

Abstract
Hepatocellular carcinoma (HCC) is an aggressive cancer with a poor prognosis. The specific cellular gene alterations responsible for hepatocarcinogenesis are not well known. Cytokine-induced antiapoptotic molecule (CIAPIN1), a recently reported antiapoptotic molecule which plays an essential role in mouse definitive hematopoiesis, is considered a downstream effecter of the receptor tyrosine kinase–Ras signaling pathway. However, the exact function of this gene in tumors is not clear. In this study, we reported that CIAPIN1 is highly expressed in HCC as compared with non-tumor hepatic tissue (P < 0.05). We employed adenovirus-mediated RNA interference technique to knock down CIAPIN1 expression in HCC cells and observed its effects on HCC cell growth in vitro and in vivo. Among the four HCC and one normal human liver cell lines we analyzed, CIAPIN1 was highly expressed in HCC cells. Knock down of CIAPIN1 could inhibit HCC cell proliferation by inhibiting the cell cycle S-phase entry. Soft agar colony formation assay indicated that the colony-forming ability of SMMC-7721 cells decreased by ∼70% after adenovirus AdH1-small interfering RNA (siRNA)/CIAPIN1 infection. In vivo experiments showed that adenovirus AdH1-siRNA/CIAPIN1 inhibited the tumorigenicity of SMMC-7721 cells and significantly suppressed tumor growth when injected directly into tumors. These results suggest that knock down of CIAPIN1 by adenovirus-delivered siRNA may be a potential therapeutic strategy for treatment of HCC in which CIAPIN1 is overexpressed.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer of men and 11th one of women worldwide, especially in several areas of Asia and Africa. There were 600 000 estimated new cases annually and almost as many deaths (1). Despite advances in therapy for HCC such as recent modifications in chemotherapy and modern surgical innovations, overall patient outcome has not substantially improved. The majority of patients with HCC have inoperable disease with very poor prognosis (2–4). The 5 year survival rate is limited to 25–39% after surgery (5) and much lower elsewhere (6,7). Long-term survival is uncommon because of the frequent presence of recurrence, metastasis or the development of new primaries (8,9). Current adjuvant or palliative treatment modalities have not been conclusively shown to prolong survival in HCC (10). Although much is known about the development and causes of HCC (11,12), no effective therapy has been found for the vast majority of HCC patients. Therefore, novel approaches to treat liver cancers are needed (12). For example, potential novel gene and immunotherapeutic strategies for HCC are worthy of study (13,14).
CIAPIN1, a novel antiapoptotic molecule, which does not show any homology to known apoptosis regulatory molecules such as Bcl-2 family, caspase family or signal transduction molecules, is proven to be a mediator of the RAS signaling pathway and plays a vitally important role in fetal liver (FL) hematopoiesis (15). Microarray analysis revealed reduced expression of Bcl-xL and Jak2 in FL of CIAPIN1 null mice, suggesting that CIAPIN1 might exert its function by upregulating the expression of Bcl-xL and Jak2. In addition, mouse CIAPIN1 protects Ba/F3 cells against etoposide, γ radiation and stauroporine in vitro (15). Our previous studies demonstrated that CIAPIN1 confers multidrug resistance in gastric cancer cells possibly by upregulating multidrug resistance gene-1 and multidrug-related protein-1 expression (16). Furthermore, CIAPIN1 was found to act as an antiapoptotic molecule in vivo because CIAPIN1−/− mice die in late gestation due to defective definitive hematopoiesis in the FL; however, the researchers did not observe the liver defect pertaining to HCC in mice with these mutant strains (15). Whether both loss and/or gain of CIAPIN1 function contribute to cancer development remains unclear and warrant further research. Moreover, the prognostic value of altered CIAPIN1 expression and its use as a potential target for cancer therapy are completely unknown and the molecular basis for its effect on cancer biology is unclear.
In this study, we established that CIAPIN1 is highly expressed in HCC tissues and HCC cell lines. Then, we employed the newly developed adenovirus-delivered small interfering RNA (siRNA) technique (17) to study the effects of knock down of CIAPIN1 on HCC cell growth in vitro and in vivo.
Materials and methods
Tissue samples, cell lines and cell culture conditions
Forty-four HCC and adjacent benign liver (distant from tumor/cirrhosis 1.5 cm) samples were obtained from patients who underwent surgical treatment for HCC at the Department of Hepatobiliary Surgery in Xijing hospital, Xi'an, China. Tissue samples were obtained intraoperatively from tumors and tissues during liver resection for HCC in 46 patients at the Singapore General Hospital. All samples were obtained from patients who gave informed consent to use excess pathological specimens for research purposes only. Specimens were frozen in liquid nitrogen and stored until use. The protocols used in the study were approved by the Hospital’s Protection of Human Subjects Committee. BEL7402, SMMC-7721, HepG2, HHCC and Chang liver (transformed liver cell line) were preserved in our institute and HEK293 (Ad5 E1-transformed human embryo kidney cell line) cells were purchased from American Type Culture Collection (Manassas, VA). All the cell lines were cultured in Dulbecco’s modified Eagle's medium supplemented with 10% fetal calf serum (Sigma Chemical Co., St Louis, MO) and maintained at 37°C in a humidified chamber with 5% CO2.
