

Abstract
We describe a combinatorial virtual screening approach of predicting high specificity heparin / heparan sulfate sequences using the well-studied antithrombin-heparin interaction as a test case. Heparan sulfate hexasaccharides were simulated in the ‘average backbone’ conformation, wherein the inter-glycosidic bond angles were held constant at the mean of known solution values, irrespective of their sequence. Molecular docking utilized GOLD with restrained inter-glycosidic torsions and intra-ring conformations, but flexible substituents at the 2-, 3-, and 6-positions and explicit incorporation of conformational variability of the iduronate residues. The approach reproduces the binding geometry of the sequence-specific heparin pentasaccharide to within 2.5 Å. Screening of a combinatorial virtual library of 6,859 heparin hexasaccharides using a dual filter strategy, in which predicted antithrombin affinity was the first filter and self-consistency of docking was the second, resulted in only 10 sequences. Of these, 9 were found to bind antithrombin in a manner identical to the natural pentasaccharide, while a novel hexasaccharide bound the inhibitor in a unique but dramatically different geometry and orientation. This work presents the first approach on combinatorial library screening for heparin/heparan sulfate GAGs to determine high specificity sequences and opens up huge opportunities to investigate numerous other physiologically relevant GAG-protein interactions.
Introduction
Glycosaminoglycans heparin and heparan sulfate play diverse roles in a number of physiological and pathological processes including coagulation,1,2 angiogenesis,3,4 immune response5,6 and viral infection.7,8 These functions originate from their interaction with numerous proteins, which include serpins, antithrombin and heparin cofactor II,1,2,9 coagulation proteinases,10 fibroblast growth factors and their receptors,3,4,11 platelet factor-4,5,12 interleukins,8,13 and viral cell envelope glycoproteins.7 It is commonly assumed that these roles arise from an optimal combination of specificity and affinity, yet, except for the interaction with antithrombin (AT),1,2,14,15 and perhaps with glycoprotein D of herpes simplex virus-1,16,17 specificity of heparin and heparan sulfate (HS)# interactions remain ill-defined and poorly understood.
A major reason for the ill-defined structure-activity relationships in these heparin/HS interactions is the phenomenal structural diversity of heparin/HS glycosaminoglycans (GAGs), and its concomitant difficulties, especially synthesis, purification, and structure identification.18 Both heparin and heparan sulfate are complex, highly anionic polysaccharides composed of alternating 1→4-linked glucosamine and uronic acid residues, which are variously modified through sulfation, acetylation and epimerization.19 These modifications can produce 48 different building blocks, or disaccharides, of which 23 have been found to date. Further, the iduronic acid residue (IdoAp) can exist in multiple conformations, especially 1C4 and 2SO for internal locations, that can inter-convert relatively easily.20,21 Thus, combination of structural and conformational variability generates millions of sequences, of which few are expected to recognize a target protein. This expectation is further borne out by the growing evidence that the heparan sulfate biosynthetic apparatus has considerable specificity and organization.22
On the protein front, considerable effort has been made in trying to deduce consensus binding sequences that recognize heparin structures. Whereas Cardin and Weintraub suggested linear consensus sequences with specific repeat pattern to be important for heparin binding, others have suggested a spatial distance relationship.23-25 Although it is clear that arginine and lysine residues lining protein surfaces dominate heparin-binding sites, their optimal 3D orientation that generates high specificity and affinity remains unclear. The situation is further compounded because not all arginine and lysine interactions with sulfate and carboxylate groups of heparin/HS are identical. While heparin interaction with antithrombin and basic fibroblast growth factor involves ∼40 and 30% ionic binding energy,26,27 respectively, that with thrombin involves ∼80%.28 Thus, identifying the precise heparin-binding site on proteins, although expected to be straightforward, has not been an easy task.
In the past decade fifteen heparin-protein co-crystal or NMR structures have become available. These include complexes with thrombin,29 growth factor and growth factor receptors,30-32 annexin V,33 heparan sulfate 3-O-sulfotransferase,34 RANTES35 and antithrombin. Of these, antithrombin is the most studied protein with five co-crystal structures detailing the interaction of a five-residue sequence with the inhibitor in either a binary or a ternary complex.36-39 Yet, these represent a small fraction of the vast number of physiologically active complexes. Further, except for with antithrombin, the ‘heparin’ structure reported in these complexes is the fully sulfated saccharide, the most common structure found in heparin, which may not be the optimal sequence.
