventeen structurally related congeners have since been isolated and identified, and the family remains of significant interest to the biological, medical, and synthetic communities.2 Bryostatin 1 exhibits an impressive array of biological properties including anticancer activity, synergistic anticancer activity with established therapeutic agents such as vincristine,3 and activity against Alzheimer's disease.4 Bryostatin 1 is known to bind to PKCα with nanomolar affinity, but this elicits different biological responses than those associated with binding by the tumor-promoting phorbol esters.5 The reasons for these differences and the mechanisms by which bryostatin 1 affects the aforementioned areas of therapeutic interest remain unclear.
In 1990, Masamune and co-workers disclosed the first total synthesis of bryostatin 7.6 More recently, both the Evans7 and Yamamura8 groups have reported bryostatin total syntheses. In addition, the promising biological profile of bryostatin 1, coupled with its scarcity from natural sources, have encouraged a number of other synthetic efforts in this area.9 Wender and co-workers have also reported on the synthesis and biological studies of several analogues of bryostatin 1.10
The synthetic strategy chosen for implementation is detailed in Figure 1. Methodology developed in these laboratories for the construction of 2,6-disubstituted-4-methylene tetrahydropyrans11, 9g, 9h was envisioned to join an A-ring hydroxy allylsilane 2 and C-ring enal 3 with concomitant formation of the B-ring. The A-ring containing the necessary pendant allylsilane could be derived from methyl ester 4 through application of the Bunnelle reaction.12 Unraveling 4 to linear synthon 5 reveals the C5 oxygenation to be 1,3-anti to both of the flanking protected hydroxyl groups, which suggested that this hydroxyl stereocenter could be exploited in the installation of both the C3 and C7 stereocenters via 1,3-asymmetric induction. Accordingly, a PMB ether was deemed to be an appropriate protecting group for this hydroxyl on the basis of its ability to participate in chelation-controlled processes,13 and its ease of removal under very specific conditions. This disconnection would, however, require addition of a nucleophile such as 6 to set the C7 stereocenter and install the gem-dimethyl moiety. Little precedent exists for such a transformation. In the context of bryostatin synthesis, with the exception of one report,14 most A-ring approaches commence with material that already contains the gem-dimethyl group. Finally, a Mukaiyama aldol reaction was envisioned for stereoselective introduction of the C3 stereocenter and the required masked carboxylic acid functionality at C1.
Figure 1.

Retrosynthetic analysis.
The synthesis of allylstannane 6 commenced with a catalytic asymmetric allylation (CAA)15 reaction on commercially available α,β-unsaturated aldehyde 9 to afford the desired homoallylic alcohol in exceptional yield and enantioselectivity (Scheme 1). This particular allylation deserves comment. Complete consumption of the aldehyde was observed after just 12 h; typically, the CAA process requires approximately 72 h to reach completion. The rapidity of this reaction is likely to be associated with the electron withdrawing unsaturated ester moiety; i.e., the substrate is a vinylogous glyoxalate. This seemingly superfluous unsaturation was deemed necessary due to previous observations made by Brown and co-workers16 in which a boron-mediated allylation into the saturated aldehyde corresponding to 9 was accompanied by significant lactonization of the product.
Scheme 1.
