

Abstract
Dopaminergic receptors are found on bovine adrenal chromaffin cells and have been implicated in the facilitation of an inward calcium current [Artalejo et al., (1990) Nature 348, 239–242] that could enhance release. However, previous studies using incubations of long duration (minutes) with dopaminergic receptor antagonists have found instead an inhibition of catecholamine release. In this work we used brief (subsecond) chemical depolarizing stimuli to reexamine the role of dopaminergic receptors on exocytosis from bovine adrenal chromaffin cells. Responses to consecutive depolarizing stimuli were compared using amperometry to monitor vesicular release events and intracellular fura-2 to examine Ca2+ dynamics within individual cells. Restoration of intracellular Ca2+ levels to their initial values following exposure to 60 mM K+ was found to be prolonged unless the exposure was brief (0.5 s) and the cells were maintained at 37ºC. However, with these optimum conditions, a second stimulation evoked more exocytotic events than the first. This effect was blocked by SCH-23390, a D1 antagonist, in a dose dependent fashion, but not by raclopride, a D2 antagonist. The D1 agonist, SKF-38393, enhanced the number of exocytotic events as did prior exposure of the cell to epinephrine. Taken together, the data indicate that released catecholamines can enhance their own release by interaction with a D1-like receptor on bovine adrenal chromaffin cells.
Exocytotic release of chemical messengers is a common mechanism for communication between biological cells. This process is tightly regulated and several controlling mechanisms have been identified including the influx of Ca2+ from the extracellular space, the normal trigger for exocytosis, and the regulation of the number of vesicles in the readily releasable pool. At many cells that undergo exocytosis, autoreceptors play an important regulatory role. For example, at most neurons autoreceptor activation inhibits further release. For example, activation of serotonin somatodendritic autoreceptors inhibits subsequent synthesis and release (1). Autoreceptors on dopaminergic neurons are D2-like (the D2 receptor class is comprised of the D2, D3, and D4 dopaminergic receptors), and dopamine autoreceptor effects are absent in mice with a genetic deletion of the D2 receptor (2). Similar autoreceptor control has been shown for norepinephrine (3). An exception to the general inhibitory nature of neuronal autoreceptors appears to be glutamatergic synapses in the entorhinal cortex of juvenile rats that contain NMDA autoreceptors that enhance release (4).
Nonneuronal cells that secrete by exocytosis also have autoreceptors. Aspinwall et al. demonstrated that insulin-stimulated pancreatic β-cells secrete insulin (5). Deletion of insulin receptors from pancreatic β-cells results in a dramatically lowered level of insulin secretion (6). Thus, in contrast to neurons, these autoreceptors appear to promote increased release. Inhibitory autoreceptor regulation has been reported at bovine chromaffin cells from the adrenal gland. Early investigations, prompted by the phylogenetic relationship between chromaffin cells and sympathetic neurons that are well established to have autoreceptors, determined that autoreceptors on bovine chromaffin cells had properties that pharmacologically resemble dopaminergic receptors (7, 8), and their activation inhibited release. Subsequent research showed that D2 dopamine receptors are located on bovine chromaffin cells and their activation inhibited release (9). A D1-like receptor (the D1 receptor class is comprised of the D1 and D5 dopaminergic receptors) has also been identified on bovine chromaffin cells (10). Activation of this receptor facilitates an inward Ca2+ current that could promote exocytosis (10). However, experimentally it was found that D1 receptor activation actually inhibited catecholamine secretion (11).
In this work we describe a reinvestigation of the action of the D1 receptor on exocytosis from bovine chromaffin cells. All of the prior secretion experiments that investigated autoreceptors on bovine chromaffin cells exposed them to secretagogues for multiple minutes. In contrast, the D1-mediated facilitation Ca2+ currents were measured on a subsecond time scale. Therefore, in this work we evaluated release from bovine chromaffin cells using similar subsecond stimuli. This was accomplished by first optimizing the temperature and timing for K+-based depolarization of individual chromaffin cells, while monitoring in real time release via amperometry and internal Ca2+ with fura-2. With release evoked by brief stimuli at 37ºC we found clear evidence for facilitation of exocytotic release mediated by a D1-like receptor on the surface of bovine chromaffin cells.
Experimental Procedures
Cultured Chromaffin Cells
Bovine chromaffin cells were prepared from adrenal glands obtained from a local abattoir as previously described (12). Chromaffin cells were obtained from the adrenal medulla via digestion with collagenase (Worthington Biochemical Corporation, Lakewood, NJ). Density gradient centrifugation using Renografin was used to obtain a culture solution epinephrine-enriched fraction of cells. Cells were plated at a density of 3 × 105 cells/35-mm diameter plate (Becton Dickinson, Franklin Lakes, NJ) containing a 25-mm glass coverslip (Carolina Biological, Burlington, NC). Cell culture plates were kept at 37ºC in a 5% CO2-supplemented incubator (Fisher Scientific, Hampton, NH). Cells were used days 3–7 postplating.
Electrodes and Electrochemistry
Carbon-fiber disk microelectrodes were prepared from T-650 fibers as previously described (13). Carbon fibers were aspirated into glass capillaries. A pipette puller (Narishige, Long Island, NY) was used to seal the glass around the carbon fiber. The carbon fibers were cut at the glass seal, and the seal was reinforced with epoxy (15% m-phenylenediamine mixed with Epon 828 resin (Miller-Stephenson, Danbury, CT)) heated to 80ºC. The electrodes were cooled at room temperature for 12 h, cured at 100ºC for 12 h, followed by curing at 150ºC for 12 h. Before use, electrodes were beveled at 45º on a diamond polishing wheel (Sutter Instruments, Novato, CA) using impedance monitoring and soaked in isopropyl alcohol for a minimum of 20 min (14). Constant potential amperometry was used to monitor exocytotic events and was measured using an Axopatch 200B (Axon Instruments, Molecular Devices, Union City, CA). The microelectrode was held at 0.650 V vs. an Ag/AgCl reference (Bioanalytical Systems, West Lafayette, IN); individual release events were detected as current spikes. The resulting signal was filtered at 5 kHz with a low-pass Bessel filter and collected at 10 kHz. Amperometric recordings were then filtered at 400 Hz using a Gaussian filter.