Recombinant adenovirus generation
The complementary DNA sequence of CIAPIN1 was obtained from GenBank (accession no. BC024196). The potential target sequences for RNA interference (RNAi) were scanned with the siRNA Target Finder and Design Tool available at the Ambion Web site. The target sequence selected, 5′-CCAAAGTCAGCTTGTGGAATTCAAGAGATTCCACAAGCTGACTTTGGTTTTT-3′ (sense), corresponds to a region 349–369 bp after the CIAPIN1 start codon. The target sequence was subcloned into pShuttle-H1 according to the method of Shen et al. (17). The pShuttle-H1-siRNA/CIAPIN1 was recombined with backbone pAdEasy-1 in BJ5183 bacteria. Adenovirus generation, amplification and titer were done according to the simplified system described by He et al. (18). Viral particles were purified by cesium chloride density gradient centrifugation.
Adenovirus infection
Cells were incubated with adenovirus in a small volume of serum-free medium at 37°C. After adsorption for 4 h, fresh complete growth medium was added and cells were placed in the incubator for additional time as indicated for the following experiments. To acquire the equal infection efficiency in different HCC cell lines, the adenovirus 5 (the same virus type as in the virus-mediated RNAi system) packaging of green fluorescent protein (GFP) (Ad5-GFP; Stratagene, La Jolla, CA) was infected into four HCC cell lines and Chang liver and showed the same as up to 90% infection efficiency in BEL7402, HepG2 and HHCC, and Chang liver cells at a multiplicity of infection (MOI) of 20, in SMMC-7721 cells at a MOI of 30, at which concentrations no virus toxicity effect on cells was found. Thus, the following experiments were performed using viruses at such MOIs except for special indications.
Reverse transcription–PCR analysis
Total RNA was extracted using TRIZOL reagent (Invitrogen, Carsbad, CA) according to the protocol of the manufacturer. Two micrograms of RNA was subjected to reverse transcription. The polymerase chain reaction (PCR) primers used were as follows: for CIAPIN1, 5′-CGG AAT TCA TGG CAG ATT TTG GGA TCT C-3′ (forward), 5′-GGT CGA CCT AGG CAT CAA GAT TGC TAT C-3′ (reverse) and for β-actin, 5′-ATG ATA TCG CCG CGC TCG TC-3′ (forward), 5′-CGC TCG GTG AGG ATC TTC A-3′ (reverse). PCR products were separated on a 1% agarose gel, visualized and photographed under ultra violet light.
Western blot analysis
For protein detection, the cell lysates were harvested and analyzed by western blots. The antibodies used were monoclonal antibodies against CIAPIN1 prepared by our group (19), antipoly adenosine diphosphate ribose polymerase (Cell Signaling Technology), anti-cyclinD1, anti-p27, anti-Bcl-2, anti-Bax, anti-Bcl-xL, (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-β-actin (Sigma) antibodies. Densitometry analysis of protein levels was performed by using Scion-Image 4.0.2 software (Scion Corporation, Frederick, MD).
Cell viability and proliferation
Cell viability was examined by routine 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay 3 days after virus infection at the indicated MOI. A cell proliferation assay was done by counting cell number. Cells were plated at a density of 5.0 × 104 cells/ml in 24-well plates in triplicate and infected with the virus at the indicated MOIs. Cells were harvested daily and counted using a hemocytometer.
Colony formation assay and ex vivo tumor inhibition
A soft agar colony formation assay was used to assess the anchorage-independent growth ability of cells. Specifically, SMMC-7721 cells were infected with virus for 24 h at an MOI of 30. Cells were then plated on a 0.6% agarose base in six-well plates (1.0 × 104 per well) in 1 ml of Dulbecco’s modified Eagle's medium containing 10% fetal bovine serum and 0.3% agarose. Colonies >50 μm were counted 15 days after plating.
Four days after infection, 3.0 × 106 infected or mock cells were injected subcutaneously into male athymic nude mice (4 weeks of age, five mice per group; BiKai Co., Shanghai, China). The tumors were monitored with a caliper every week over a 6 week period after initial cells were injected. Then, the tumor-bearing mice were killed and the tumors were removed and weighed.
Cell cycle analysis
Standard fluorescence-activated cell sorter analysis was used to determine the distribution of cells in cell cycle or apoptosis of the cells. Briefly, the cells were infected with AdSiCIAPIN1 or AdSiGFP virus for 2–4 days. Adherent cells then were collected by trypsinization and fixed with 70% ethanol overnight at 4°C. After washing with phosphate-buffered saline (PBS), the cells were treated with 100 μg/ml RNase A (Roche Diagnostics), 50 μg/ml of propidium iodide (Sigma) and 0.05% (vol/vol) Triton X-100 and incubated for 45 min at room temperature. The samples were analyzed using a FACScalibur flow cytometer (Becton Dickinson, San Jose, CA). The cell cycle distribution was established by plotting the intensity of the propidium iodide signal, which reflects the cellular DNA content. Apoptotic cells were identified as a hypodiploid DNA peak representing cells (sub-G1). Findings from at least 20 000 cells were collected and analyzed with CellQuest software (Becton Dickinson).
4′,6-Diamidino-2-phenylindole staining
After infection for 4 days, virus-treated and untreated (mock) cells were fixed with 4% paraformaldehyde (wt/vol) in PBS, pH 7.4, at room temperature for 10 min. Then the cells were stained with 4′,6-diamidino-2-phenylindole (Sigma) at 1 μg/ml in PBS for 2 min, washed with PBS and mounted using PBS:glycerol (3:1, vol/vol). Fluorescence was visualized with an Olympus standard fluorescence microscope (Olympus, Tokyo, Japan). Normal nuclei were identified as non-condensed chromatin dispersed over the entire nucleus. Apoptotic nuclei were identified by condensed chromatin, contiguous to the nuclear membrane, and nuclear fragmentation of condensed chromatin.
