

SUMMARY
The induction of opioid receptor adaptation by mixed agonist-antagonists such as buprenorphine has not been investigated. To this end, neonatal rats were given injections of buprenorphine (0.1-2.5 mg/kg/day) and μ binding (Kd and Bmax) to brain membranes was measured with [3H][D-Ala2,MePhe4,Gly-ol5]enkephalin. At doses of buprenorphine of ≥0.5 mg/kg, μ sites were reduced 47-75%, without changes in affinity. Chronic administration of the structurally related partial agonist diprenorphine (2.5-75 mg/kg) failed to alter μ binding. Apparent loss of sites due to receptor blockade by residual buprenorphine was ruled out by several lines of evidence. Bmax values for δ ([3H][D-Ser2,L-Leu5]enkephalyl-Thr) and κ ([3H]U69593) binding were elevated 1.9–4.2-fold by buprenorphine treatment. In adult rats buprenorphine (0.5-2.5 mg/kg) reduced μ-opioid binding to forebrain membranes dose dependently, by 25-77%. [3H][D-Ser2,L-Leu5] Enkephalyl-Thr-labeted δ subtype receptors and κ sites in adult forebrain membranes were up-regulated 2-3-fold. The δ subtype receptors that bind [3H][D-Pen2,D-Pen5]enkephalin in neonatal or adult brain membranes were unaffected by 0.5-2.5 mg/kg buprenorphine treatment. Down-regulation (70-74%) of μ sites and up-regulation (1.9-6.7 fold) of δ and κ receptors were also observed in synaptic plasma membrane-enriched and microsomal fractions from buprenorphine-treated adult rat brain. Because agonist-induced opioid receptor down-regulation is difficult to elicit in adult mammalian brain, these data indicate that buprenorphine is a useful tool to study brain opioid receptor adaptation in vivo.
The opioid mixed agonist-antagonist buprenorphine is presently undergoing clinical evaluation for its ability to antagonize the reinforcing effects of both heroin and cocaine in humans (1-3). Its advantages include minimal adverse side effects and less potential for abuse liability than opioid agonists presently used for drug rehabilitation (4-6). Nevertheless, buprenorphine has 25-40 times the analgesic potency of morphine in humans (5) and rodents (4) after parenteral administration.
Initial behavioral investigations revealed that buprenorphine acts as a mixed agonist (4, 7). Subsequently, μ and possibly δ and κ agonist activity of buprenorphine was reported in a number of other pharmacological studies (8-10). However, with the availability of more κ-selective ligands as competitors, buprenorphine has proven to be a potent κ-opioid receptor antagonist (11-13). Further clarification of the interaction of buprenorphine with various opioid receptor types and subtypes awaits additional pharmacological investigations as well as systematic competition binding assays with selective ligands.
The purpose of this study was to define the in vivo effects of buprenorphine on μ−, δ-, and κ-opioid binding using selective ligands. In contrast to well documented brain opioid receptor up-regulation, agonist-induced down-regulation has been observed infrequently in vivo and was restricted to selected brain regions, depending on the agonist (Refs. 14-18 and references cited therein). In contrast, agonist-induced opioid receptor down-regulation has been readily demonstrated in cell cultures (19-21) and in neonatal rat brain (22-24). Little is known of the in vivo effects of mixed agonist-antagonists on opioid receptor adaptation. Here we measured opioid binding in brain membrane preparations from P7 and adult rats treated with buprenorphine.
Materials and Methods
Chemicals
DAMGE, DSLET and DPDPE were obtained from Multiple Peptide Systems (San Diego, CA) and U69593, nor-BNI, and diprenorphine from National Institute on Drug Abuse Drug Supply (Research Triangle, NC). ICI174864 was purchased from Cambridge Research Biochemicals (Wilmington, DE) and CTOP from Bachem (Torrance, CA). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Buprenorphine treatment of rats
P1 Sprague-Dawley rats were given subcutaneous injections of either buprenorphine (0.01-2.5 mg/kg of body weight), diprenorphine (2.5-75 mg/kg), or saline alone, daily for 6 days. P7 rats were sacrificed and brains (with cerebellum) were removed. In some experiments a single injection of buprenorphine was given to P6 rats and brains were collected 20 hr later. Adult male Sprague-Dawley rats were given intraperitoneal injections of either buprenorphine (0.5-2.5 mg/kg) or saline, and 20 hr later the animals were killed. In preliminary experiments a 6-day regimen of buprenorphine (as used for neonates) gave the same results as the 20-hr treatment. Adult brains without cerebellum were collected, immediately frozen in dry ice, and stored at −70° until binding assays were performed.
Measurement of in vivo opioid receptor occupation by [3H] buprenorphine
Doses of [3H]buprenorphine (30-50 μCi, 43.8 Ci/mmol; National Institute on Drug Abuse Drug Supply) combined with unlabeled buprenorphine at a concentration of 0.5 mg/kg were injected subcutaneously into P1, P3, and P7 rats. After a period of 20 hr, rats were sacrificed and membranes were prepared.
Washing procedure to remove buprenorphine
Membrane preparations from neonatal or adult rat brains were washed at least five times with 50 mM Tris- HCl, pH 7.4. In some experiments a second washing procedure was adopted for adult rat brain membranes. This entailed incubation for 2 hr at 25° with 50 mM Tris · HCl, pH 7.4, buffer containing 100 mM NaCl and 50 μM Gpp(NH)p, followed by four additional washes with Tris buffer.
Subcellular fractionation
Subcellular fractions (SPMs and microsomes) were prepared from adult rat forebrain homogenates by sequential differential and sucrose density gradient centrifugation, according to the method of Roth et al. (25).
Opioid receptor binding assays
Rat membrane preparations and Subcellular fractions were assayed for opioid binding activity as described (26). Membrane preparations (300-800 μg of protein) were incubated in duplicate with 1 nM [3H]U69593 (53 Ci/mmol; Amersham, Arlington Heights, IL), [3H]DAMGE (35 Ci/mmol; Multiple Peptide Systems), [3H]buprenorphine, or [3H]DSLET (35 Ci/mmol; Multiple Peptide Systems) at 25° for 1 hr, 1-6 nM [3H]diprenorphine (31 Ci/mmol; National Institute on Drug Abuse) at 37° for 20 min, or 2 nM [3H]DPDPE (24 Ci/mmol; Multiple Peptide Systems) at 25° for 3 hr. Bmax and Kd values were estimated from homologous competition binding assays performed in the presence of 12-16 different concentrations of the corresponding unlabeled ligand. [3H]Buprenorphine (1 nM) binding to μ-opioid sites in the presence of 100 mM NaCl and 50 μM Gpp(NH)p was estimated by using 100 nM ICI174864 or nor-BNI, as specific blockers for δ and κ sites, respectively. Nonspecific binding was determined with 0.1 μM etorphine.
[3H]Diprenorphine specific binding in the presence of selective opioid antagonists (CTOP, ICI174864, or nor-BNI, 100 nM) was measured to obtain the relative proportions of μ, κ and δ sites. Bmax, values were determined with 6 nM [3H]diprenorphine, as described (27).