Release Measurements
Culture plates containing cells in buffer (in mM) 145 NaCl, 5 KCl, 1 MgCl2, 11.2 glucose, 10 HEPES, and 2 CaCl2) were placed on the stage of a Zeiss 35 inverted microscope (Zeiss, Thornwood, NY). The position of the carbon-fiber microelectrode and stimulating pipette was controlled with piezoelectric micromanipulators (Burleigh Instruments, Exfo, Plano, TX). Exocytosis was evoked with 60 mM K+ delivered by pressure ejection from the stimulating pipettes that were constructed with a horizontal puller (Sutter Instruments, Novato, CA). The diameter of the pipette tip was adjusted to 10 μm using a microforge (Narishige, Long Island, NY). Ejection of the high K+ solution was accomplished with a Picospritzer set at 7 psi (General Valve Corporation, Parker Hannifin, Fairfield, NJ) using either a 0.5 or 2-s bolus. The stage holding the cell plate was either kept at room temperature, 25ºC, or heated with a water bath (Fisher Scientific, Hampton, NH) to 37ºC.
Ca2+ Measurements
Ca2+-imaging employed the fluorescent dye, fura-2, and an imaging system (Empix, Inc., Mississauga, ON, Canada) attached to a microscope (Nikon, Lewisville, TX). An initial fura-2 solution was diluted with 40 μL DMSO and 10 μL of 10% pluronic. This fura-2 solution was diluted to 1 μM in experimental buffer containing 0.10 % BSA (Molecular Probes, Invitrogen, Carlsbad, CA). Cells were incubated in this solution for 20 min at 25ºC, followed by a rinse for 20 min at 25ºC in the experimental buffer. Ca2+-imaging experiments were conducted either simultaneously with amperometry or alone. Intracellular fura-2 bound to Ca2+ was excited at 340 nm while free fura-2 was excited at 380 nm. Emission was monitored at 510 nm using a digital CCD camera and software (Empix Imaging, Inc, Mississauga, ON, Canada). The ratio of the emission with 340 nm excitation and 380 nm excitation was determined. The ratiometric measurements were then converted to [Ca2+]i as previously described (15, 16). The area under the Ca2+ response curve was determined with MiniAnalysis (Synaptosoft, Decatur, GA). The intracellular level directly before a stimulation was taken as the baseline.
Autoreceptor experiments
Experiments were conducted at 37ºC using an S2/S1 protocol. The number of exocytotic events following an initial delivery (S1) of 60 mM K+ for 0.5-s was compared to a second delivery 60 mM K+ (S2) 10 or 30 s later. To evaluate D1 receptors, the D1 antagonist, SCH-23390 (1 μM, 10 μM, 100 μM Sigma-Aldrich, St. Louis, MO), was present in the buffer. To evaluate D2 receptors, the D2 antagonist, raclopride (1 μM, 10 μM, 100 μM Sigma-Aldrich, St. Louis, MO), was present in the buffer. To evaluate D1 receptors via a D1 agonist, SKF-38393 was introduced to the buffer. Paired 0.5-s boluses spaced 30 s apart were used to deliver the 60 mM K+ secretagogue to cells. At 10 s after the initial stimulus, 100 uM SKF-38393 (Sigma-Aldrich, St. Louis, MO), a D1 agonist, was delivered to the buffer and remained present during the second stimulus.
To evaluate the endogenous ligand, a 3-s bolus of 50 μM epinephrine was applied to the cell 22 s after S1 and S2 was applied 5 s later. As a control, the experiment was repeated at separate cells with ejection of experimental buffer substituted for epinephrine.
Data analysis and statistics
Spike analysis was performed using MiniAnalysis. For selection as spikes, the amplitude had to exceed the root-mean-square current noise by a factor of 5. Student’s t-test and one-way ANOVA were used to determine significance among data sets. A value of p < 0.05 was taken to indicate a significant difference.
Results
Vesicular release and Ca2+ dynamics evoked by 0.5-s and 2-s stimuli at 25ºC
At 25 ºC, a 0.5-s pressure ejection of 60 mM K+ onto a chromaffin cell was sufficient to cause Ca2+ influx into the cell, measured by fura-2 fluorescence, and this was accompanied by exocytotic spikes measured by amperometry (example traces in left panel of Figure 1). Intracellular [Ca2+] reached a maximum after the 60 mM K+ ejection was terminated and then slowly returned to baseline. The decay of the [Ca2+]i was sufficiently slow that it failed to reach the original level within 10 s of the initial K+ exposure. A second, 0.5-s pressure ejection of 60 mM K+, 10 s after the first, caused a smaller increase in [Ca2+]i that returned to baseline. Again, exocytosis was observed during the time [Ca2+]i was elevated.
Fig. 1.

Comparison of 0.5-s and 2-s exposures to 60 mM K+ at 25ºC. Stimulations were paired with a 10-s interstimulus interval. Uppermost traces show bolus of 10 μM dopamine applied to electrode to show ejection profile. Middle traces show [Ca2+]i collected with fura-2; bottom traces show amperometric traces of release. Right and left panels show 0.5-s and 2-s stimulations, respectively.
To quantify the effects of consecutive 0.5-s stimuli on the occurrence of exocytotic events at 25ºC, we examined the ratio of the number of spikes evoked on a second K+ exposure (S2) to those evoked on the first (S1). If preceding stimuli do not affect subsequent exocytotic events, this spike number ratio should be unity. The ratios were examined with different intervals between the stimuli to allow increasing recovery times (Table 1). With a 10-s or 20-s interval between 0.5-s, 60 mM K+ exposures, the spike number ratios were approximately unity, but they exceeded unity with 30 or 40 s between stimuli. The ratio of the areas under the [Ca2+]i curves was also calculated as an S2/S1 ratio. With a 10-s interval between stimuli, the S2/S1 [Ca2+]i area ratio was 0.45 ± 0.06, but increased to a value between 0.7 and 0.8 with longer intervals between stimuli (Table 2). The average [Ca2+]i area ratio measured across the four intervals was 0.71 ± 0.04 and was significantly depressed from unity (p < 0.05).
Table 1.