HCC xenograft tumor model in nude mice
The SMMC-7721 cells were grown to subconfluency in complete medium. Viable cells (2.0 × 106 in PBS/100 μl) were injected subcutaneously into the flanks of 4-week-old nude mice. Two weeks after tumor cell inoculation, mice were divided randomly into three groups (six mice per group) and were treated every other day for 10 days by way of multiple-center intratumor injection of 100 μl of AdSiCIAPIN1 or AdSiGFP at 2.0 × 108 plaque-forming units per animal or PBS as a control. The tumors were monitored with a caliper every week over an 8 week period after cell inoculation. Tumor volume for each mouse was determined (in cubic millimeter) by measuring in two directions and calculated as tumor volume = length × (width)2/2. One week after the last virus injection, one of the mice in each group was killed and the xenografted tumors were excised and prepared with a routine pathological procedure. Tumor sections were deparaffinized and subjected to immunohistochemistry analysis as described in the Adenovirus infection and Immunohistochemistry.
Immunohistochemistry
Tissue samples were fixed in 10% buffered formalin solution and embedded in paraffin. For CIAPIN1 immunostaining, mouse anti-CIAPIN1 monoclonal antibody (16) was used at 1:3000 dilution. The tumor sections were visualized using a streptavidin peroxidase kit (Beijing Zhongshan Company, Beijing, China). In situ apoptosis assay was performed with the DeadEnd colorimetric terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling system (Promega, Madison, WI). Apoptotic nuclei were stained dark brown.
Statistical analysis
Each experiment was repeated at least three times. Bands from western blot or reverse transcription–PCR were quantified by Quantity One software (Bio-Rad). Relative protein or messenger RNA (mRNA) levels were calculated by referring them to the amount of β-actin. The difference between means was performed with analysis of variance and then a post hoc test. All statistical analyses were performed using SPSS11.0 software (Chicago, IL). P < 0.05 was considered as statistically significant.
Results
CIAPIN1 expression is elevated in majority HCC patient samples and HCC cell lines
The pathologic data are summarized as follows: concomitant liver cirrhosis occurred in 17 (39%) of 44 patients and dysplasia within the cirrhotic liver was diagnosed in 9 (21%) of 44 patients. Sixteen of 44 (36%) HCC samples had a single tumor and 28 of 44 (64%) had multiple HCC nodules. Satellite formation occurred in 23 (52%) of 44 patients. No patients claimed that they have exposed to benzene hexachloro- HCB or afratoxin. Twenty-four of 44 (55%) patients have infected the hepatitis B virus and/or hepatitis C virus virus before. The CIAPIN1 expression in the HCC samples with the hepatitis B virus and/or hepatitis C virus infection was similar to that in the uninfected ones (33 versus 25%, P > 0.05), and we did not find any relationship between the hepatitis B virus and/or hepatitis C virus infection with the CIAPIN1 expression (P > 0.05).
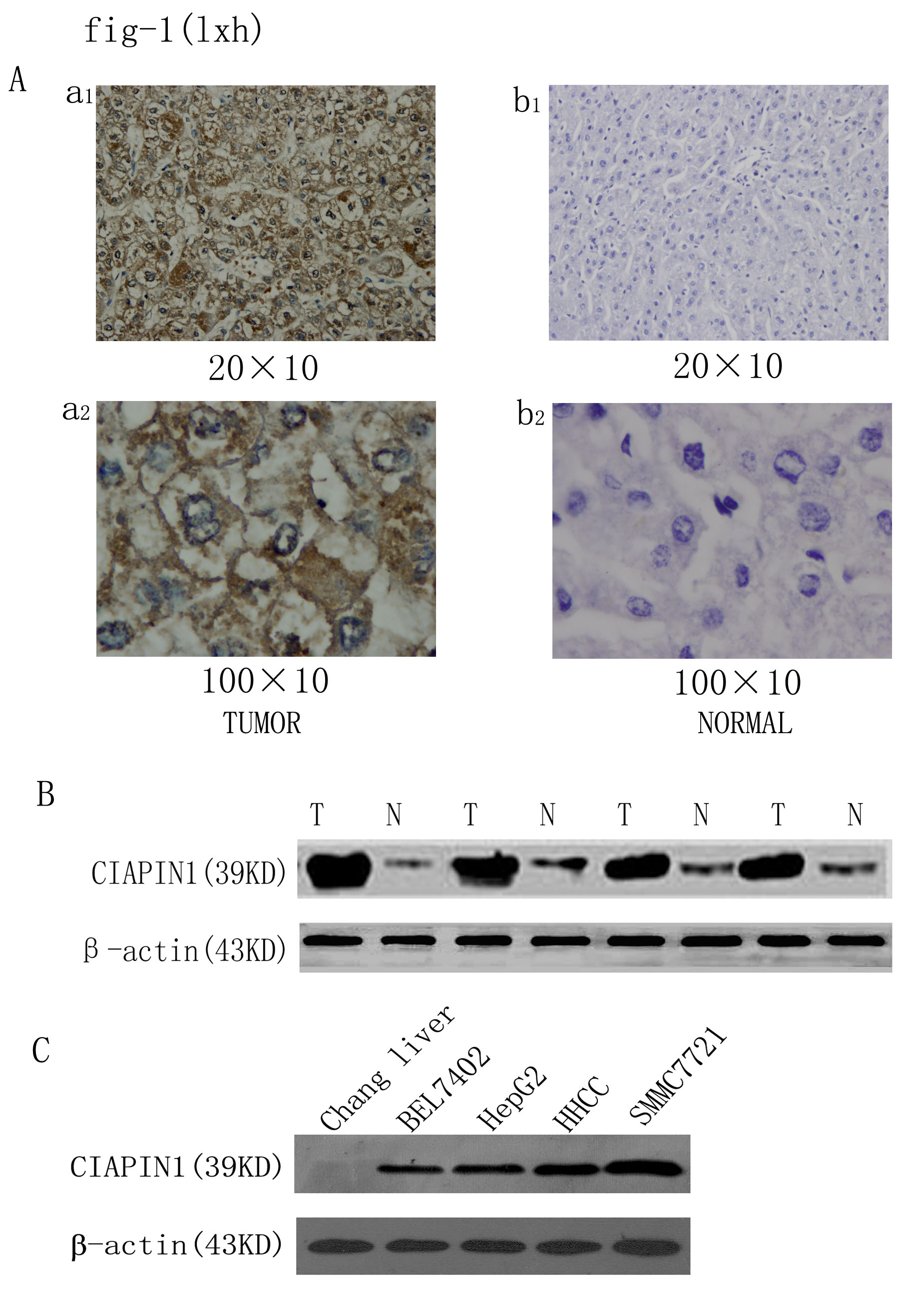
Here, we investigated the expression of CIAPIN1 in 44 samples of patients with HCC in China. CIAPIN1 expression was classified as negative, weak and strong in 13 (29.6%), 10 (22.7%) and 21 (47.7%) patients, respectively (Table I). The CIAPIN1 expression in HCC tumors was significantly higher than that in the surrounding non-tumor liver tissues (P < 0.05, Table I) and the representative pictures were presented in Figure 1A. To further confirm these observations, western blot assay was done using four paired human normal liver tissues and their tumor tissue specimens with known levels of CIAPIN1 expression by immunohistochemical staining (Figure 1B). It was clear that the tumor tissue specimens had a drastic increase of CIAPIN1 expression as compared with the normal liver tissue, which was consistent with the level of CIAPIN1 protein expression determined by immunohistochemical staining. Tumorigenesis is associated with multiple factors and CIAPIN1 overexpression may be an important factor in HCC; thus, we hypothesize that knock down of CIAPIN1 expression may inhibit the growth of HCC in which CIAPIN1 is overexpressed.