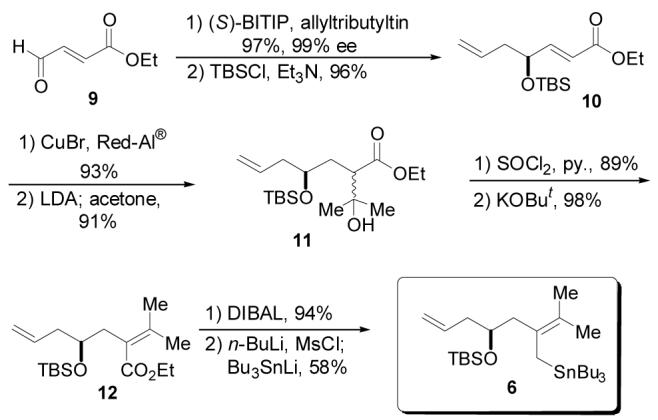
Synthesis of Tetrasubstituted Allylstannane 6
After considerable experimentation, conjugate reduction of the α,β-unsaturated ester was accomplished efficiently by application of Semmelhack's Cu(I)/Red-Al® protocol.17 Introduction of the gem-dimethyl moiety was accomplished by condensation of ester 10 with acetone to provide tertiary alcohol 11. Elimination of the hydroxyl group by treatment with SOCl2/pyridine yielded the terminal olefin which subsequently underwent base-mediated olefin migration to afford 12.18 Full reduction of the ester proceeded without difficulty to afford the corresponding alcohol, setting the stage for installation of the stannyl group. A one-pot mesylation/Bu3SnLi displacement19 ensued to provide allylstannane 6. The 37% overall yield for this 8 step sequence provided expedient access to this stannane.
Attention was next turned to the synthesis of the C1-C7 subunit. This first required the generation of Mukaiyama aldol substrate 16 (Scheme 2). A CAA reaction was relied upon once again to provide homoallylic alcohol 14 in 90% yield and 93% ee. Conversion of the alcohol to PMB ether 15 was accomplished by reaction with p-methoxybenzyl trichloroacetimidate and catalytic CSA. It is worth noting that unavoidable silyl migration was observed under the typical KH/PMBBr conditions. Additionally, the use of CSA was found to offer superior results than those obtained with other commonly employed acids such as TfOH, PPTS, and BF3·OEt2. Deprotection of the primary TBDPS ether with TBAF and subsequent Parikh-Doering oxidation gave the requisite aldehyde 16 in 92% yield over the two reactions.
Scheme 2.
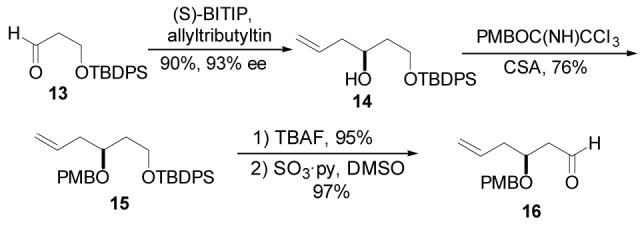
Synthesis of Aldehyde 16
A variety of Lewis acids and conditions were screened in the Mukaiyama aldol reaction (Table 1). The use of MgBr2·OEt2 afforded moderate diastereoselectivities for this transformation (entries 1 and 2). A slight improvement in aldehyde facial selectivity was observed when the monodentate Lewis acid BF3·OEt2 was employed (entry 3). While it is noteworthy that the aldehyde proved stable to exposure to TiCl3(Oi-Pr), no enhancement in selectivity resulted from the use of this Lewis acid (entry 4). A dramatic improvement in selectivity was realized when TiCl2(Oi-Pr)2 was used in place of TiCl3(Oi-Pr), but the conversion was modest as 40% of aldehyde 16 was recovered (entry 5). However, a further increase in aldehyde facial selectivity and a significant improvement in conversion was observed when the number of equivalents of the mixed titanium Lewis acid was increased (entry 6). Optimization of this reaction revealed that the use of 2.5 equivalents of the TiCl2(Oi-Pr)2 afforded a 95% yield of aldol adduct 17, as a 41:1 mixture of diastereomers, as ascertained by HPLC analysis (entry 7).20 The suspected relative stereochemical relationship between the C3 and C5 stereocenters was confirmed by application of Rychnovsky's acetonide NMR method.21
Table 1.