Spike number ratios, S2/S1, are shown at 25ºC for 0.5-s at 10-, 20-, 30-, and 40-s intervals (n = 5, 7, 13, 12, respectively) and 2-s stimuli for 10-, 20-, 30-, and 40-s intervals (n = 5, 22, 23, 13, respectively). Spike number ratios are also shown at 37ºC for 0.5-s at 10-, 20-, 30-, and 40-s intervals (n = 10, 9, 16, 10, respectively) and 2-s stimuli for 10-, 20-, 30-, and 40-s intervals (n = 13, 11, 17, 10, respectively).
| Interstimulus interval (s) | 25ºC | 37ºC | ||
|---|---|---|---|---|
| 0.5-s stimulus | 2-s stimulus | 0.5-s stimulus | 2-s stimulus | |
| 10 | 1.1 ± 0.3 | 1.0 ± 0.1 | 2.0 ± 0.1 | 1.2 ± 0.2 |
| 20 | 1.0 ± 0.2 | 0.69 ± 0.10 | 1.6 ± 0.4 | 1.7 ± 0.2 |
| 30 | 1.8 ± 0.3 | 1.2 ± 0.1 | 1.6 ± 0.1 | 1.3 ± 0.2 |
| 40 | 1.3 ± 0.2 | 0.76 ± 0.10 | 1.7 ± 0.4 | 1.5 ± 0.2 |
Table 2.
[Ca2+]i area ratios, S2/S1, are shown at 25ºC for 0.5-s at 10-, 20-, 30-, and 40-s intervals (n = 5, 7, 9, 12, respectively) and 2-s stimuli for 10-, 20-, 30-, and 40-s intervals (n = 5, 13, 17, 14, respectively). [Ca2+]i area ratios are also shown at 37ºC for 0.5-s at 10-, 20-, 30-, and 40-s intervals (n = 6, 6, 6, 7, respectively) and 2-s stimuli for 10-, 20-, 30-, and 40-s intervals (n = 5 for all intervals).
| Interstimulus interval (s) | 25ºC | 37ºC | ||
|---|---|---|---|---|
| 0.5-s stimulus | 2-s stimulus | 0.5-s stimulus | 2-s stimulus | |
| 10 | 0.45 ± 0.06 | 0.18 ± 0.05 | 1.2 ± 0.1 | 0.82 ± 0.04 |
| 20 | 0.88 ± 0.03 | 0.57 ± 0.10 | 1.2 ± 0.1 | 0.65 ± 0.06 |
| 30 | 0.79 ± 0.07 | 0.51 ± 0.05 | 1.1 ± 0.1 | 0.61 ± 0.04 |
| 40 | 0.71 ± 0.08 | 0.47 ± 0.05 | 0.87 ± 0.05 | 0.52 ± 0.06 |
Similar experiments were done at 25ºC with 2-s pressure ejections of 60 mM K+. With the longer stimulus, the [Ca2+]i reached a maximum during the stimulus, evoking exocytosis, but again was slow to return to prestimulus values (see right panel of Figure 1). A second exposure 10 s later did not evoke much of a change in [Ca2+]i, but did evoke further exocytosis. Surprisingly, the number of spikes evoked on the first exposure with 2-s exposure to 60 mM K+ (21.5 ± 1.8) was not significantly different from that evoked with a 0.5-s pulse (15.1 ± 2.5; p ≥ 0.05; Figure 2A). However, the [Ca2+]i area with the 2-s pressure ejection (44.95 ± 6.29 arbitrary units) was significantly greater than with a 0.5-s exposure (25.08 ± 1.54 arbitrary units; p < 0.05; Figure 2B).
Fig. 2.

Effects of stimulus time and temperature on vesicular release and Ca2+ influx. A, Number of spikes elicited using 0.5-s (n = 53) and 2-s stimuli (n = 42) at 25ºC were not statistically significant. There was a significant difference between the number of spikes elicited at the two stimuli at 37ºC (p < 0.05). Temperature did not show an effect on number of evoked spikes at a particular stimulus duration. B, Ca2+ dynamics in response to temperature and stimulus conditions. Ca2+ area during evoked Ca2+ influx using 0.5-s (n = 5) and 2-s stims (n = 7) at 25ºC was statistically significant (p < 0.05); 0.5-s stimuli (n = 25) and 2-s stimuli (n = 20) at 37ºC also evoked average Ca2+ areas that were significantly different (p < 0.05) Ca2+ dynamics are more sensitive to temperature and stimulus duration.
The spike number ratios for consecutive stimuli using the 2-s stimulus were also found to be variable and near unity, but no consistent trend with interval between the stimuli was seen (Table 1). The [Ca2+]i area ratio was very low with a 10-s interval between 2-s exposures to 60 mM K+, and it increased for longer intervals (Table 2). The average measured over the four intervals was 0.43 ± 0.05, which was significantly lower than unity (p < 0.05), again indicating insufficient time for recovery before the second stimulus. Clearly, the 2-s stimulus at 25ºC sufficiently perturbs the mechanisms that regulate [Ca2+]i such that the responses on the second exposure differ considerably from those on the first exposure.
Vesicular release and Ca2+ dynamics evoked by 0.5-s and 2-s stimuli at 37ºC
Identical experiments were done at 37ºC. At this temperature, the Ca2+ responses were more rapid (Figure 3). With a 0.5-s delivery of 60 mM K+, [Ca2+]i was maximal soon after the stimulus, and returned to near baseline within 10 s, and multiple exocytotic spikes were observed. With a 10-s interval between stimuli, the spike number ratio approached 2, and it was above 1.5 with longer intervals between stimuli (Table 1). These S2/S1 spike number ratios evoked during 0.5 s exposures were significantly higher than unity (p < 0.05) indicating facilitation of release on the second stimulation at 37ºC. The [Ca2+]i area ratios were above unity with a 10-s interval between stimuli, and remained high until the stimuli were separated by 40 s (Table 2).
Fig. 3.