In view of the limited structural knowledge on GAG-protein interactions and the phenomenal structural diversity of HS, computational docking approaches represent a powerful means of assessing binding affinity and specificity. In fact, the computational literature is replete with numerous GAG studies, yet no approach has been devised that predicts high specificity sequences. Modeling GAGs is challenging because of their high negative charge density, which induces recognition of practically any collection of positively charged residues, and their surface-exposed, shallow binding sites on proteins.29-35 With respect to antithrombin in particular, two computational attempts have been made to understand heparin binding with mixed results.40,41 Both Grootenhuis and van Boeckel40 and Bitomsky and Wade41 attempted to derive the heparin pentasaccharide binding site on antithrombin using molecular dynamics and docking, but their modeled geometry turned up to be significantly different from the co-crystal structure. Similar challenging results were noted with our small, sulfated molecules, which were rationally designed to recognize the pentasaccharide-binding site in antithrombin but were found to preferentially interact with the extended-heparin binding site.42-44 Thus, we reasoned that a robust docking protocol for GAGs in general, and heparin/HS in particular, would be very useful for understanding specificity of their interactions.
This paper describes our novel approach of predicting high specificity GAG sequences with the well-studied antithrombin-heparin interaction as a test case. Our approach is based on a two-filter strategy, with the first step involving an affinity filter followed by a geometry convergence filter using the genetic algorithm-based docking tool, GOLD.45 The two-filter strategy rapidly sorted a combinatorial virtual library of nearly 7,000 heparin hexasaccharides into specific and non-specific sequences, thus suggesting its potential use for identifying ‘needle(s) in a haystack’.
Computational Methods
Software/Hardware
SYBYL 6.9.2 (Tripos Associates, St. Louis, MO) was used for molecular visualization, minimization and for adding hydrogens to protein structures from the Protein DataBank. All modeling was performed on MIPS R16K or R14K IRIX 6.5-based SGI Tezro and Fuel graphical workstations. GOLD, version 2.2,45 was used for docking experiments. Combinatorial GAG structures were built in an automated manner using in-house SPL (SYBYL Programming Language) scripts.
Energy Minimizations
Energy minimization of modeled structures was performed to optimize the geometric conformation of GAG and AT. Except where stated, energy minimization was performed using the Tripos forcefield with Gasteiger-Hückel charges, a fixed dielectric constant of 80 and a non-bonded cutoff radius of 8 Å. Minimization was carried out for a maximum of 5,000 iterations subject to a termination gradient of 0.05 kcal/(mol·Å).
Protein Co-ordinates
The coordinates for the activated form of AT were extracted from the crystal structure of the ternary AT-pentasaccharide-thrombin complex (PDB entry 1TB6).38 Hydrogen atoms were added in SYBYL 6.9.2 and the structure minimized with fixed heavy-atom co-ordinates using the Tripos forcefield for 1,000 iterations subject to a termination gradient of 0.05 kcal/(mol·Å). Single-point mutants of AT, including K114Q, K125M and R129H, were generated using the residue mutation protocol in SYBYL. The side-chains of the mutated residues were optimized through energy minimization in which the all other side chains were held rigid.
Co-ordinates for synthetic pentasaccharide H5CRYS
The 1TB6 antithrombin-heparin-thrombin ternary crystal structure38 has a heparin chain containing a synthetic pentasaccharide sequence, H5CRYS, which is a variant of the natural pentasaccharide DEFGH (Fig. 1). This synthetic pentasaccharide H5CRYS has six O-sulfate groups in residues D (position 6), F (positions 2,3 and 6) and H (positions 2 and 6), has glucose residues, instead of glucosamines, and has all available -OH groups in residues D (positions 2 and 3), E (positions 2 and 3), G (positions 2 and 3) and H (positions 1 and 3) protected in the form of -OCH3 groups (Fig. 1). In SYBYL, the atom type of sulfur and oxygen atoms in -SO3 groups was modified to S.o2 and O.co2, respectively, and the bond type between these atoms was modified to aromatic bond. Hydrogen atoms, absent in the PDB structure, were added in SYBYL and the resultant structure minimized to optimize geometry of hydrogen atoms only (no change in non-H atoms).