Table I.
Summary of immunohistochemical analysis of CIAPIN1 in adjacent benign liver and HCCs
| Immunostaining | Adjacent benign liver | HCCs (n = 44) |
|||
| Stage I (n = 6) | Stage II (n = 18) | Stage III (n = 9) | Stage IV (n = 11) | ||
| Negative | 26/44 (59.1%) | 3/6 (50%) | 6/18 (30%) | 2/9 (22%) | 2/11 (18%) |
| Weak | 12/44 (27.3%) | 1/6 (17%) | 4/18 (22%) | 2/9 (22%) | 3/11 (27%) |
| Strong | 6/44 (13.6%) | 2/6 (33%) | 8/18 (45%) | 5/9 (56%) | 6/11 (55%) |
Fig. 1.

Immunohistochemical analysis of CIAPIN1 protein expression in non-tumor liver samples and HCC tissues (44 cases). (A) Representative photographs were taken at different magnifications (×200 versus ×1000 for inserts). (a1) HCC tissues ×200 magnification, (a2) HCC tissues ×1000 magnification; (b1) non-tumor liver samples ×200 magnification, (b2) non-tumor liver samples ×1000 magnification. (B) Western blot analysis of whole-cell protein extracts prepared from five paired non-tumor liver (N) and HCC (T) specimens. The level of CIAPIN1 protein expression was significantly higher in tumor tissue than in normal tissue. (C) CIAPIN1 protein expression is drastically increased in HCC cells compared with the normal human liver cells, Chang liver, determined by western blot analysis. β-actin expression levels were used as internal controls.
CIAPIN1 expression in four HCC cell lines was examined to assess the biological activities by western blot analysis. Chang liver was used as references for CIAPIN1 expression. Our data indicate that compared with the Chang liver cells, the expression of CIAPIN1 was significantly increased in all four HCC cell lines (Figure 1C).
Construction of recombinant adenovirus Ad-CIAPIN1 and its effects on CIAPIN1 expression
We examined the possibility that siRNA expressed from an adenoviral vector can be used to specifically inhibit target gene expression in HCC cells. The adenoviruses packaging of hairpin siRNA targeted against CIAPIN1 or GFP were used to infect four HCC cell lines. As shown in Figure 2A, the suppression of CIAPIN1 expression was detected as early as 48 h after infection, and the suppression was up to 80% in the cells infected with AdSiCIAPIN1 at 96 h. The inhibitory effect of the AdSiCIAPIN1 was shown to be specific because a control AdSiGFP had no effect on CIAPIN1 expression level. In addition, AdSiCIAPIN1 did not cause a non-specific downregulation of gene expression, as shown by the β-actin control.
Fig. 2.

Adenovirus packaging of vectors capable of producing short hairpin RNA-targeting CIAPIN1 was constructed and proved to be specific and potent in silencing CIAPIN1 expression in four HCC cell lines. (A) Indicated cells were infected with AdSiGFP (control) or AdSiCIAPIN1. Cell lysates were harvested at 48 or 96 h after infection and detected by anti-CIAPIN1 and anti-β-actin antibodies. (B) Real-time reverse transcription–PCR of CIAPIN1 mRNA from SMMC-7721 cells harvested 2 days after infection at the indicated MOIs showed a dose-dependent decrease in mRNA levels. At an MOI of 30, the AdSiCIAPIN1 suppresses the expression of CIAPIN1 mRNA by up to 80% compared with the mock group.