Mukaiyama Aldol Survey
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acid | equiv | temp (°C) | solvent | dr |
| 1 | MgBr2·OEt2 | 2.0 | −20 | CH2Cl2 | 4 : 1 |
| 2 | MgBr2·OEt2 | 2.0 | −78 to −20 | CH2Cl2 | 4.5 : 1 |
| 3 | BF3·OEt2 | 1.1 | −78 | CH2Cl2 | 5 : 1 |
| 4 | TiCl3(OiPr) | 1.0 | −78 | PhCH3 | 5 : 1 |
| 5 | TiCl2(OiPr)2 | 1.0 | −78 | PhCH3 | 32 : 1a |
| 6 | TiCl2(OiPr)2 | 2.0 | −78 | PhCH3 | 37 : 1b |
| 7 | TiCl2(OiPr)2 | 2.5 | −78 | PhCH3 | 41 : 1c |
40% of 16 recovered.
10% of 16 recovered.
95% of 17 isolated.
Preliminary coupling studies suggested that the C3 hydroxyl protecting group might remotely influence the facial bias in the stannane addition to the aldehyde. Thus, two differentially protected aldehydes were synthesized in preparation for the coupling studies (Scheme 3). Secondary alcohol 17 was protected as the TBS ether by treatment with TBSOTf and lutidine. Ozonolytic cleavage of the terminal olefin provided aldehyde 18 in an 82% yield over the two steps. The analogous C3 TBDPS protected aldehyde was synthesized by silylation with TBDPSCl followed by OsO4/NMO dihydroxylation and cleavage of the resulting diol by Pb(OAc)4.22 Aldehyde 19 was accessed in 91% yield from alcohol precursor 17.
Scheme 3.
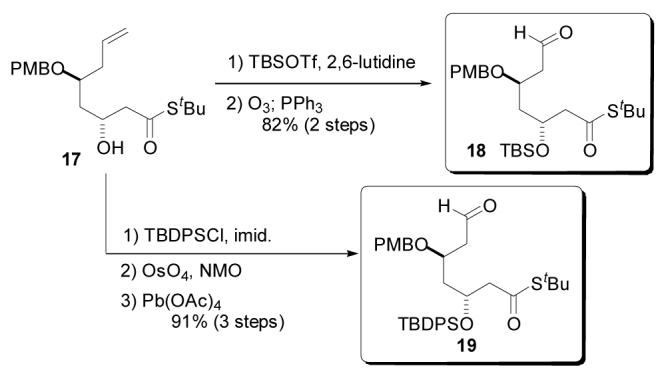
Synthesis of Two Aldehyde Coupling Partners
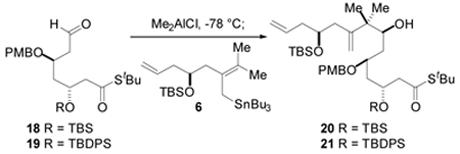
With both aldehyde and stannane coupling partners in hand, attention was directed towards the critical coupling reaction. Surprisingly, both MgBr2·OEt2 and TiCl2(Oi-Pr)2 failed to promote this addition. It was reasoned that aldehyde 18 may need increased activation in order for the addition of the relatively hindered stannane 6 to occur. Dimethylaluminum chloride appeared to be a suitable Lewis acid candidate to explore as it is recognized for its exceptional chelating ability.23
In the event, subjection of aldehyde 18 to Me2AlCl (2.5 equiv) in CH2Cl2 gave the desired addition product 20 in good yield but with modest 3.5:1 diastereoselectivity (Table 2, entry 1).24 An increase in the amount of Lewis acid used to 5.0 equivalents improved the level of stereoselectivity observed (entry 2). Reactions using the TBDPS ether containing aldehyde 19 confirmed our suspicions that the C3 protecting group might influence the stereochemical outcome; under identical conditions to those employed in entry 1, the desired adduct was obtained in comparable yield but with 7:1 diastereoselectivity (entry 3). A dramatic improvement in diastereoselectivity was realized by a change of solvent (entry 4). When the same reaction was performed in toluene a single addition product was obtained in 79% yield. As entry 5 demonstrates, when the reaction was carried out using 5.0 equiv of Me2AlCl in toluene, an 88% yield of 21 resulted under these optimized conditions. The expected stereochemistry was corroborated via NOE data obtained after closure of the A-ring (Scheme 4).