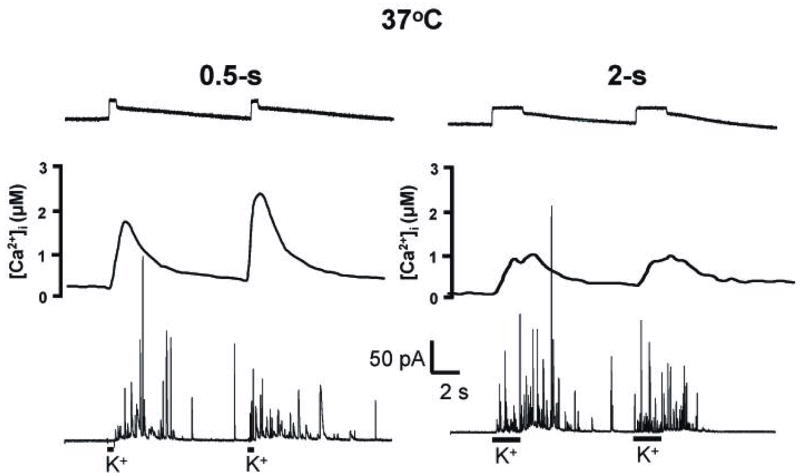
Comparison of 0.5-s and 2-s exposures to 60 mM K+ at 37ºC. Stimulations were paired with a 10-s interstimulus interval. Uppermost traces show bolus of 10 μM dopamine applied to electrode to show ejection profile. Middle traces show [Ca2+]i collected with fura-2; bottom traces show amperometric traces of release. Right and left panels show 0.5-s and 2-s stimulations, respectively.
At 37ºC, a 2-s stimulus of 60 mM K+ caused Ca2+ influx that was maximal during the ejection, and which decayed to base line within 10 s. In addition, there were significantly more spikes evoked with a 2-s bolus (25.5 ± 2.5) than with a 0.5-s bolus (14.2 ± 1.7; p < 0.05, Figure 2).
The spike number ratio was 1.2 with 10 s between 2-s ejections of 60 mM K+ at 37ºC (Table 1). With longer intervals between stimuli, the spike number ratio hovered between 1.3 and 1.7; these spike number ratios were significantly higher than unity, also indicating the presence of facilitation (p < 0.05). With all time intervals, the average [Ca2+]i area ratio with 2-s pressure ejections was less than unity (Table 2). The average ratio over the 10-, 20-, 30-, and 40-s intervals was 0.65 ± 0.01, which is significantly different from unity (p< 0.05). Thus, even at 37ºC, 2-s exposures to 60 mM K+ perturbed the cell sufficiently that [Ca2+]i could not achieve the initial evoked value on subsequent stimuli, at least with the inter-stimulus times investigated. Note that while there was little change in the number of spikes evoked at the two different temperatures, the [Ca2+]i area ratios were significantly smaller at 37ºC with both ejection durations (Figure 2). This is because [Ca2+]i is more rapidly restored to low levels at the higher temperature.
Facilitation and depression of vesicular release via the D1 receptor
To investigate whether the elevated number of spikes seen following a second exposure to 0.5-s exposure to 60 mM K+ at 37ºC was due to specific receptor interactions, we examined the effects of selective pharmacological agents. In the first experiment, the effect of different concentrations of the D1-receptor antagonist, SCH-23390, on the spike number ratio was examined with 0.5 s K+ pressure ejections that were 10 s apart. The average S2/S1 spike number ratio without drug for the 10-s interval yielded a mean of 2.2 ± 0.2. This facilitation decreased progressively with increasing concentrations of SCH-23390 (Figure 4). Facilitation was significantly decreased related to experiments without drug (p < 0.05) in the presence of SCH-23390 at concentrations of 10 and 100 μM.
Fig. 4.
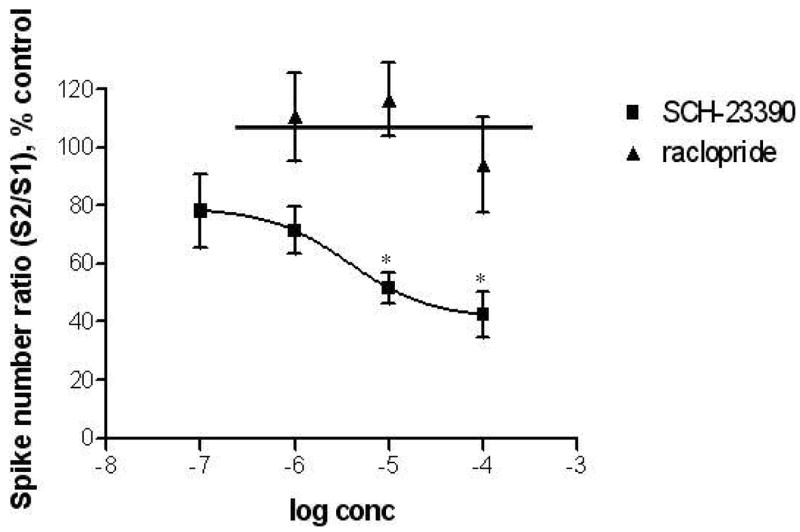
Dose response curve of D1 and D2 antagonists. Data was obtained using 0.5-s K+ stimuli at 37ºC with 10-s intervals. To investigate the presence of a D2-receptor mediated effect on facilitation, raclopride, a D2 antagonist, was incubated with cells at 1, 10, and 100 μM (n = 5, 10, 7, respectively). The S2/S1 responses were compared to control values measured at the same cell. Various concentrations of SCH-23390 (0.1, 1, 10, 100 μM; n = 8, 7, 7, 8, respectively) were also used to examine its effect on facilitation. Spike number ratios obtained at 10 and 100 μM SCH-23390 were significantly different from control values (p < 0.05).
To test whether D2 receptors were participating in this effect on release, the D2 antagonist, raclopride, was used in an analogous study at doses of 1 μM, 10 μM, and 100 μM (Figure 4). For this set of experiments, the average spike number ratio, S2/S1 without drug was 2.2 ± 0.3 with 0.5-s exposures 10 s apart. Changes in spike number ratios were not observed at any of the concentrations tested.
Next, the effect of a D1-receptor agonist was evaluated. In these experiments, the K+ stimuli were 30 s apart because, under control conditions (Table 1), facilitation was not as pronounced as with the shorter intervals. SKF-38393 (100 μM), a D1-receptor agonist, was introduced into the buffer 10 s after the first 0.5 s K+ pressure ejection. The spike number ratio was 2.5 ± 0.4 following the agonist compared to 1.2 ± 0.2 (p < 0.05) in the absence of the agonist (Figure 5). The facilitation caused by SKF-38393 was blocked when 10 μM of the D1-receptor antagonist, SCH-23390, was included in the buffer (p < 0.05; Figure 5).
Fig. 5.