To examine the reduction level of target mRNA induced by the AdSiRNA, a real-time reverse transcription–PCR was performed with the prepared mRNA from the infected SMMC-7721 cells. As shown in Figure 2B, AdSiCIAPIN1 caused a dramatic reduction in the level of CIAPIN1 mRNA whereas the mock or AdSiGFP could not. In addition, we found a progressive decrease in the mRNA level of CIAPIN1 in SMMC-7721 cells infected with increasing levels of the AdSiCIAPIN1 virus (increasing MOI). Thus, the AdSiRNA expression was correlated with a specific reduction in the level of target mRNA. Altogether, these data indicated that the AdSiRNA could suppress effectively the CIAPIN1 overexpression in HCC cells.
Inhibition of cell growth and S-phase entry in HCC cells by depletion of CIAPIN1
We first tested the effect of AdSiCIAPIN1 on the cell viability of BEL7402, SMMC-7721, HepG2 and HHCC in cell culture. Chang liver infected with adenovirus at an MOI of 20 was taken as a negative control. As shown in Figure 3A, after virus infection, AdSiCIAPIN1 significantly inhibited cell growth of all four HCC cell lines as compared with AdSiGFP or mock treatment. The difference was more pronounced after day 3, a time at which essentially all the cells established cell–cell contacts. In contrast, we did not observe any effect of AdSiCIAPIN1 on the cell viability of Chang liver. The inhibitory effect on SMMC-7721 cell growth also was confirmed by colony formation assay (Figure 3B), suggesting that autonomous growth potential also is decreased. The number of colonies was not increased when measurements were made at a later time, indicating that this is not solely a reflection of the reduced growth rate. Additionally, the suppression effect of AdSiCIAPIN1 on HCC growth was confirmed in ex vivo assay. We compared the growth of SMMC-7721 cells with different treatment as tumors in the subcutaneous tissue of male nude mice. As shown in Figure 3C and D, AdSiCIAPIN1 significantly suppressed tumor growth in mice as compared with control. To determine whether the cell cycle distribution has changed after depletion of CIAPIN1 in HCC cells, we performed flow cytometry at 96 h after virus infection. Our results showed a decreased cell population in the S phase after AdSiCIAPIN1 infection in all four cell lines compared with control, as shown in Figure 3E. Taken together, these data indicate that the AdSiRNA-targeting CIAPIN1 exhibited a specific inhibitory effect on the HCC cell growth as well as inhibition of S-phase entry in HCC cells.
Fig. 3.

Adenovirus-delivered CIAPIN1 siRNA inhibits HCC cell growth, tumorigenesis and S-phase entry. (A) The growth of HCC cells and normal Chang liver infected with adenovirus was measured by 3-(4,5-dimethylthiazole-2-yl)-2,5-biphenyl tetrazolium bromide assay. Values were given at the indicated time points as the mean absorbance with a standard deviation of four wells. (B) Colony formation assay was performed by using SMMC-7721 cells infected with adenovirus for 96 h. The cells without any infection (mock) were used as control. The data were the averages from three independent triplicate experiments. *P < 0.05 relative to the mock group. (C) Ex vivo assay for the tumor suppression effect of AdSiCIAPIN1. SMMC-7721 cells were infected with adenoviruses or untreated cells (mock) in culture plates for 96 h. Then viable cells (3.0 × 106) were injected subcutaneously into the dorsal flanks of five mice in each group. Photographs of a representative mouse in each group are shown. (D) Quantitation of tumor volume shown in (C). Tumor formation was scored weekly. Data were expressed as means ± standard deviation (n = 5). (E) AdSiCIAPIN1 decreased S-phase population in HCC cells after infection for 96 h. The S-phase population in the indicated cells was determined by flow cytometry and experiments were repeated three times. Error bars: standard deviation. (F) Cell cycle-related molecules were determined by western blot analysis. In the western blot analysis, depletion of CIAPIN1 expression could downregulate cyclinD1 but upregulate p27 expression in SMMC-7721-infected cells.
In order to explore the underlying molecular mechanism of CIAPIN1 affecting the cell cycle progression, we detected the expression of cell cycle-related molecules in AdSiCIAPIN1-infected SMMC-7721 cells, such as cyclinD1, p27, cdk2, cdk4 and p21 by western blot and found cdk2, cdk4 and p21 were not affected (data not show), except CyclyinD1 was downregulated and P27 was upregulated (Figure3F). Therefore, we may infer that G1 to S arrest induced by knocking down CIAPIN1 in HCC cells was at least partly mediated by downregulation of cyclinD1 and upregulation p27 expression.
Induction of apoptosis in HCC cells by adenovirus-delivered CIAPIN1 siRNA
Two days after infection of AdSiCIAPIN1, we observed the appearance of some HCC cells being shrunk, rounded up and detached from plates, suggesting apoptosis had occurred. In contrast, in the AdSiGFP-treated or mock group the cells remained attached to the dishes and showed normal morphology. To determine whether the depletion of CIAPIN1 promotes tumor cell death, flow cytometry was performed after infection of adenovirus into four HCC cells. The cells were analyzed at different time points (48 and 96 h) after infection and significant sub-G1 (apoptotic) populations were observed at 96 h (Figure 4A). We found that variable apoptotic populations existed in different HCC cells, 11.6% of BEL7402 and 21.3% of HHCC cells underwent apoptosis in the AdSiCIAPIN1-treated group versus 1.7 and 3.3% in the AdSiGFP-treated group, respectively. Even higher apoptotic populations were found when SMMC-7721 and HepG2 cells were tested (58.0 and 47.5%, respectively). We confirmed the apoptosis of HCC cells by 4′,6-diamidino-2-phenylindole staining (Figure 4B1). To test if apoptosis can be triggered in normal cells that do not overexpress CIAPIN1, we also infected normal Chang liver with AdSiRNA. In contrast to HCC cells, we did not observe significant apoptosis of Chang liver at 48 or 96 h after infection (Figure 4B1). Better quantitation of apoptosis has been shown in Figure 4B2, which was similar to the flow cytometer results shown in Figure 4A. These data together suggested that depletion of CIAPIN1 specifically triggered apoptosis of CIAPIN1-overexpressing HCC cells.