Table 2.
Optimization of the Coupling Reaction
 | |||||
|---|---|---|---|---|---|
| entry | R | equiva | solvent | yield (%)b | dr |
| 1 | TBS | 2.5 | CH2Cl2 | 83 | 3.5 : 1 |
| 2 | TBS | 5.0 | CH2Cl2 | 73 | 7 : 1 |
| 3 | TBDPS | 2.5 | CH2Cl2 | 78 | 7 : 1 |
| 4 | TBDPS | 2.5 | PhCH3 | 79 | single isomerc |
| 5 | TBDPS | 5.0 | PhCH3 | 88 |
single isomerc |
Refers to Lewis acid.
All reactions were performed with 1.3 equiv of stannane, yield based on aldehyde.
By 1H and 13C NMR analysis.
Scheme 4.
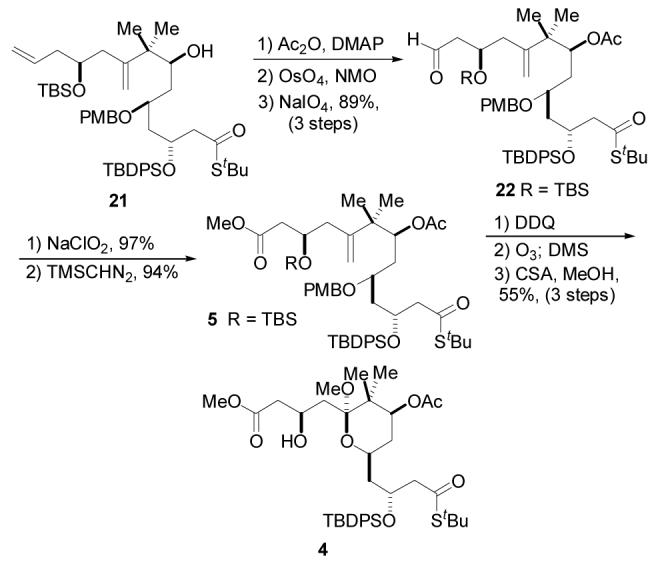
Completion of the A-ring Synthesis
The only remaining tasks were formation of the A-ring and elaboration of the terminal olefin to the methyl ester. Toward this end, acylation of the newly formed hydroxyl group was accomplished by exposure to Ac2O and DMAP. It was anticipated that oxidative cleavage could be carried out simultaneously on both the C9 and terminal olefins in order to minimize the number of chemical operations required to reach the A-ring target. Unfortunately, no conditions were found which would effect this transformation. Independent oxidative operations on the olefins were thus carried out as follows. Dihydroxylation of the terminal olefin followed by NaIO4 cleavage of the resulting diol was accomplished in good yield to provide aldehyde 22. No product resulting from the dihydroxylation of the C9 olefin was detected, undoubtedly as a consequence of the considerable steric demand imposed the proximal gem-dimethyl group. Pinnick oxidation25 of the aldehyde and methylation of the resulting carboxylic acid with (trimethylsilyl) diazomethane provided methyl ester 5 in 91% yield over the two steps. With the methyl ester in hand, oxidative cleavage of the PMB group was accomplished in 91% yield by reaction with DDQ. Finally, ozonolysis of the remaining olefin afforded a mixture of ketol and open-chain keto-alcohol. This equilibrating mixture underwent conversion to the cyclic methyl ketal with concomitant deprotection of the TBS ether under acidic methanolic conditions to give A-ring target 4 in 55% yield from alkene 5.