In the [3H]DPDPE and [3H]diprenorphine experiments, filters were presoaked in incubation buffer containing 0.02% polyethylenimine. Protein concentrations were determined by the method of Lowry et al (28), with bovine serum albumin as standard. Statistical analyses of data were performed using the Student t test. Binding parameters (Kd, Ki, and Bmax) were estimated by the LIGAND (29) and INPLOT4 (GraphPad Software Inc., San Diego, CA) programs. Heterologous competition curves were generated with SigmaPlot (JANDEL Scientific, Corte Madera, CA), using an equation from the ALLFIT program.
Results
Effects of in vivo administration of buprenorphine on μ-, δ-, and κ-opioid binding to neonatal and adult rat brain membranes
P1 rat pups were treated daily with buprenorphine for 6 days and opioid binding to whole-brain membrane preparations was determined 20 hr after the last injection. Selective μ ([3H]DAMGE), δ ([3H]DPDPE and [3H] DSLET), and κ ([3H]U69593) agonists were used as radioligands. Dose-dependent effects of buprenorphine on corresponding Bmax values are shown in Fig. 1. A decrease in μ sites and an up-regulation of κ binding was observed. Up-regulation of δ sites was also evidenced with [3H]DSLET as radioligand, whereas [3H]DPDPE binding was unaffected by buprenorphine treatment. Buprenorphine at concentrations of ≥0.5 mg/kg was sufficient to affect μ and κ binding, whereas δ-opioid binding was up-regulated at ≥0.75 mg/kg drug. A single injection of 0.5 mg/kg buprenorphine given to P6 rats elicited a 55% down-regulation of μ sites after 20 hr, comparable to that produced by the 6-day treatment (Fig. 1). Administration of a range of doses (2.5, 5, 50, and 75 mg/kg/day) of the partial agonist diprenorphine to rat pups for 6 days (P1-P6) failed to alter μ-opioid binding densities or affinities (data not shown).
Fig. 1.
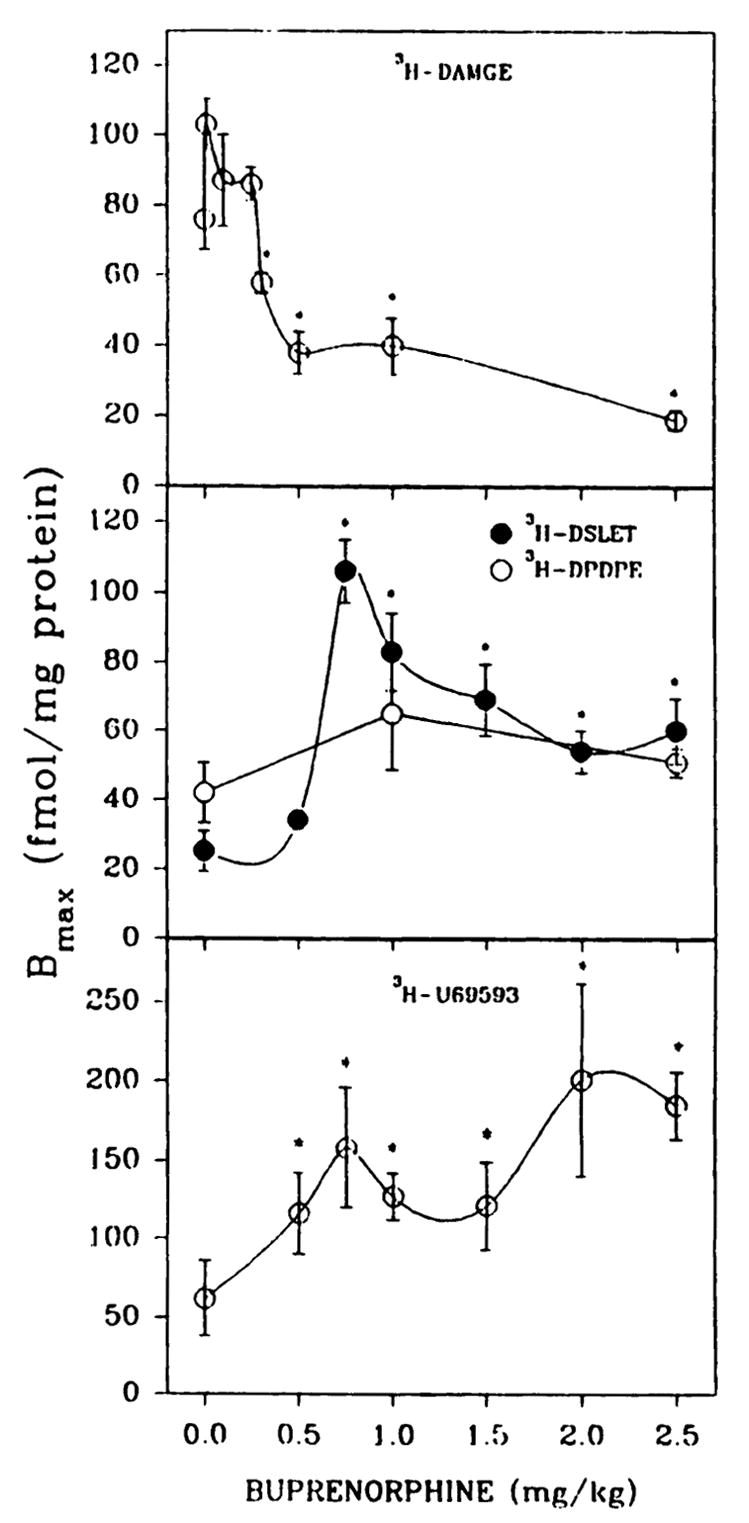
Opioid binding to brain membrane preparations from P7 rats treated in vivo with various concentrations of buprenorphine. Pups were given subcutaneous injections of saline or buprenorphine daily for 6 days and were sacrificed 20 hr after the last administration. Brain membranes were washed five times with 25–30 ml of 50 mM Tris·HCl, pH 7.4, before binding assays. Opioid binding was measured with 1 nM [3H]DAMGE, [3H]DSLET, or [3H]U69593 or 2 nM [3H]DPDPE in 12-point homologous competition assays. Bmax, and Kd values were estimated using the LIGAND program. The affinity of [3H]DAMGE (μ) is shown in Fig. 3. In the order of increasing buprenorphine concentrations from 0 to 2.5 mg/kg, Kd values were 3.3 ± 0.6, 3.3 ± 0.2, 7.6 ± 1.2, 6.2 ± 1.1, 2.8 ± 0.2, 6.0 ± 0.9, and 5.1 ± 0.7 nM ([3H]DSLET); 3.8 ± 0.6, 5.3 ± 1.3, and 3.4 ± 0.2 nM ([3H]DPDPE); and 3.1 ± 0.7, 5.1 ± 0.9, 4.2 ± 0.8, 6.4 ± 0.9, 4.9 ± 1.2, 5.8 ± 1.4, and 4.9 ± 0.3 nM ([3H]U69593). *, Significantly different from saline treated, p < 0.05. Results are from four to seven separate experiments.
Adult rats were given injections of 0.5–2.5 mg/kg buprenorphine and sacrificed 20 hr later. Again μ binding was down-regulated by 25–77%, whereas κ binding was up-regulated 2–3-fold at buprenorphine concentrations of ≥1.0 mg/kg (Fig. 2). In agreement with the neonatal data, δ binding measured with [3H]DPDPE did not change upon buprenorphine treatment. When δ binding was estimated with [3H]DSLET, a moderate receptor down-regulation was observed after 0.5–1.0 mg/kg buprenorphine treatment, whereas 2.5 mg/kg drug induced a 2-fold up-regulation of δ-opioid binding (Fig. 2).