Figure 1. Structure of natural pentasaccharide H5 and its derivative H5CRYS extracted from crystal structure study of Li et al38 and used in docking studies.

D, E, F, G, and H refer to historical label of residues from the non-reducing end. Sulfate groups in H5 highlighted as filled ovals  are critical for high-affinity interaction with antithrombin.
are critical for high-affinity interaction with antithrombin.
Natural Pentasaccharide DEFGH co-ordinates
The sequence-specific pentasaccharide H5, or sequence DEFGH (Fig. 1), was modeled by introducing the necessary changes to the 1TB6 pentasaccharide. The methylated anomeric reducing end was retained to simulate the environment of the +1 residue. Several research groups have reported ϕH/ψH inter-glycosidic torsion angles for HL-GAGs in the free and bound conformations.20,36,46-49 These torsion angles fall within a relatively narrow range and suggest that they remain fairly constant irrespective of a change in substitution pattern around the inter-glycosidic bond. Thus, we utilized an average value of bond torsion for each inter-glycosidic linkage, which was the mean of the two extremes reported in literature. The natural sequence H5 was minimized at the average ϕH/ψH values, subject to a restraining force constant of 0.01 kcal·mol-1·deg-2. The final ϕH/ψH values, following minimization, deviated by not more than ±7° from the initial values. The minimized structure retained the initial 2SO IdoAp conformation and was used for docking studies.
Co-ordinates for Variant Pentasaccharides
The ‘average-backbone’ natural pentasaccharide DEFGH structure was used as a template for the generation of H5 variants. These variants include truncated forms, tetrasaccharides H5-H and H5-D, trisaccharides H5-GH and H5-DE, and functional group variants H5+3SH, H5+3SE, H5+3SG, H5-6SD, H5-3SF and H5-2SH (Fig. 2). For each of these structural variants, following appropriate structural modifications in the natural pentasaccharide sequence, minimization was performed with constraints that retain the average ϕH/ψH values. In addition to these structural variants of the natural sequence, a DEFGH sequence was also prepared in which the iduronic acid residue G was in the 1C4 conformation, labeled as H5/G*, rather than the 2SO form. The 1C4 IdoAp ring co-ordinates were taken from the crystal structure of FGF2-GAG (PDB entry 1BFC)30 and the structure minimized as described above.
Figure 2. Structures of H5 and its truncated variants studied for docking.

D, E, F, G, and H refer to residue labels. Variations in structure (V, W, X, Y, Z, and Z’) to give variants with either one more sulfate group (H5+3SH, H5+3SE and H5+3SG) or one less sulfate group (H5-6SD, H5-3SF, H5-2SH) are highlighted as filled ovals  . Truncated pentasaccharides H5-H, H5-GH, H5-D, and H5-DE refer to one or two residue deletion variants from the either end.
. Truncated pentasaccharides H5-H, H5-GH, H5-D, and H5-DE refer to one or two residue deletion variants from the either end.
Co-ordinates for HL-GAG Virtual Library
The co-ordinates for the HL-GAG sequences of the virtual HS hexasaccharide library were generated with a series of SPL scripts and a set of nineteen HS disaccharide building blocks (Fig. 3). Although the number of possible HL-GAG UAp(1→4)GlcNp disaccharides is 48, only 23 have been experimentally observed. Based on sequence-specific natural pentasaccharide sequence, we restricted our library to include GlcAp sequences that have GlcNp3S and IdoAp sequences that do not contain GlcNp3S. Because IdoAp residues in heparin can exist either in the 2SO or 1C4 conformations, each IdoAp residue was modeled explicitly in these two different states. Thus, our virtual library consisted of 16 IdoAp- and 3 GlcAp-containing disaccharides generating a total of 19 building blocks (Fig. 3).
Figure 3. Disaccharide sequences used to build virtual library of 6,859 hexasaccharides.

Although theoretically 48 disaccharides are possible, 23 have been experimentally observed.22 Based on the sequence of H5, our library included GlcAp sequences with GlcNp3S and IdoAp sequences without GlcNp3S, with IdoAp in either 2SO or 1C4 conformation. These variations made up 19 disaccharide sequences to produce 19×19×19 = 6,859 hexasaccharides.