In conclusion, A-ring subunit 4 was accessed from aldehyde 13 in 17 linear steps in 15% overall yield. This connective fragment assembly approach provides an efficient means for introduction of both the gem-dimethyl group and the C7 stereocenter in a highly stereoselective manner. Efforts to utilize this approach in a total synthesis program are in progress.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (through GM-28961) is gratefully acknowledged. Dr. Michael R. Wiley of Eli Lilly is acknowledged for helpful discussions regarding the synthesis of allylstannane 6.
Footnotes
Supporting Information Available: Full experimenal details as well as spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pettit GR, C. L, Douobek DL, Herald DL. J. Am. Chem. Soc. 1982;104:6846. [Google Scholar]
- 2.(a) Hale KJ, Hummersome MG, Manaviazar S, Frigerio M. Nat. Prod. Rep. 2002;19:413. doi: 10.1039/b009211h. For reviews, see: [DOI] [PubMed] [Google Scholar]; (b) Mutter R, Wills M. Bioorg. Med. Chem. 2000;8:1841. doi: 10.1016/s0968-0896(00)00150-4. [DOI] [PubMed] [Google Scholar]
- 3.Al-Katib AM, Smith MR, Kamanda WS, Pettit GR, Hamdan M, Mohamed AN, Chelladurai B, Mohammad RM. Clin. Cancer Res. 1998;4:1305. [PubMed] [Google Scholar]
- 4.Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc. Natl. Acad. Sci., U S A. 2004;101:1141. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dell'Aquila ML, Herald CL, Kamano Y, Pettit GR, Blumberg PM. Cancer Res. 1988;48:3702. [PubMed] [Google Scholar]
- 6.Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J. Am. Chem. Soc. 1990;112:7407. [Google Scholar]
- 7.Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540. [Google Scholar]
- 8.Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew. Chem. Int. Ed. 2000;39:2290. [PubMed] [Google Scholar]
- 9.(a) Voight EA, Seradj H, Roethle PA, Burke SD. Org. Lett. 2004;6:4045. doi: 10.1021/ol0483044. For recent synthetic work in this area, see: [DOI] [PubMed] [Google Scholar]; (b) Voight EA, Roethle PA, Burke SD. Org. Lett. 2004;6:4534. [Google Scholar]; (c) Hale KJ, Frigerio M, Manaviazar S, Hummersome MG, Fillingham IJ, Barsukov IG, Damblon CF, Gerscher A, Roberts GCK. Org. Lett. 2003;5:499. doi: 10.1021/ol027392u. [DOI] [PubMed] [Google Scholar]; (d) Hale KJ, Frigerio M, Hummersome MG, Manaviazar S. Org. Lett. 2003;5:503. doi: 10.1021/ol027393m. [DOI] [PubMed] [Google Scholar]; (e) Ball M, Baron A, Bradshaw B, Omori H, MacCormick S, Thomas EJ. Tetrahedron Lett. 2004;45:8747. [Google Scholar]; (f) Keck GE, Yu T, McLaws MD. J. Org. Chem. 2005;70:2543. doi: 10.1021/jo048308m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Keck GE, Truong AP. Org. Lett. 2005;7:2149. doi: 10.1021/ol050511w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Keck GE, Truong AP. Org. Lett. 2005;7:2153. doi: 10.1021/ol050512o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ball M, Bradshaw BJ, Dumeunier R, Gregson TJ, MacCormick S, Omori H, Thomas EJ. Tetrahedron Lett. 2006;47:2223. [Google Scholar]