Fig. 4.

AdSiRNA-targeting CIAPIN1 induces cell apoptosis. (A) Downregulation of CIAPIN1 promoted apoptosis of the four indicated HCC cell lines. Ninety-six hours after the virus infection, the adherent cells were collected by trypsinization and determined by flow cytometry. Three individual experiments were performed, and the cell distribution in the cell cycle was determined by standard fluorescence-activated cell sorter analysis. The cell population in sub-G1 was shown. The x-axis and y-axis represent DNA content and the cell number, respectively. (B1) 4′,6-Diamidino-2-phenylindole staining showed the apoptotic cells in HCC cells infected with AdSiCIAPIN1 for 96 h, whereas this apoptosis cannot be seen in normal Chang liver being infected. The arrows indicate the blue color in apoptotic nuclei with condensed chromatin or nuclear membrane contiguousness morphology. (B2) Apoptosis cell numbers were counted in 100 cells under fluorescent microscopy, and results were representative of three independent experiments. (C) Western blot analysis of Bcl-2 family mediators of apoptosis showed that depletion of CIAPIN1 could downregulate Bcl-2 and Bcl-XL and upregulate Bax in AdSiCIAPIN1-transduced cells, suggesting that the apoptotic effect of CIAPIN1 could be partly mediated by the Bcl-2 family.
Furthermore, analysis of Bcl-2 family mediators of apoptosis, such as Bcl-2, Bax and Bcl-XL, showed that depletion of CIAPIN1 could downregulate Bcl-2, Bcl-XL and upregulate Bax in AdSiCIAPIN1-transduced cells, suggesting that the apoptotic effect of CIAPIN1 could be partly mediated by the Bcl-2 family (Figure 4D).
Suppression of established tumor growth in nude mice by use of AdSiCIAPIN1
To determine whether the adenovirus-delivered CIAPIN1 siRNA could serve as a therapeutic agent against HCC tumor formation, we established a tumor model in nude mice bearing SMMC-7721 xenografts. Two weeks after inoculation of SMMC-7721, AdSiCIAPIN1, AdSiGFP or PBS was administered locally by way of multiple-center intratumor injection. This kind of treatment causes infection of >90% of tumor cells, which was confirmed by the injection of Ad5-GFP as a control. After treatment with AdSiCIAPIN1, the tumor growth obviously was inhibited compared with the AdSiGFP or PBS animal group (Figure 5A). To investigate the mechanism of AdSiCIAPIN1-induced inhibition of tumor growth, tumor specimens were obtained from tumor-bearing animals after death. The specimens were subjected to the in situ apoptosis terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay. As shown in Figure 5B, treatment with AdSiCIAPIN1 dramatically induced apoptotic cell death inside the tumors, whereas treatment with AdSiGFP or PBS did not cause any tumor cell apoptosis. Taken together, these data indicate that targeting CIAPIN1 by AdSiRNA can exert a strong antitumor effect in vivo on HCC and that AdSiCIAPIN1 inhibits tumor growth by inducing apoptosis of CIAPIN1-overexpressing tumor cells.
Fig. 5.

Local injection of adenovirus-delivered CIAPIN1 siRNA suppresses the established tumor growth in nude mice. (A) Two weeks after subcutaneous inoculation of SMMC-7721 cells, three groups of nude mice were treated every other day for 10 days by intratumor injection of AdSiCIAPIN1 or AdSiGFP at 2.0 × 108 plaque-forming units per animal or mock as a control (arrow). Representative photographs of a mouse in each group 6 weeks later are shown. (B) Quantitation of tumor volume shown in (A). The tumor size was measured and the tumor volume was calculated weekly. Data are presented as mean ± standard deviation (n = 6). Error bars: standard deviation. (C) In situ terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling apoptosis analysis of tumor sections derived from mice receiving different treatments. The apoptotic nuclei were stained dark brown (original magnification ×200).
Discussion
HCC is the fourth most common cause of death from cancer and China alone accounts for 53% of all liver cancer deaths worldwide (20). However, even among patients detected earlier, there are very few candidates for surgery because they generally lack a hepatic reserve as a result of the coexisting advanced cirrhosis (21). Moreover, clinical observations have shown that tumor recurrence rates are very high in patients with HCC who receive medical or surgical treatments. Thus, new treatment modalities must be pursued. With the expectation of increasing therapeutic efficacy, gene therapy is being investigated as an effective treatment modality (22,23). Cancer gene therapies can be broadly classified into two groups: (i) those that modify the host responses to a tumor and (ii) those that induce direct antitumor action by introducing genetic material that directly affects the cancer cell and halts its growth (24). Replacement therapies involving tumor suppressor genes are the prototypes of gene therapies with direct antitumor action. CIAPIN1 is a recently reported antiapoptosis molecule, but its biological function is far from being fully elucidated.
In this study, we detected that CIAPIN1 expression is elevated in majority HCC patient samples and HCC cell lines, and consequently, we hypothesized that overexpression of CIAPIN1 may be associated with hepatocarcinogenesis and knock down of CIAPIN1 may inhibit tumorigenic growth of HCC in which CIAPIN1 is overexpressed.