Fig. 2.
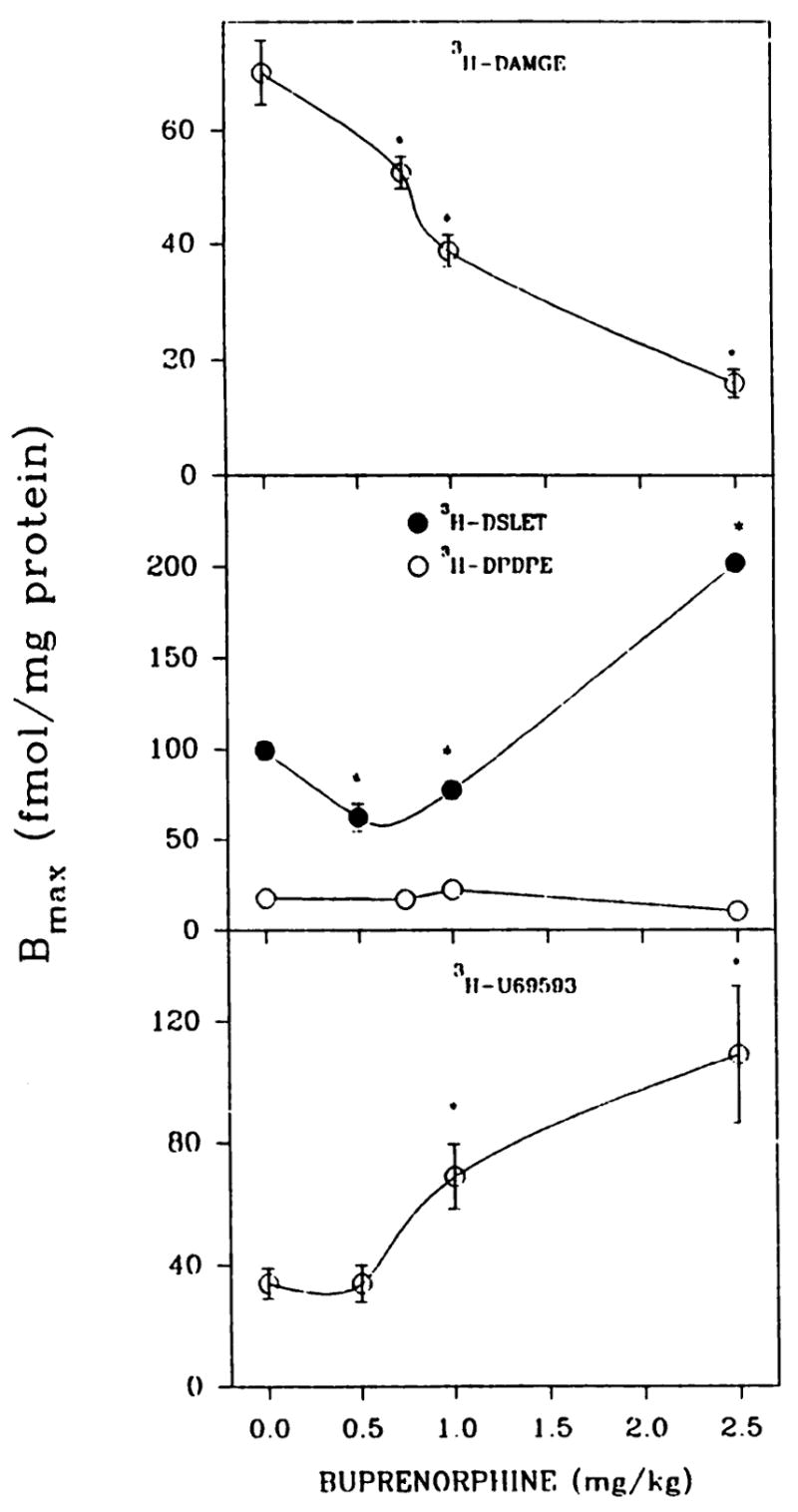
Optoid binding in brain membrane preparations from adult rats treated in vivo with various concentrations of buprenorphine. Adult male rats were administered (intraperitoneally) either buprenorphine (0.5–2.5 mg/kg) or saline, and 20 hr later forebrains were collected. The affinity of [3H]DAMGE (μ) is shown in Fig. 3. In the order of increasing buprenorphine concentrations from 0 to 2.5 mg/kg, Kd values were 2.2 ± 0.2, 2.0 ± 0.3, 2.2 ± 0.3, and 4.6 ± 0.6 nM ([3H]DSLET); 2.0 ± 0.2, 2.0 ± 0.3, 2.4 ± 0.2, and 1.6 ± 0.3 nM ([3H]DPDPE); and 3.3 ± 0.6,6.2 ± 1.2, 4.3 ± 0.5, and 5.7 ± 1.5 nM ([3H]U69593. *, Significantly different from saline-treated, p < 0.05. Results are from three to six separate experiments.
Evidence to demonstrate that buprenorphine induces down-regulation, rather than blocking μ-opioid receptors
Because buprenorphine is lipophilic, we addressed the question of whether receptor occupancy by this drug causes an apparent down-regulation. The introduction of five membrane washes with 50 mM Tris·HCl, pH 7.4, and the absence of increases in Kd values for down-regulated μ sites tend to rule out the possibility of blockade of sites 20 hr after administration of buprenorphine (Fig. 3). However, we found that 1-hr exposure of rats to buprenorphine (0.5 and 2.5 mg/kg) completely blocked tritiated DAMGE, DSLET, and U69593 binding to membrane preparations washed five to 10 times with the Tris·HCl buffer. To further probe the prospect of receptor blockade, several approaches were taken.
Fig. 3.
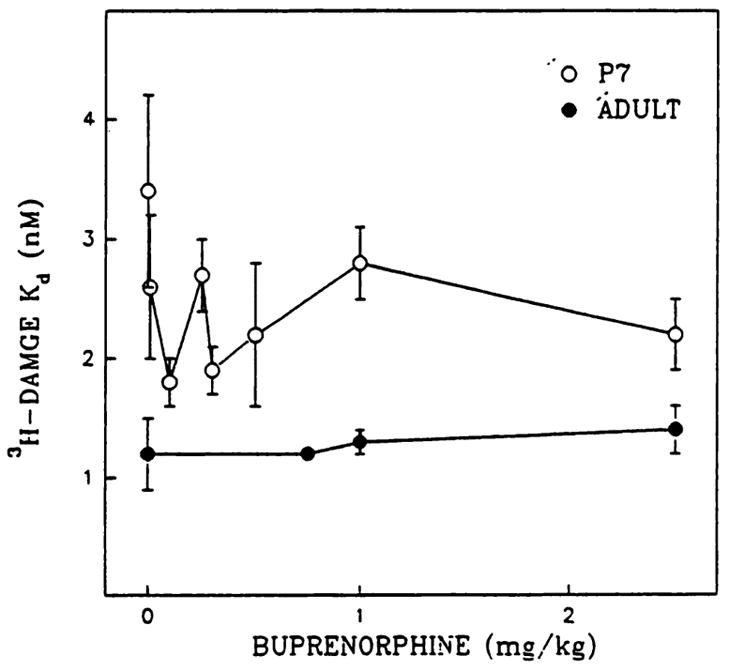
Ka values for [3H]DAMGE binding to membrane preparations from P7 and adult rats treated with buprenorphine. Rats were given injections of buprenorphine as described for Figs. 1 and 2. [3H]DAMGE Kd estimations were obtained from 12-point homologous competition binding assays. Note that P7 control Ka values were consistently higher than the Kd values for buprenorphine-treated rats. Results are from six or seven separate experiments.