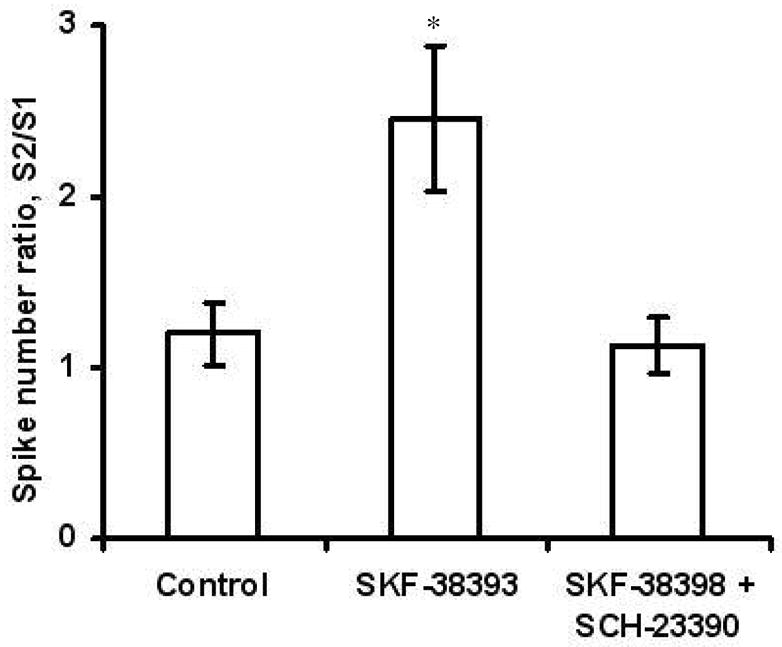
D1 receptor-mediated facilitation. Experiments were conducted with a 30-s interstimulus interval. Spike number ratios showed recovery in control cells (n = 9) and facilitation in cells incubated with 100 μM SKF-38393, a D1 agonist (n = 9). Co-incubation of 100 μM SKF-38393 and 10 μM SCH-23390 showed inhibition of the facilitation effect (n = 5). Incubation with SKF-38393 alone was significantly different from control value and co-incubation of SKF-38393 and SCH-23390 (p < 0.05).
Epinephrine-induced facilitation
To test whether the physiological ligand that is secreted by chromaffin cells would elicit facilitation, the experiment was repeated with a transient application of 50 μM epinephrine. Epinephrine was applied as a 3-s bolus 22 s after the first 0.5-s bolus of K+; 5 s after completion of the epinephrine application, the second 0.5-s bolus of K+ was applied to the cell. Epinephrine evoked facilitation in release as was seen with the D1 agonist, SKF-38393 (Figure 6). Transient application of 50 μM epinephrine to chromaffin cells yielded a spike number ratio of 2.7 ± 0.4 (Figure 6, inset) which was 265 ± 44 % of the control ratio that was obtained by pressure ejecting experimental buffer instead of epinephrine (1.1 ± 0.1; p < 0.05; Figure 6).
Fig. 6.
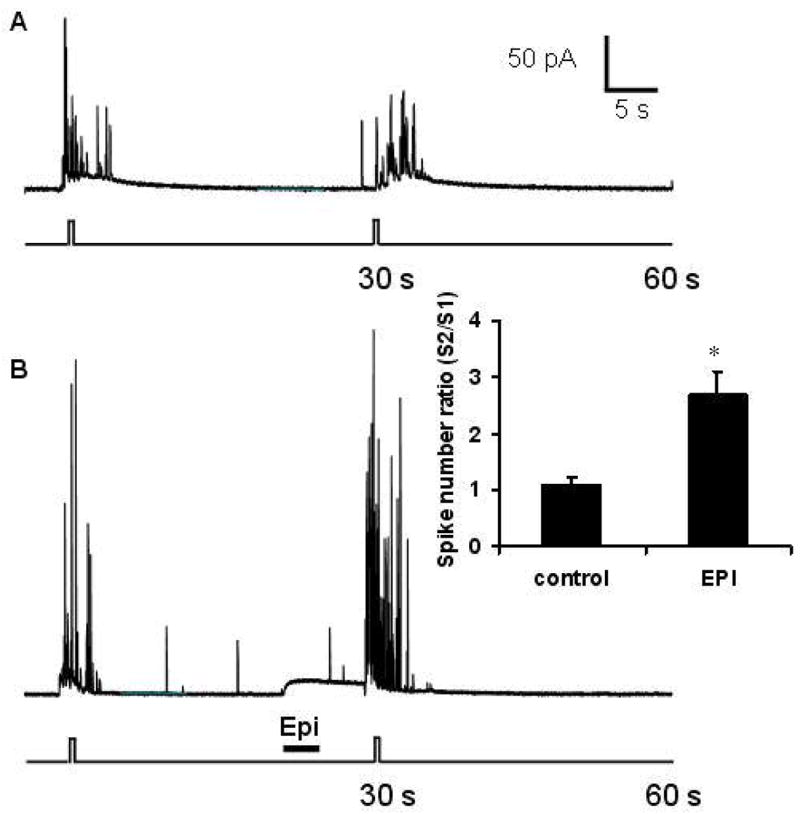
Epinephrine-induced facilitation. Experiments were conducted with a 30-s interstimulus interval. A, Control trace where experimental buffer was applied to the cell for 3-s records recovery of release upon the second stimulus, S2. B, Release was recorded with 3-s application of 50 μM epinephrine. S2 in these cells showed a greater number of elicited spikes. Note the baseline change due to detection of the pressure ejection of epinephrine. Inset shows average S2/S1 values for control cells and cells that received brief applications of epinephrine (n = 12). The values were shown to be significantly different (p < 0.05).
To test whether the effect of epinephrine was D1 receptor-mediated, two consecutive S2/S1 protocols were used. In the first, epinephrine was introduced 5 s before S2. Then, the experiment was repeated 5 min later, with or without a D1-receptor antagonist. The second epinephrine application in the absence of drug showed facilitation, but its S2/S1 spike number ratio was 79 ± 17 % of the first (Figure 7). This may indicate desensitization that is occurring at the receptor. However, in the presence of SCH-23390 (10 μM) the facilitation effect was blocked. The ratio of S2/S1 spike number ratios was 37.4 ± 8.7%, which was significantly different than the response in the absence of drug (p < 0.05; Figure 7).
Fig. 7.