- 10.(a) Stone JC, Stang SL, Zheng Y, Dower NA, Brenner SE, Baryza JL, Wender PA. J. Med. Chem. 2004;47:6638. doi: 10.1021/jm0495069. See, for example: [DOI] [PubMed] [Google Scholar]; (b) Wender PA, Baryza JL, Brenner SE, Clarke MO, Craske ML, Horan JC, Meyer T. Curr. Drug Disc. Tech. 2004;1:1. doi: 10.2174/1570163043484888. [DOI] [PubMed] [Google Scholar]; (c) Wender PA, Mayweg AVW, VanDeusen CL. Org. Lett. 2003;5:277. doi: 10.1021/ol0272390. [DOI] [PubMed] [Google Scholar]; (d) Wender PA, Baryza J, Bennett C, Bi C, Brenner SE, Clarke M, Horan J, Kan C, Lacote E, Lippa B, Nell P, Turner T. J. Am. Chem. Soc. 2002;124:13648. doi: 10.1021/ja027509+. [DOI] [PubMed] [Google Scholar]
- 11.Keck GE, Covel JA, Schiff T, Yu T. Org. Lett. 2002;4:1189. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Bunnelle WH, Narayanan BA. Tetrahedron Lett. 1987;28:6261. [Google Scholar]; (b) Bunnelle WH, Narayanan BA. Org. Syn. 1990;8:89. [Google Scholar]
- 13.(a) Keck GE, Castellino S, Wiley MR. J. Org. Chem. 1986;51:5478. [Google Scholar]; (b) Evans DA, Duffy JL, Dart MJ. Tetrahedron Lett. 1994;35:8537. [Google Scholar]; (c) Reetz MT. Acc. Chem. Res. 1993;26:462. and references cited therein. [Google Scholar]
- 14.Yadav JS, Bandyopadhyay A, Kunwar AC. Tetrahedron Lett. 2001;42:4907. [Google Scholar]
- 15.(a) Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467. [Google Scholar]; (b) Keck GE, Krishnamurthy D. Org. Syn. 1998;75:12. [Google Scholar]
- 16.Ramachandran P, Krzeminksi MP, Reddy RV, Brown HC. Tetrahedron: Asymmetry. 1999;10:11. [Google Scholar]
- 17.Semmelhack MF, Stauffer RD, Yamashita A. J. Org. Chem. 1977;42:3180. [Google Scholar]
- 18.Hirai K, Ooi H, Esumi T, Iwabuchi Y, Hatakeyama S. Org. Lett. 2003;5:857. doi: 10.1021/ol0275264. For a similar synthetic sequence involving introduction of a 1,1-dimethyl unsaturated olefin, see: [DOI] [PubMed] [Google Scholar]
- 19.Weigand S, Brückner R. Synthesis. 1996:475. [Google Scholar]
- 20.For a related chelation-controlled Mukaiyama addition involving a PMB protected β-hydroxy aldehyde, see reference 7.
- 21.Rychnovsky SD, Rogers BN, Richardson TI. Acc. Chem. Res. 1998;31:9. [Google Scholar]
- 22.This two-step procedure for olefin cleavage was used in lieu of ozonolysis due to the fact that aldehyde 19, unlike 18, suffered from deleterious elimination of the PMB group during chromatographic purification which proved necessary following reductive workup of the ozonide. Conversely, subjection of the purified diol to Pb(OAc)4 followed by filtration and removal of benzene under reduced pressure afforded the aldehyde which could be carried onto the coupling without need for further manipulation. The origin of the difference in stability to chromatography between aldehydes 18 and 19 is unclear at present.
- 23.Evans DA, Allison BA, Yang MG, Masse CE. J. Am. Chem. Soc. 2001;123:10840. doi: 10.1021/ja011337j. [DOI] [PubMed] [Google Scholar]
- 24.Due to the disproportionation of Me2AlCl following complexation with aldehyde, a minimum of 2.5 equivalents of Lewis acid are generally employed in this transformation. It is possible that the reaction proceeds more efficiently with 5.0 equivalents due to the sequestration of Lewis acid by the thiol ester and/or electron rich PMB group. Moreover, TBS ethers have been shown to bind with Me2AlCl (TBDPS ethers do not) and therefore could be a source of chelate disruption, leading to lower diastereoselectivity than that observed with the TBDPS containing aldehydes. See ref. 23.
- 25.Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.