First, P1, P3, or P7 rats were given a single injection of a 0.5 mg/kg dose of a mixture of unlabeled buprenorphine and [3H] buprenorphine. A sufficient amount of label was used for detection of a possible buprenorphine blockade of μ sites. Twenty hours later brain membranes were harvested and residual radioactivity associated with membrane preparations before and after five washes with 50 mM Tris·HCl, pH 7,4, was measured. In pup brains of all ages studied, the amount of radioactivity retained in membrane preparations after the washes was 70–147 fmol/mg of protein (10% of the original amount detected in cell homogenates before centrifugation). However, a similar level of radioactivity (45–109 fmol/mg of protein) was found in P7 cerebellar tissue as well, despite the presence of few opioid receptors in cerebellum at this developmental stage (22). Originally we found that 0.5 mg/kg (unlabeled) buprenorphine induced a decrease in μ site density to 38 fmol/mg of protein in P7 membrane preparations (Fig. 1). Although the cerebellar data suggest that buprenorphine is nonspecifically bound to membranes, rather than being bound to opioid sites, the residual binding detected in the [3H]buprenorphine experiments could account for the observed decreases in μ binding.
To assess the effect of different levels of buprenorphine on binding of 1 nM [3H]DAMGE, 2 nM [3H]DPDPE, and 1 nM [3H]U69593, heterologous competition curves were generated (Fig. 4). Experiments were performed with adult membrane preparations (Fig. 4, lower) and [3H]DAMGE was tested in P1 rat membranes (Fig. 4, upper). The residual 147 fmol/mg of protein is equivalent to 0.1 nM buprenorphine in our assays, when calculated on the basis of the amount of protein and total volume used. As seen in Fig. 4, 0.1 nM buprenorphine did not displace μ binding in adult or P1 membrane preparations. Corresponding Ki and Bmax values for μ-, δ-, and κ-opioid binding are given in Table 1. Buprenorphine displayed the highest affinity for μ-opioid sites and lower affinities for δ and κ sites.
Fig. 4.
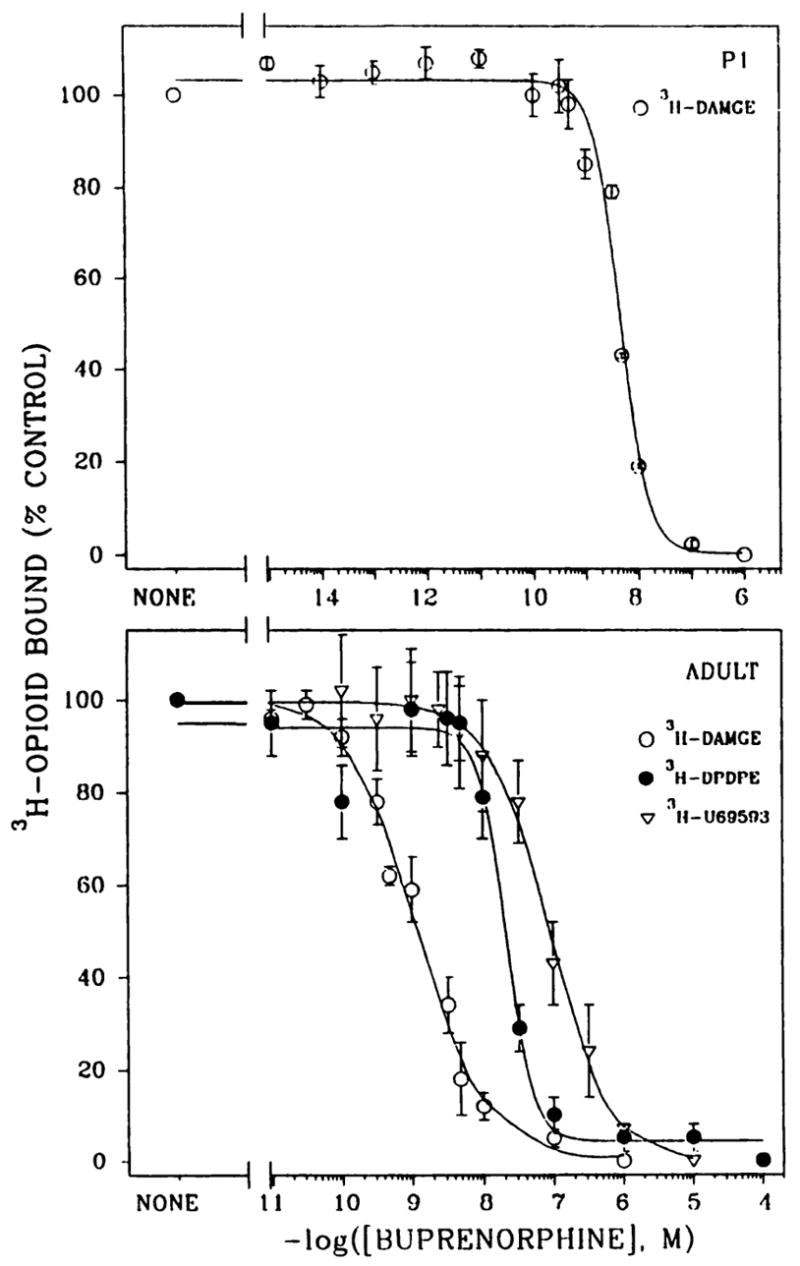
Heterologous competition binding curves with buprenorphine as competitor for μ, δ, and κ receptor sites. [3H]DAMGE (1 nM) binding was determined in P1 (upper) and adult (lower) brain membranes in the presence of 12–16 concentrations of buprenorphine. [3H]DPDPE (2 nM) and [3H]U69593 (1 nM) binding was conducted with adult brain membranes. Corresponding Bmax and Ki values are given in Table 1. Results are from three to nine separate experiments.
TABLE 1. In vitro binding parameters of buprenorphine for μ-, δ-, and κ-opioid receptors.
Membranes from P1 and adult rat brains were prepared as described in Materials and Methods. Heterologous competition binding curves were generated with 1 nM [3H]DAMGE (μ), 2 nM [3H]DPDPE (δ), or 1 nM [3H]U69593 (κ) in the presence of 12–16 concentrations of buprenorphine (Fig. 4). Control specific binding ranged from 750 to 1166 dpm/tube. Results are from three to nine separate experiments.
| Receptor type | Adult
|
P1
|
||
|---|---|---|---|---|
| Ki | Bmax | Ki | Bmax | |
| nM | fmol/mg of protein | nM | fmol/mg of protein | |
| μ | 1.2 ±0.9 | 95 ±11 | 2.2 ± 0.03 | 140 ±27 |
| δ | 22 ± 4.0 | 21 ±1.5 | ||
| κ | 40 ±15 | 80 ±11 | ||
When homologous competition [3H]DAMGE binding assays were performed with adult rat brain membranes in the presence of varying concentrations of buprenorphine (preincubated for 1 hr), evidence for competitive inhibition by this drug was obtained. Although at 0.1 nM buprenorphine no change in Kd was observed (1.2 ± 0.1 versus 1.2 ± 0.3 nM for controls), at 5 nM buprenorphine the Kd increased to 19.6 ± 4.4 nM. Bmax values for [3H]DAMGE binding did not change with 0.1 nM buprenorphine but decreased by 37% with 5 nM buprenorphine. At buprenorphine concentrations of 0.1 and 1.0 μM, no μ binding was detected, in agreement with the data shown in Fig. 4.