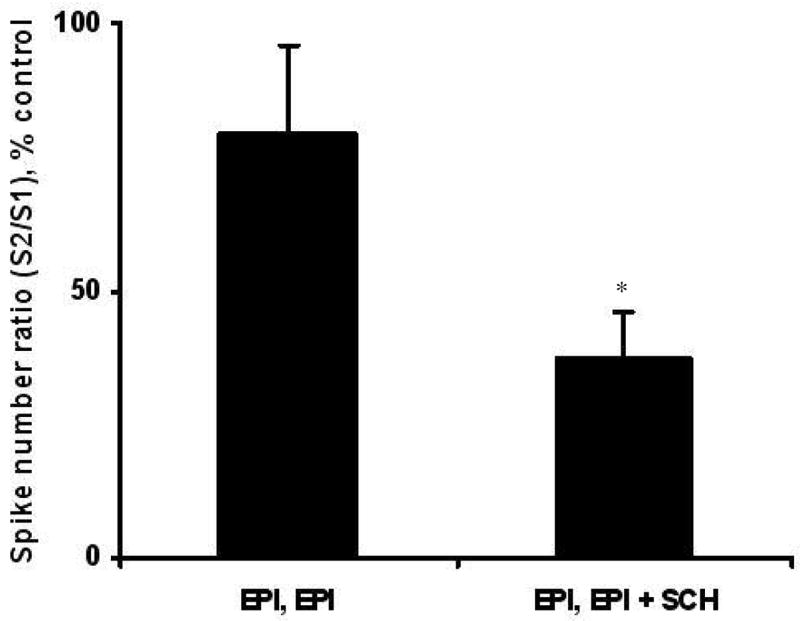
Epinephrine-induced facilitation is D1-modulated. Experiments were conducted with 30-s interstimulus interval. To investigate if the epinephrine-induced facilitation was mediated by a D1-like receptor, SCH-23390 was used to attempt to block the effect. A pair of S2/S1 stimuli were evaluated at each cell with 50 μM epinephrine applied during S2; after 5 min, a second set of identical paired stimuli was used with transient application with (n = 5) or without 10 μM SCH-23390 (n = 7). Values are shown as the percent of the initial S2/S1 spike number ratios.
Discussion
The results described here demonstrate that there is an autoreceptor on bovine chromaffin cells that facilitates release of epinephrine. Previous work has demonstrated the presence of dopaminergic D1 (8, 10) and D2 (9) receptors on bovine chromaffin cells. Consistent with those findings, chromaffin cells have been shown to express RNA for the D4 and D5 dopaminergic receptors (11). Additionally, it has been shown that D1 receptor activation (presumably through activation of the D5 receptor) causes facilitation of Ca2+ currents (10). Since it was postulated that the Ca2+ influx linked to the D1 receptor was sufficient to evoke further release (17), we tested for this effect. Clear evidence for facilitation of release was found at physiological temperature and with subsecond exposure to small amounts of K+. This facilitation was blocked by a D1 receptor antagonist in a dose-dependent manner. The facilitation could be mimicked by a D1 agonist as well as the endogenous secreted species, epinephrine. Thus, we have established a functional role for the previously identified D1 receptor on bovine chromaffin cells.
Crucial to our observation of facilitated autoreceptor-dependent release was the optimization of the stimulus conditions. Rapid application of secretagogues was enabled by the fabrication of ejection pipettes that permit delivery of a relatively sharp concentration profile even on a 0.5-s timescale (18). The pipette tips have a diameter of 10 μm or less, which minimizes leakage of the secretagogue from the tip. The paired stimuli approach that employed a 0.5-s or 2-s exposure to 60 mM K+ was sufficient to promote release of multiple vesicles with each exposure. Since the electrode samples release from approximately 6% of the cell surface, each 0.5-s exposure can be estimated from the data in Figure 2 to release approximately 250 vesicles. Because one estimate of the readily releasable pool is approximately 175 (19) of the total 10,000 vesicles in the cell (20), the majority of our release is believed to come from this compartment.
The spike number ratio was always unity or greater with the 0.5-s stimuli. In contrast, with 2-s stimuli, the spike number ratio was often lower than unity at 25ºC suggesting that the cell (or the vesicle pools and their associated release mechanisms) on the second stimulus was recovering from the first stimulus (21). Consistent with prior work, the number of spikes initially evoked by the same stimulus duration was similar at both temperatures (22, 23). The strongest evidence that the cell is dramatically perturbed with the 2-s stimuli comes from the [Ca2+]i area ratios. Only with 0.5-s stimuli at 37ºC were [Ca2+] area ratios near unity achieved. The cells were able to recover more effectively at 37ºC as the [Ca2+]i was restored to near resting levels even within a 10-s interstimulus interval. Intracellular Ca2+ levels are primarily maintained by uptake into mitochondria (24, 25), an energy dependent process that is more effective at physiological temperature (26). Thus, we used the 0.5-s stimulus at 37ºC to investigate further receptor-mediated facilitation of release. With short (10 s) intervals between stimuli under these conditions, the [Ca2+] area ratios exceeded unity, although the ratios were not as great as the facilitated Ca2+ currents reported by Artalejo and coworkers (27). As well as reporting on cytoplasmic Ca2+, fura-2 also buffers it and, because our measurements are of whole cell fluorescence, this average measure of Ca2+ is expected to be much lower than the enhanced Ca2+currents measured with patch clamp technology (28).
To test the hypothesis that facilitation of exocytosis is mediated by interaction of a released substance with a D1 dopamine receptor (17), the conditions that showed facilitation were repeated in the presence of SCH-23390, a D1 antagonist. While this agent blocked facilitated release in a dose-dependent manner, facilitated release was unaffected by the D2 antagonist, raclopride. The doses of SCH-23390 needed to inhibit facilitation are quite high. Two possibilities may contribute to this finding. First, the binding of SCH-23390 to sites on chromaffin cell is considerably weaker than to other tissues that have D1 receptor sites (29). Because of this, we are probably more correct to refer to this receptor as a “D1-like receptor.” Second, the assay used involves the competition between released epinephrine and the antagonist. Further evidence that a D1-like receptor is involved in facilitation was obtained with the D1 agonist, SKF-38393. This agent induced exocytotic facilitation in a 30-s paired pulse interval, a paradigm that normally shows limited facilitation.