The human adenovirus serotype 5 was used in our system. This is a replication-defective adenovirus with deletion of E1 and E3 genes, which render this virus incapable of replication itself (18). Moreover, this system makes use of the efficient homologous recombination in HEK293 cells, to produce recombinant adenovirus by a double recombination event between cotransfected adenoviral backbone plasmid pacAd5 and a shuttle plasmid. Our studies here were designed to test whether the more advantageous AdSiRNA can be applied to target HCC cells selectively with overexpression of a recently reported antiapoptosis gene, CIAPIN1, which is overexpressed in a higher percentage of HCC patients.
Our results show that AdSiRNA can downregulate CIAPIN1 expression effectively by up to 80% in HCC cells with great specificity, indicating the potency of adenovirus-based RNAi as an effective strategy for cancer therapy. Also, the blockage of proliferation and induction of apoptosis in HCC cells and the tumor suppression effect in nude mice additionally support the effectiveness of this treatment. There is no induced apoptosis observed in normal Chang liver, which is absent from CIAPIN1 expression, and this specificity should increase the therapeutic index of RNAi-based therapies.
After cells were treated with AdSiCIAPIN1 for 96 h, we observed a significant decrease of S-phase population in all tested HCC cells. This blockage of S-phase entry combined with the enhanced cyclinD1 and decreased P27 expression in SMMC-7721 cells conversely substantiated the effect of CIAPIN1 overexpression on HCC, by regulating cyclinD1 and P27 to activate expression of genes necessary for entering into the S phase. Interestingly, in addition to the inhibition of cell growth, depletion of CIAPIN1 for 96 h triggered apoptosis in all tested HCC cells. Previous microarray analysis revealed that the expressions of Bcl-xL and Jak2 were downregulated in FL of CIAPIN1-null mice (15). In this study, we tested the expression level of three Bcl-2 family members (Bcl-2, Bcl-xL and Bax) in this CIAPIN1 depletion-triggered apoptosis in HCC cells. We found that depletion of CIAPIN1 could downregulate Bcl-2 and Bcl-XL and upregulate Bax in AdSiCIAPIN1-transduced tumor cells, suggesting that Bcl-2 family members may contribute partially to the apoptosis of HCC cells with CIAPIN1 depletion. Because the precise molecular mechanism for the CIAPIN1 apoptosis signaling pathway is still unclear, there might be other different mechanisms to trigger apoptosis in different cell lines depending on the unique genetic context in the individual lines. In order to explore these differences with the known molecular differences among the four cell lines. We further explored the role of p53 in AdSiCIAPIN1-mediated apoptosis. HepG2 and SMMC-7721 cells that contained the wild-type p53 gene were more sensitive to AdSiCIAPIN1-induced apoptosis than HHCC cells lacking the wild-type p53 gene and BEL7402 cells containing a mutant p53 gene. The expression of p53 protein was greatly enhanced in AdSiCIAPIN1-infected HepG2 and SMMC-7721 cells (supplementary Figure A is available at Carcinogenesis Online). In contrast, no p53 protein could be detected in HHCC cells, and no obvious change was detected in BEL7402 cells.
To explore whether AdSiCIAPIN1-induced cell death in HepG2 was p53 dependent, we generated a HepG2 cell line stably expressing antisense p53. The reduced p53 expression was confirmed by western blotting and real-time PCR analysis (supplementary Figure B is available at Carcinogenesis Online). AdSiCIAPIN1-induced cell death was significantly more attenuated in PsiP53-expressing cells than in mock or Psilencer3.1-transfected cells (supplementary Figure C is available at Carcinogenesis Online). However, AdSiCIAPIN1-induced cell death was not completely abolished. This confirmed our hypothesis that AdSiCIAPIN1 might induce apoptosis through different mechanisms in different genetic context cell lines.
In addition to showing the successful use of AdSiRNA in silencing CIAPIN1 overexpressed in HCC cells, our results also indicate that CIAPIN1 might serve as a novel promising therapeutic target for HCCs. CIAPIN1, a newly reported antiapoptosis molecule, is proven to be a mediator of the RAS signaling pathway and plays a vitally important role in FL hematopoiesis (15). Previous study indicated that CIAPIN1 does not show any homology to known apoptosis regulatory molecules such as Bcl-2 family, caspase family or signal transduction molecules. Here, we showed that when overexpressed CIAPIN1 was depleted in HCC, it resulted in multiple antitumor effects in cancer cells such as inhibition of cell growth and induction of apoptosis. These results suggest that CIAPIN1 overexpression may be essential for maintaining cell proliferation as well as cell survival in HCC. More importantly, we showed the local injection of AdSiRNA for CIAPIN1 into established tumors in nude mice significantly could inhibit the tumor growth in vivo by inducing apoptosis of HCC cells. This HCC-targeted therapy is realized by two means. One is the adoption of local injection delivery of AdSiRNA into the tumor mass by way of multiple-center injections. The other is to take advantage of the fact that normal liver cells express very little CIAPIN1. We show that silencing antiapoptosis molecule CIAPIN1 by AdSiRNA is a potential effective approach to treat HCCs. In conclusion, our results show that the significant downregulation of CIAPIN1 expression in HCC cells inhibits cell growth in vitro and in vivo and induces the cancer cell apoptosis. Therefore, our studies suggest that knock down of CIAPIN1 by adenovirus-delivered siRNA may be a potential therapeutic strategy in the treatment of HCC in which CIAPIN1 is overexpressed.
Supplementary material
Supplementary Figures A, B and C can be found at http://carcin.oxfordjournals.org/
Funding
The National Foundation of Natural Sciences, China (30471989).