Second, because NaCl and GTP analogs reduce opioid agonist binding (30), membrane preparations from brains of adult rats treated with 0.5 mg/kg buprenorphine were also washed with a combination of this cation and nucleotide. In prior control assays, the sensitivity of buprenorphine binding to these agents was tested with P1, P7, or adult rat brain membrane preparations. [3H]Buprenorphine together with unlabeled δ and κ suppressors (antagonists) were incubated in the presence of 100 mM NaCl and 50 μM Gpp(NH)p. Consistent with agonist behavior, [3H]buprenorphine binding to μ -opioid sites was reduced by 51–62%. Buprenorphine-treated adult rat brain membrane preparations either were washed five times with 50 mM Tris·HCl, pH 7.4, or were first incubated for 2 hr at 25° with the same buffer containing 100 mM NaCl and 50 μM Gpp(NH)p, followed by four additional washes with Tris·HCl buffer. Opioid binding densities were then measured with [3H]diprenorphine (Fig. 5). Diprenorphine was used because (a) the Na+/Gpp(NH)p treatment apparently shifts the receptor into an antagonist conformation that binds agonists such as DAMGE poorly, (b) selective μ antagonist radioligands were not commercially available, and (c) diprenorphine binding to μ, δ, and κ sites is insensitive to Na+/Gpp(NH)p. No significant changes were observed in Bmax values after the two washing procedures, suggesting that the five-step washing with Tris·HCl buffer is sufficient to remove unbound buprenorphine from binding sites.
Fig. 5.
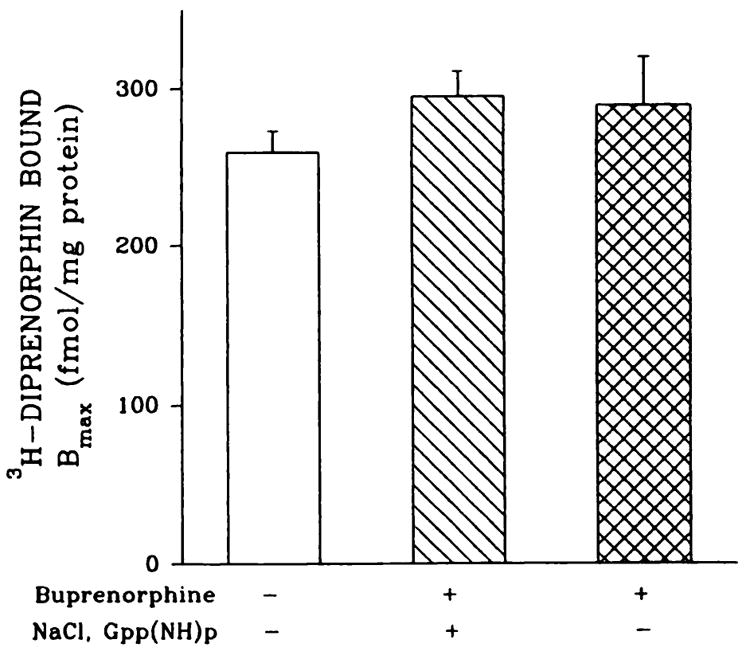
Optoid binding to NaCl/Gpp(NH)p-washed brain membranes from buprenorphine-treated adult rats. Adult rats were given injections of 0.5 mg/kg buprenorphine. Forebrains were divided into two hemispheres; one, used for membrane preparations, was washed with 50 mM Tris·HCl, pH 7.4, buffer and the other was washed with 50 mM Tris·HCl, pH 7.4, buffer containing 100 mM NaCl and 50 μM Gpp(NH)p, followed by four additional washes with buffer alone. Homologous displacement assays were performed with 1 nM [3H]diprenorphine and Bmax values were determined with the INPLOT4 program. Kd values varied from 1.1 ± 0.1 to 4.4 ± 0.9 nM. Results are from four or five separate experiments.
To determine whether the same down- and up-regulation of opioid receptors could be detected after the Na+/Gpp(NH)p washings, binding assays were performed with saturating amounts of the partial agonist [3H]diprenorphine in the presence of μ, δ, and κ suppressors (Fig. 6). Using this method adult rat membrane preparations from rats given injections of buprenorphine (0.5 mg/kg, 20 hr) showed a 26% decrease in the levels of μ-opioid receptors. These data are consistent with the observed 25% decline in [3H]DAMGE binding (Fig. 2). The 40–47% increases in δ- and κ-opioid binding seen with diprenorphine and suppressors were greater than those observed with tritiated DSLET and U-69593, respectively (Fig. 2). Because receptor density changes were observed when binding was measured with an opioid agonist and partial agonist, receptor-G protein uncoupling effects are ruled out.
Fig. 6.
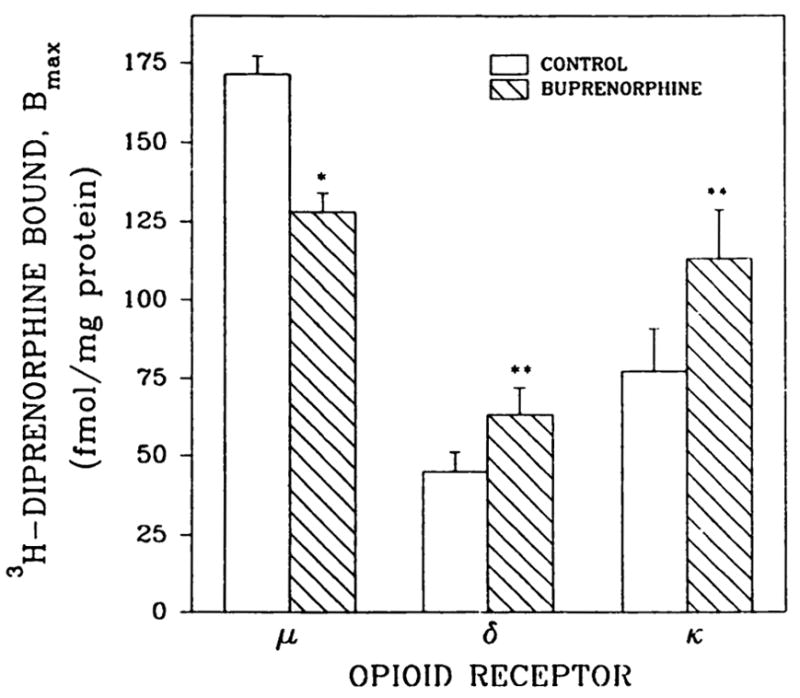
[3H]Diprenorphine binding to brain membranes from buprenorphine-treated adult rats. Animals were given one intraperitoneal injection of 0.5 mg/kg buprenorphine 20 hr before sacrifice. Membranes were incubated for 2 hr with 50 mM Tris·HCl, pH 7.4, buffer containing 100 mm NaCl and 50 μM Gpp(NH)p, followed by four additional washes with buffer alone. Opioid binding was measured with 1 or 6 nM [3H]diprenorphine, and unlabeled diprenorphine (5 μM) was used to determine non-specific binding, as described in Materials and Methods. *, p < 0.05; **, p < 0.01. Results are from three separate experiments.