Consistent with the concept that facilitation is autoreceptor-mediated, epinephrine induced robust facilitation of the number of vesicular release events at a modest concentration compared to the surface concentration upon its release (30). Desensitization appears to occur because the epinephrine-induced facilitation was somewhat diminished upon repeating the application (Figure 7). However, the facilitation induced by epinephrine appears receptor-mediated because it was inhibited by the D1-receptor antagonist, SCH-23390. Previously, it was proposed that intracellular or plasma dopamine was the primary species responsible for activating dopaminergic receptors on bovine chromaffin cells (9, 10). This work shows that released epinephrine could be the catecholamine activating the D1-like receptor. Indeed, epinephrine is well known to have a high affinity for the D1 receptor (IC50 = 68 nM) (31). Epinephrine is in abundance in this preparation since the majority of chromaffin cells package and release epinephrine; the ratio of epinephrine- to norepinephrine-containing cells is approximately 70%:30% (32). Moreover, the experiments performed in this work were conducted on the epinephrine-enriched fraction of cells obtained from the cell culture preparation. Therefore, the observed facilitation originates by binding of epinephrine to a functional D1-like autoreceptor.
Several earlier studies demonstrated a D2 receptor-mediated inhibition of catecholamine release (7–9). Furthermore, previous reports of D1 receptor-mediated secretion at chromaffin cells suggested a decrease in release (11). However, many of these were done at room temperature, a condition not explored here. Both D1 and D2 mediated-effects were ascribed to inhibition of sodium uptake that would cause an attenuated membrane depolarization (33). We attribute the difference in responses obtained here to differences in the time scale of secretagogue application. Prior studies of catecholamine release from bovine chromaffin cells used incubation times of minutes. In this work each secretagogue exposure was transient and attempted to mimic physiological conditions of a brief burst of action potentials. D1 receptor activation may have predominantly short-term effects, as indicated by our results, while the effect of D2 receptor activation may become predominant over longer time periods.
The D1 receptor-mediated facilitation that occurs in chromaffin cells points to a positive feedback cycle that operates with paired stimuli that are spaced closely together. Previously, using a paired stimuli approach, an overfilling of the releasable pool of vesicles was noted at relatively short interstimulus intervals (approximately 2–10 s) in bovine chromaffin cells (21). The autoreceptor that we have functionally characterized in this work may be the origin of this apparent overfilling. By activating facilitating Ca2+ currents, more Ca2+ is available to stimulate greater release. Autoreceptor-mediated positive feedback occurs in other systems as well. In addition to the positive feedback observed with application of insulin to β-pancreatic cells (5), another positive feedback regulator is gonadotropin-releasing hormone (GnRH). GnRH potentiates the pulsatile secretion of GnRH neurons in vitro (34). GnRH agonists were also shown to potentiate the pulsatile neuronal response while GnRH antagonists had an inhibitory effect. Moreover, oxytocin (OT) and its agonist enhance bursting activity in OT neurons while the OT antagonist inhibits this same response (35). In the chromaffin cell, which is part of the “fight-or-flight” mechanism, a positive feedback system is desirable. In this way, the body is flooded with adrenaline in order to be prepared for high stress situations. Closely spaced stimuli would signal to these cells the urgent need for the rapid release of large amounts of catecholamine into the bloodstream and initiate the positive feedback cycle necessary for an immediate response to the presented threat or stress.
Acknowledgments
The authors acknowledge the NIH for support of this work (NS 38879).
Abbreviations
- SCH-23390
1H-3-benzazepin-7-ol, 8-chloro-2,3,4,5-tetrahydro-3-methyl-5-phenyl-, (5R)
- SKF-38393
1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol
- GnRH
gonadotropin-releasing hormone
- HEPES
1-[4-(2-hydroxyethyl)-1-piperazinyl]ethane-2-sulfonic acid
- NMDA
N-Methyl-D-aspartic acid
- OT
oxytocin
References
- 1.Blier P, Pineyro G, el Mansari M, Bergeron R, de Montigny C. Role of somatodendritic 5-HT autoreceptors in modulating 5-HT neurotransmission. Annals of the New York Academy of Sciences. 1998;861:204–216. doi: 10.1111/j.1749-6632.1998.tb10192.x. [DOI] [PubMed] [Google Scholar]
- 2.Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Starke K. Presynaptic autoreceptors in the third decade: focus on alpha2-adrenoceptors. J Neurochem. 2001;78:685–693. doi: 10.1046/j.1471-4159.2001.00484.x. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci. 2006;26:406–410. doi: 10.1523/JNEUROSCI.4413-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aspinwall CA, Lakey JRT, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic beta cells. Journal of Biological Chemistry. 1999;274:6360–6365. doi: 10.1074/jbc.274.10.6360. [DOI] [PubMed] [Google Scholar]
- 6.Ueki K, Okada T, Hu J, Liew CW, Assmann A, Dahlgren GM, Peters JL, Shackman JG, Zhang M, Artner I, Satin LS, Stein R, Holzenberger M, Kennedy RT, Kahn CR, Kulkarni RN. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat Genet. 2006;38:583–588. doi: 10.1038/ng1787. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez MC, Artalejo AR, Montiel C, Hervas PP, Garcia AG. Characterization of a dopaminergic receptor that modulates adrenomedullary catecholamine release. Journal of Neurochemistry. 1986;47:382–388. doi: 10.1111/j.1471-4159.1986.tb04513.x. [DOI] [PubMed] [Google Scholar]
- 8.Artalejo AR, Garcia AG, Montiel C, Sanchez-Garcia P. A dopaminergic receptor modulates catecholamine release from the cat adrenal gland. Journal of Physiology. 1985;362:359–368. doi: 10.1113/jphysiol.1985.sp015683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bigornia L, Allen CN, Chung-Ren J, Lyon RA, Titeler M, Schneider AS. D2 dopamine receptors modulate calcium channel currents and catecholamine secretion in bovine adrenal chromaffin cells. Journal of Pharmacology and Experimental Therapeutics. 1990;252:586–592. [PubMed] [Google Scholar]