Supplementary Material
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- FL
fetal liver
- GFP
green fluorescent protein
- HCC
hepatocellular carcinoma
- mRNA
messenger RNA
- MOI
multiplicity of infection
- PCR
polymerase chain reaction
- PBS
phosphate-buffered saline
- RNAi
RNA interference
- siRNA
small interfering RNA
References
- 1.Parkin D, et al. Estimates of the world-wide incidence of 18 major cancers in 1985. Int. J. Cancer. 1993;54:594–606. doi: 10.1002/ijc.2910540413. [DOI] [PubMed] [Google Scholar]
- 2.Levin B, et al. Therapy of unresectable hepatocellular carcinoma. N Engl. J. Med. 1995;332:1294–1296. doi: 10.1056/NEJM199505113321910. [DOI] [PubMed] [Google Scholar]
- 3.Okuda K. Hepatocellular carcinoma. J. Hepatol. 2000;32(suppl.):225–237. doi: 10.1016/s0168-8278(00)80428-6. [DOI] [PubMed] [Google Scholar]
- 4.Lou WY. Primary liver tumors. Semin. Surg. Oncol. 2000;19:135–144. doi: 10.1002/1098-2388(200009)19:2<135::aid-ssu6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 5.Colombo M. Hepatocellular carcinoma. J. Hepatol. 1992;15:225–236. doi: 10.1016/0168-8278(92)90041-m. [DOI] [PubMed] [Google Scholar]
- 6.Lai EC, et al. Hepatic resection for hepatocellular carcinoma. An audit of 343 patients. Ann. Surg. 1995;221:291–298. doi: 10.1097/00000658-199503000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takenaka K, et al. Results of 280 liver resections for hepatocellular carcinoma. Arch. Surg. 1996;131:71–6. doi: 10.1001/archsurg.1996.01430130073014. [DOI] [PubMed] [Google Scholar]
- 8.Huguet C, et al. Primary hepatocellular cancer: Western experience. In: Blumgart L, editor. Surgery of the Liver and Biliary Tract. London: Churchill Livingstone; 2000. pp. 1365–1369. [Google Scholar]
- 9.Lai E, et al. Hepatocellular carcinoma: the Asian experience. In: Blumgart L, editor. Surgery of the Liver and the Biliary Tract. London: Churchill Livingstone; 1994. pp. 1349–1363. [Google Scholar]
- 10.Chan ES, et al. Neoadjuvant and adjuvant therapy for operable hepatocellular carcinoma. Cochrane Database Syst. Rev. 2000 doi: 10.1002/14651858.CD001199. CD001199. Cochrane Database Syst Rev. (2):CD001199. Review. [DOI] [PubMed] [Google Scholar]
- 11.Thorgeirsson SS, et al. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 12.Bruix J, et al. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology. 2002;35:519–524. doi: 10.1053/jhep.2002.32089. [DOI] [PubMed] [Google Scholar]
- 13.Qian C, et al. The potential of gene therapy in the treatment of hepatocellular carcinoma. J. Hepatol. 2000;32:344–351. doi: 10.1016/s0168-8278(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz J, et al. Gene therapy of viral hepatitis and hepatocellular carcinoma. J. Viral Hepat. 1999;6:17–34. doi: 10.1046/j.1365-2893.1999.6120136.x. [DOI] [PubMed] [Google Scholar]
- 15.Hirohiko Shibayama, et al. Identification of a cytokine-induced antiapoptotic molecule anamorsin essential for definitive hematopoiesis. J. Exp. Med. 2004;199:581–92. doi: 10.1084/jem.20031858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao Z, et al. CIAPIN1 confers multidrug resistance by upregulating the expression of MDR-1 and MRP-1 in gastric cancer cells. Cancer Biol. Ther. 2006;5:261–266. doi: 10.4161/cbt.5.3.2381. [DOI] [PubMed] [Google Scholar]
- 17.Shen C, et al. Gene silencing by adenovirus-delivered siRNA. FEBS Lett. 2003;539:111–114. doi: 10.1016/s0014-5793(03)00209-6. [DOI] [PubMed] [Google Scholar]
- 18.He TC, et al. A simplified system for generating recombinant adenoviruses. Proc. Natl Acad. Sci. USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hao Z, et al. Preparation and characterization of a specific monoclonal antibody against CIAPIN1. Hybridoma. 2005;24:141–145. doi: 10.1089/hyb.2005.24.141. [DOI] [PubMed] [Google Scholar]
- 20.Pisani P, et al. Estimates of the worldwide mortality from 25 cancers in 1990. Int. J. Cancer. 1999;83:18–29. doi: 10.1002/(sici)1097-0215(19990924)83:1<18::aid-ijc5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 21.Liver Cancer Study Group of Japan. Primary liver cancer in Japan. Clinicopathologic features and results of surgical treatment. Ann. Surg. 1990;211:277–287. [PMC free article] [PubMed] [Google Scholar]
- 22.Friedmann T. Progress toward human gene therapy. Science. 1989;244:1275–1281. doi: 10.1126/science.2660259. [DOI] [PubMed] [Google Scholar]
- 23.Strauss M. Liver-directed gene therapy: prospects and problems. Gene Ther. 1994;1:156–164. [PubMed] [Google Scholar]
- 24.Lee C-T, et al. Gene therapy. In: Pass HI, Mitchell JB, Johnson DH, Turrissi AT, Minna JD, editors. Lung Cancer. 2nd edn. 2000. pp. 318–333. Philadelphia, PA: Lippincott Williams & Wilkins. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}