Third, methanol completely removed the radioactivity remaining after Na+/Gpp(NH)p washing of membranes from an in vivo administration of [3H]buprenorphine. These results eliminate the possibility that residual radioligand or its degradation products were covalently bound to membrane proteins.
Effects of buprenorphine on opioid binding in subcel-lular fractions from adult rat brain membranes
[3H] DAMGE (μ), [3H]DSLET (δ), and [3H]U69593 (κ) binding was measured in subcellular fractions from brains of rats treated with buprenorphine (1 mg/kg). No changes in Kd values were observed for the SPMs and microsomes from control and buprenorphine-treated rats. As shown in Fig. 7, there was a 70–-74% decrease in μ-opioid binding in both microsomes and SPMs from buprenorphine-treated rats. Up-regulation (1.9–6.7-fold) of microsomal and SPM δ and κ sites was also evidenced. Microsomal sites constituted a smaller (37, 22, and 40% for μ, δ, and κ, respectively) population than SPM receptors in control brain. Intracellular δ sites were up-regulated to a greater extent than were those in SPMs.
Fig. 7.
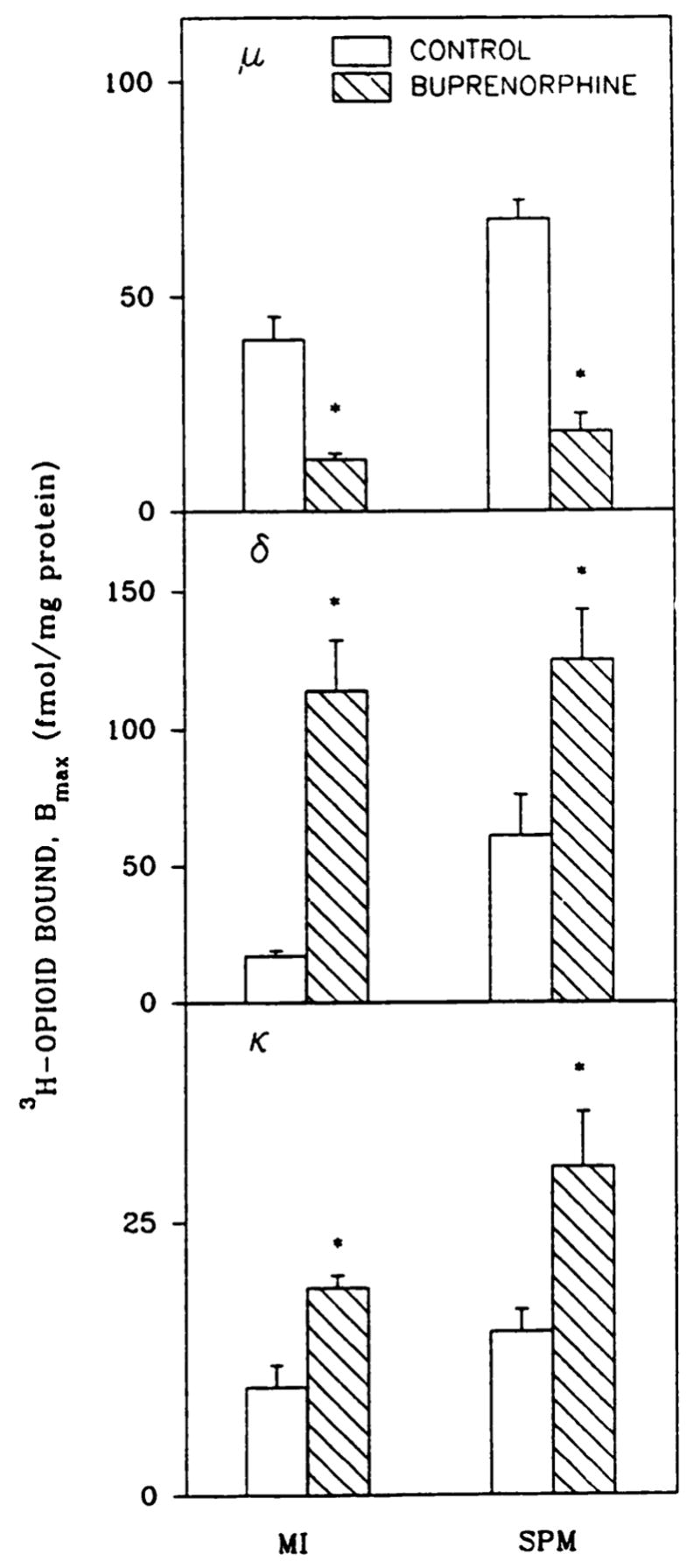
Down- and up-regulation of opioid receptors in subcellular fractions from brain of buprenorphine-treated adults. SPMs and microsomes (MI) from adult rats treated with saline or buprenorphine (1 mg/kg, 20 hr) were prepared as described in Materials and Methods. Bmax values were estimated with 1 nM [3H]DAMGE (μ), [3H]DSLET (δ), and [3H] U69593 (κ) in homologous competition assays. For microsomes and SPMs, control Kd values were 1.8 ± 0.1 and 1.4 ± 0.07 nM (μ), 1.3 ± 0.2 and 3.3 ± 0.6 nM (δ), and 1.9 ± 0.4 and 1.2 ± 0.1 nM (κ), whereas those for buprenorphine-treated membranes were 5.5 ± 2.5 and 1.9 ± 0.3 nM (μ), 3.7 ± 0.5 and 3.9 ± 0.5 nM (δ), and 3.5 ± 0.1 and 3.3 ± 0.4 nM (κ), respectively. *, Significantly different from saline-treated rats, p < 0.05. Results are from three to five separate experiments.
Discussion
The present findings demonstrate that buprenorphine administration to neonatal or adult rats engendered a down-regulation of forebrain μ-opioid binding and up-regulation of κ-opioid and one subtype of δ-opioid binding. These significant and relatively unique agonist and antagonist properties of buprenorphine ensue upon administration of similar doses. The conflicting agonist and antagonist actions of buprenorphine previously seen in behavioral and in vitro bioassays have been attributed to dose dependency effects (4, 7–9). However, the type- and subtype-specific receptor adaptation observed here may also explain the pharmacological data.