- 10.Artalejo CR, Ariano MA, Perlman RL, Fox AP. Activation of facilitation calcium channels in chromaffin cells by D1 dopamine receptors through a cAMP/protein kinase A-dependent mechanism. Nature. 1990;348:239–242. doi: 10.1038/348239a0. [DOI] [PubMed] [Google Scholar]
- 11.Dahmer MK, Senogles SE. Dopaminergic inhibition of catecholamine secretion from chromaffin cells: evidence that inhibition is mediated by D4 and D5 receptors. Journal of Neurochemistry. 1996;66:222–232. doi: 10.1046/j.1471-4159.1996.66010222.x. [DOI] [PubMed] [Google Scholar]
- 12.Leszczyszyn DJ, Jankowski JA, Viveros OH, Diliberto JEJ, Near JA, Wightman RM. Nicotinic receptor-mediated catecholamine secretion from individual chromaffin cells. Journal of Biological Chemistry. 1990;265:14736–14737. [PubMed] [Google Scholar]
- 13.Kawagoe KT, Zimmerman JB, Wightman RM. Principles of voltammetry and microelectrode surface states. Journal of Neuroscience Methods. 1993;48:225–240. doi: 10.1016/0165-0270(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 14.Bath BD, Michael DJ, Trafton BJ, Joseph JD, Runnels PL, Wightman RM. Subsecond adsorption and desorption of dopamine at carbon-fiber micro-electrodes. Analytical Chemistry. 2000;72:5994–6002. doi: 10.1021/ac000849y. [DOI] [PubMed] [Google Scholar]
- 15.Finnegan JM, Wightman RM. Correlation of real-time catecholamine release and cytosolic Ca2+ at single bovine chromaffin cells. Journal of Biological Chemistry. 1995;270:5353–5359. doi: 10.1074/jbc.270.10.5353. [DOI] [PubMed] [Google Scholar]
- 16.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 17.Artalejo CR, Dahmer MK, Perlman RL, Fox AP. Two types of Ca2+ currents are found in bovine chromaffin cells: facilitation is due to the recruitment of one type. J Physiol. 1991;432:681–707. doi: 10.1113/jphysiol.1991.sp018406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hochstetler SE, Puopolo M, Gustincich S, Raviola E, Wightman RM. Real-time amperometric measurements of zeptomole quantities of dopamine released from neurons. Anal Chem. 2000;72:489–496. doi: 10.1021/ac991119x. [DOI] [PubMed] [Google Scholar]
- 19.Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- 20.Ungar A, Phillips JH. Regulation of the adrenal medulla. Physiological Reviews. 1983;63:787–843. doi: 10.1152/physrev.1983.63.3.787. [DOI] [PubMed] [Google Scholar]
- 21.Dinkelacker V, Voets T, Neher E, Moser T. The readily releasable pool of vesicles in chromaffin cells is replenished in a temperature-dependent manner and transiently overfills at 37 degrees C. J Neurosci. 2000;20:8377–8383. doi: 10.1523/JNEUROSCI.20-22-08377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pihel K, Travis ER, Borges R, Wightman RM. Exocytotic Release from Individual Granules Exhibits Similar Properties at Mast and Chromaffin Cells. Biophysical Journal. 1996;71:1633–1640. doi: 10.1016/S0006-3495(96)79368-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker A, Glavinovic MI, Trifaro J. Temperature dependence of release of vesicular content in bovine chromaffin cells. Pflugers Arch. 1996;432:885–892. doi: 10.1007/s004240050212. [DOI] [PubMed] [Google Scholar]
- 24.Yang D-M, Kao L-S. Relative contribution of the Na+/Ca2+ exchanger, mitochondria and endoplasmic reticulum in the regulation of cytosolic Ca2+ and catecholamine secretion of bovine adrenal chromaffin cells. Journal of Neurochemistry. 2001;76:210–216. doi: 10.1046/j.1471-4159.2001.00055.x. [DOI] [PubMed] [Google Scholar]
- 25.Haynes CL, Buhler LA, Wightman RM. Vesicular Ca2+-induced secretion promoted by intracellular pH-gradient disruption. Biophysical Chemistry. 2006;123:20–24. doi: 10.1016/j.bpc.2006.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, Garcia-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- 27.Artalejo CR, Rossie S, Perlman RL, Fox AP. Voltage-dependent phosphorylation may recruit Ca2+ current facilitation in chromaffin cells. Nature. 1992;358:63–66. doi: 10.1038/358063a0. [DOI] [PubMed] [Google Scholar]
- 28.Klingauf J, Neher E. Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophys J. 1997;72:674–690. doi: 10.1016/s0006-3495(97)78704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahmer MK, Senogles SE. Atypical SCH23390 binding sites are present on bovine adrenal medullary membranes. Neurochem Res. 2000;25:321–326. doi: 10.1023/a:1007569518010. [DOI] [PubMed] [Google Scholar]
- 30.Finnegan JM, Pihel K, Cahill PS, Huang L, Zerby SE, Ewing AG, Kennedy RT, Wightman RM. Vesicular quantal size measured by amperometry at chromaffin, mast, pheochromocytoma, and pancreatic β-cells. Journal of Neurochemistry. 1996;66:1914–1923. doi: 10.1046/j.1471-4159.1996.66051914.x. [DOI] [PubMed] [Google Scholar]
- 31.Seeman P. Brain dopamine receptors. Pharmacological Reviews. 1980;32:229–313. [PubMed] [Google Scholar]
- 32.Moro MA, Lopez MG, Gandia L, Michelena P, Garcia AG. Separation and culture of living adrenaline- and noradrenaline-containing cells from bovine adrenal medullae. Analytical Biochemistry. 1990;185:243–248. doi: 10.1016/0003-2697(90)90287-j. [DOI] [PubMed] [Google Scholar]
- 33.Dahmer MK, Senogles SE. Differential inhibition of secretagogue-stimulated sodium uptake in adrenal chromaffin cells by activation of D4 and D5 dopamine receptors. Journal of Neurochemistry. 1996;67:1960–1964. doi: 10.1046/j.1471-4159.1996.67051960.x. [DOI] [PubMed] [Google Scholar]
- 34.Khadra A, Li Y-X. A model for the pulsatile secretion of gonadotropin-releasing hormone from synchronized hypothalamic neurons. Biophysical Journal. 2006 doi: 10.1529/biophysj.105.080630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jourdain P, Israel J-M, Dupouy B, Oliet SHR, Allard M, Vitiello S, Theodosis DT, Poulain DA. Evidence for a hypothalamic oxytocin-sensitive pattern-generating network governing oxytocin neurons in vitro. Journal of Neuroscience. 1998;18:6641–6649. doi: 10.1523/JNEUROSCI.18-17-06641.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]