In agreement with previous binding studies (8,9), the administration of buprenorphine to adult rats 60 min before sacrifice resulted in a total blockade of μ-, δ-, and κ-opioid sites in membrane preparations even after five washes with buffer. Here, 20 hr after administration the bulk (99.97%) of this lipophilic drug appears to be cleared from the brain. It was reported that only unchanged buprenorphine was present in brain (31). Although the residual buprenorphine (70–147 fmol/mg of protein) could account for the observed down-regulation of μ sites, several lines of evidence refute this possibility. (a) Authentic down-regulation of μ-opioid binding was corroborated by the absence of increases in the Kd values of [3H] DAMGE binding in treated P7 and adult rat brain preparations (Fig. 3). In contrast, “apparent” receptor down-regulation due to the presence of residual agonists has been detected in studies with opioid receptors (14,26) and other systems and is generally accompanied by a significant decrease in receptor affinity and little or no change in density. In the in vitro binding assays described here, 5 nM buprenorphine produced a 20-fold increase in [3H]DAMGE Kd and a 37% decrease in Bmax values, (b) When [3H]buprenorphine was administered to rats, residual amounts of radioactivity remained in forebrain membrane preparations after Tris·HCl washing. However, the amount of buprenorphine equivalent to the remaining radioactive drug did not displace [3H]DAMGE in heterologous competition assays (Fig. 4). Moreover, a comparable amount of label was localized in cerebellum, which has few opioid binding sites. The results suggest that the small amount of residual buprenorphine is nonspecifically associated with membranes and not bound to opioid sites. (c) Microsomal and SPM-enriched μ-opioid receptors proved to be down-regulated to a greater extent than crude membranes. Subcellular fractionation entails a number of differential and sucrose density gradient centrifugations, where-upon residual buprenorphine would be removed. Because there was a loss of microsomal and SPM sites, it is reasonable to assume that receptor internalization and degradation occur, consistent with in vitro findings for opioid receptors (21) and other receptor systems. Previous studies have shown that these two subcellular fractions comprise the bulk of the opioid binding sites in the 1000 × g supernatant of brain cell-free homogenates (25). Our results indicate that down-regulation, rather than a simple redistribution by internalization of cell surface opioid binding sites, occurs, and the data are consistent with cell culture experiments (Ref. 26 and references cited therein). The greater up-regulation of δ intracellular sites, compared with SPM receptors, is consistent with previous results on naltrexone induction of rat brain μ-opioid receptors, wherein a 2-fold increase in microsomal sites and a 0.5-fold elevation in SPM receptors were observed (32).
Differential buprenorphine regulation of [3H]DPDPE and [3H]DSLET binding densities in P7 and adult rat brain membrane preparations was discovered in these studies. At both ages DPDPE binding sites were not affected by the mixed agonist-antagonist. Buprenorphine up-regulated DSLET binding in pups and adults. These data can be explained by the existence of a heterogeneous population of δ receptors in brain, where DPDPE interacts with one subtype and DSLET binds to a different subtype of δ receptors, as recently proposed (33).
In addition to the in vivo effects elicited by buprenorphine, we found that this mixed agonist-antagonist possessed different affinities for the three types of opioid receptors in vitro. The affinity in the competition studies with [3H]DAMGE, [3H] DPDPE, and [3H]U69593 (Fig. 4) was highest for μ-opioid receptor sites, intermediate for δ receptors, and lowest for κ receptors. Because selective ligands were used here, the data afford an evaluation of buprenorphine binding characteristics.
Considerable evidence has accumulated to suggest that opioid receptor down-regulation does not always accompany the dependence and tolerance phenomena in animals (Refs. 34–37 and references cited therein). The demonstration here of a potent down-regulation of μ sites by buprenorphine, which displays low dependence and tolerance ability, strengthens this hypothesis. Furthermore, there are data to suggest that opioid tolerance involves receptor uncoupling from G proteins (16,17, 35–39). Here we show that similar changes in agonist ([3H] DAMGE) and partial agonist ([3H]diprenorphine) Bmax values ensue after buprenorphine treatment, arguing against the occurrence of changes in G protein coupling. Moreover, μ- and κ-opioids have opposite tonic effects on the mesolimbic dopaminergic pathway, which has been implicated in mediating motivational effects of opioids, cocaine, and other drugs of abuse. In the ventral tegmental area of this pathway, β-endorphin increases dopamine release via μ receptors by disinhibition of γ-aminobutyric acid neurons. This dopamine is released from projections in the nucleus accumbens, where dynorphins tonically inhibit the process via κ receptors (40). By down-regulation of μ sites and up-regulation of κ receptors, buprenorphine may exert a dual blockade of mesolimbic dopamine release, thereby accounting for its ability to antagonize the reinforcing effects of heroin and cocaine. Taken together, these in vivo studies demonstrate that buprenorphine represents a useful tool to explore mechanisms of opioid receptor adaptation, especially in light of its potential for the treatment of opiate and cocaine abuse.
Acknowledgments
This work was supported by Grant DA05412 from the National Institute on Drug Abuse.
ABBREVIATIONS
- P7
postnatal day 7
- P1
postnatal day 1
- P3
postnatal day 3
- P6
postnatal day 6
- CTOP
cyclic D-Phe-Cys-Tyr-D-Trp-Om-Thr-Pen-Thr amide
- DAMGE
[D-Ala2,MePhe4,Gly-ol5]enkephalin
- DPDPE
[D-Pen2,D-Pen5]enkephalin
- DSLET
[D-Ser2,L-Leu5]enkephalyl-Thr
- Gpp(NH)p
5′-guanylylimidodiphosphate
- G protein
GTP-binding regulatory protein
- nor-BNI
nor-binaltorphimine
- SPM
synaptic plasma membrane
References
- 1.Mello NK, Mendelson JH. Buprenorphine suppresses heroin use by heroin addicts. Science (Washington D C) 1980;207:657–659. doi: 10.1126/science.7352279. [DOI] [PubMed] [Google Scholar]
- 2.Kosten TR, Kleber HD, Morgan C. Treatment of cocaine abuse with buprenorphine. Biol Psychiatry. 1989;26:637–639. doi: 10.1016/0006-3223(89)90090-5. [DOI] [PubMed] [Google Scholar]
- 3.Johnson RE, Jaffe JH, Fudala PJ. A controlled trial of buprenorphine treatment for opioid dependence. JAMA. 1992;267:2750–2755. [PubMed] [Google Scholar]
- 4.Cowan A, Lewis JW, MacFarlane IR. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol. 1977;60:537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jasinski DR, Pevnick JS, Griffith D. Human pharmacology and abuse potential of the analgesic buprenorphine. Arch Gen Psychiatry. 1978;35:601–616. doi: 10.1001/archpsyc.1978.01770280111012. [DOI] [PubMed] [Google Scholar]
- 6.Strain EC, Preston KL, Liebson IA, Bigelow GE. Acute effects of buprenorphine, hydromorphone and naloxone in methadone-maintained volunteers. J Pharmacol Exp Ther. 1992;261:985–993. [PubMed] [Google Scholar]
- 7.Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine and nalorphine-like drugs in the non-dependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1986;197:517–523. [PubMed] [Google Scholar]
- 8.Dum JE, Herz A. In vivo receptor binding of the opiate partial agonist, buprenorphine, correlated with its agonistic and antagonistic actions. Br J Pharmacol. 1981;74:627–633. doi: 10.1111/j.1476-5381.1981.tb10473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadee W, Rosenbaum JS, Herz A. Buprenorphine: differential interaction with opiate receptor subtypes in vivo. J Pharmacol Exp Ther. 1982;223:157–162. [PubMed] [Google Scholar]
- 10.Kajiwara M, Aoki K, Ishii K, Numata H, Matsumiya T, Oka T. Agonist and antagonist actions of buprenorphine on three types of opioid receptor in isolated preparations. Jpn J Pharmacol. 1986;40:95–101. doi: 10.1254/jjp.40.95. [DOI] [PubMed] [Google Scholar]
- 11.Richards ML, Sadee W. Buprenorphine is an antagonist at the κ opioid receptor. Pharm Res. 1985;2:178–181. doi: 10.1023/A:1016340106299. [DOI] [PubMed] [Google Scholar]
- 12.Leander JD. Buprenorphine has potent kappa opioid receptor antagonist activity. Neuropharmacology. 1987;26:1445–1447. doi: 10.1016/0028-3908(87)90112-2. [DOI] [PubMed] [Google Scholar]
- 13.Negus SS, Picker MJ, Dykstra LA. Kappa antagonist properties of buprenorphine in non-tolerant and morphine-tolerant rats. Psychopharmacology. 1989;98:141–143. doi: 10.1007/BF00442021. [DOI] [PubMed] [Google Scholar]
- 14.Tao PL, Chang LR, Law PY, Loh HH. Decrease in δ-opioid receptor density in rat brain after chronic [D-Ala2, D-Leu5]enkephalin treatment. Brain Res. 1988;462:313–320. doi: 10.1016/0006-8993(88)90559-8. [DOI] [PubMed] [Google Scholar]
- 15.Tao PL, Lee HY, Chang LR, Loh HH. Decrease in mu-opioid receptor binding capacity in rat brain after chronic PL017 treatment. Brain Res. 1990;526:270–275. doi: 10.1016/0006-8993(90)91231-5. [DOI] [PubMed] [Google Scholar]
- 16.Loh HH, Tao PL, Smith AP. Role of receptor regulation in opioid tolerance mechanisms. Synapse. 1988;2:457–462. doi: 10.1002/syn.890020414. [DOI] [PubMed] [Google Scholar]
- 17.Werling LL, McMahon PN, Cox BM. Selective changes in μ opioid receptor properties induced by chronic morphine exposure. Proc Natl Acad Sci USA. 1989;86:6393–6397. doi: 10.1073/pnas.86.16.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhargava HN, Gulati A, Rahmani NH. Differences in the binding of [3H][D-Ser2, Thr6]leucine-enkephalin and [3H][D-Pen2, D-Pen5]enkephalin to brain membranes of morphine tolerant-dependent rats. Eur J Pharmacol. 1991;202:403–408. doi: 10.1016/0014-2999(91)90286-y. [DOI] [PubMed] [Google Scholar]
- 19.Simantov R, Baram D, Levy R, Nadler H. Enkephalin and α-adrenergic receptors: evidence for both common and differentiable regulatory pathways and down-regulation of the enkephalin receptor. Life Sci. 1982;31:1323–1326. doi: 10.1016/0024-3205(82)90372-1. [DOI] [PubMed] [Google Scholar]
- 20.Chang KJ, Eckel RW, Blanchard SG. Opioid peptides induce reduction of enkephalin receptors in cultured neuroblastoma cells. Nature (Lond) 1982;296:446–448. doi: 10.1038/296446a0. [DOI] [PubMed] [Google Scholar]
- 21.Law PY, Horn DS, Loh HH. Down-regulation of opiate receptor in neuroblastoma × glioma NG108-15 hybrid cells. J Biol Chem. 1984;259:4096–4104. [PubMed] [Google Scholar]
- 22.Tsang D, Ng SC. Effect of antenatal exposure to opiates on the development of opiate receptors in rat brain. Brain Res. 1980;188:199–206. doi: 10.1016/0006-8993(80)90568-5. [DOI] [PubMed] [Google Scholar]
- 23.Zadina JE, Kastin AJ. Neonatal peptides affect developing rats: β-endorphin alters nociception and opiate receptors, corticotropin-releasing factor alters corticosterone. Dev Brain Res. 1986;29:21–29. doi: 10.1016/0165-3806(86)90078-7. [DOI] [PubMed] [Google Scholar]
- 24.Tempel A, Habas JE, Paredes W, Barr GA. Morphine-induced down-regulation of μ-opioid receptors in neonatal rat brain. Dev Brain Res. 1988;41:129–133. doi: 10.1016/0165-3806(88)90176-9. [DOI] [PubMed] [Google Scholar]
- 25.Roth BL, Laskowski MB, Coscia CJ. Evidence for distinct subcellular sites of opiate receptors: demonstration of opiate receptors in smooth microsomal fractions isolated from rat brain. J Biol Chem. 1981;256:10117–10121. [PubMed] [Google Scholar]
- 26.Belcheva MM, Barg J, Gloeckner C, Gao XM, Chuang DM, Coscia CJ. Antagonist induced transient down-regulation of δ opioid receptors in NG108-15 cells. MoL Pharmacol. 1992;42:445–452. [PubMed] [Google Scholar]
- 27.Barg J, Levy R, Simantov RJ. Paradoxical and subtype-specific effects of opiate antagonists on the expression of opioid receptors in the rat brain cultures. J Neurosci Res. 1989;22:322–330. doi: 10.1002/jnr.490220312. [DOI] [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Fair AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Munson PJ, Rodbard D. LIGAND: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 30.Childers SR, Snyder SH. Differential regulation by guanine nucleotides of opiate agonist and antagonist receptor interactions. Life Sci. 1978;23:759–762. doi: 10.1111/j.1471-4159.1980.tb11184.x. [DOI] [PubMed] [Google Scholar]
- 31.Heel RC, Brogden RN, Speight TM, Avery GS. Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1979;17:81–110. doi: 10.2165/00003495-197917020-00001. [DOI] [PubMed] [Google Scholar]
- 32.Moudy AM, Spain JW, Coscia CJ. Differential up-regulation of microsomal and synaptic membrane μ opioid receptors. Biochem Biophys Res Common. 1985;132:735–741. doi: 10.1016/0006-291x(85)91194-5. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Q, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI, Porreca F. Differential antagonism of opioid δ antinociception by [D-Ala2, Leu5, Cys4]enkephalin and naltrindole-5′-isothiocyanate: evidence for δ receptor subtypes. J Pharmacol Exp Ther. 1991;257:1069–1075. [PubMed] [Google Scholar]
- 34.Nestler EJ. Molecular mechanisms of drug addiction. J Neurosci. 1992;12:2439–2450. doi: 10.1523/JNEUROSCI.12-07-02439.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nestler EJ, Erdos JJ, Terwilliger R, Duman RS, Tallman JF. Regulation of G-proteins by chronic morphine in the rat locus coeruleus. Brain Res. 1989;476:230–239. doi: 10.1016/0006-8993(89)91243-2. [DOI] [PubMed] [Google Scholar]
- 36.Childers SR. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- 37.Collin E, Cesselin F. Neurobiological mechanisms of opioid tolerance and dependence. Cfin Neuropharmacol. 1991;14:465–488. doi: 10.1097/00002826-199112000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci USA. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong CS, Su YF, Watkins WD, Chang KJ. Continuous intrathecal opioid treatment abolishes the regulatory effects of magnesium and guanine nucleotides on mu opioid receptor binding in rat spinal membranes. J Pharmacol Exp Ther. 1992;262:317–326. [PubMed] [Google Scholar]
- 40.Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]