GlcAp- and 2SO-IdoAp-containing disaccharides were generated using the EF and GH residues from the 1TB6 co-crystal structure as template, while the template for the 1C4-IdoAp disaccharides was obtained from the 1BFC structure.30 Appropriate side-chain modifications were made to generate the 19 building blocks. Each disaccharide was minimized at the average ϕH/ψH value subject to a restraining force constant of 0.01 kcal·mol-1·deg-2. The 19 disaccharides were then used to build a combinatorial HS hexasaccharide library using an SPL script, following which each sequence was minimized with 10,000 iterations as described above in an automated manner. Thus, the HS combinatorial library contained 19×19×19 = 6,859 hexasaccharide sequences.
Docking of HL-GAG Sequences
Docking of saccharide ligands onto the activated form of antithrombin was performed with GOLD v.2.2.45 The binding site in antithrombin was comprised of all atoms within 16 Å from the Cζ atom of Phe121 in the D helix. This definition of the binding site covers all important known heparin-binding residues including Lys11, Arg13, Arg46, Arg47, Trp49, Lys114, Phe121, Lys125, Arg129, and Arg132.1,2,36 GOLD is a “soft docking” method that implicitly handles local protein flexibility by allowing a small degree of interpenetration, or van der Waals overlap, of ligand and protein atoms. GOLD also optimizes the positions of hydrogen-bond donating atoms on Ser, Thr, Tyr and, most importantly, Lys residues as part of the docking process. Whereas all saccharide bonds were constrained for the rigid body docking experiment, only the inter-glycosidic bonds were constrained when docking structures with the average torsion angles. Unless specified otherwise, default parameters were employed during the GOLD docking runs.
For the native H5 sequence, and its truncated and variant forms (Fig. 2), docking was performed using no speed-up and a genetic algorithmic search with 100,000 iterations for each of 10 runs. In this search, GOLD starts with a population of 100 arbitrarily docked ligand orientations, evaluates them using a scoring function (the GA “fitness” function) and improves their average “fitness” by an iterative optimization procedure that is biased towards high scores. As the initial population is selected at random, several such GA runs are required to more reliably predict correct bound conformations. In this study 10 GA runs were performed with the GOLD score as the “fitness” function. Collectively, these 10 GA runs will be referred to as one docking experiment. In addition, to enhance speed, the GA was set to pre-terminate if the top two ranked solutions were within 2.5 Å RMSD. Docking experiments were performed in triplicate to ensure reproducibility and to reduce false positives. The top two solutions of each docking experiment were considered for further analysis. Thus, a typical triplicate docking experiment would yield a minimum of 6 solutions.
In contrast, when docking the HS combinatorial library made from 19 disaccharides (Fig. 3), a two-step docking protocol was utilized. The first step consisted of screening all possible sequences using 10,000 GA iterations (7-8x speed up) and GOLD score evaluation of only the top-ranked solution. This step identified the most promising sequences (∼ top 1%) that have a relatively high GOLD score. The second step consisted of docking these most interesting sequences according to the protocol described above for the natural pentasaccharide DEFGH and its variants.
Docking was driven by the GOLD scoring function. Although this scoring function correlates with the observed free energy of binding, a modified form of the scoring function has been found to be more reliable.50 This modified GOLD score, which utilizes hydrogen-bonding and van der Waals interactions (Equation 1), was used to rank the final docked solutions.
| [1] |
where HBEXT and VDWEXT are the “external” (nonbonded interactions taking place between the ligand and receptor) hydrogen bonding and van der Waals terms, respectively.
Results and Discussion
GOLD predicts the binding geometry of synthetic pentasaccharide H5CRYS to within 0.6 Å
The ability to predict the conformation of GAG in the receptor-bound state was studied using a rigid body docking procedure in GOLD. The rigid body docking procedure reduces the search space significantly and allows an assessment of GOLD’s fitness function, which comprises of four energy terms - external and internal hydrogen bonding and van der Waals energy. Docking was evolved for 100,000 iterations in each of the runs to ensure sampling of most of the conformational space available in the pre-defined binding site on antithrombin. Further, to ensure greater confidence in docked geometries, the process was repeated three times.
Successful docking is typically evaluated by RMS difference between the docked solution(s) and the X-ray geometry, if available. While a RMSD value of less than 1.0 Å would indicate near identity of the two geometries, values between 1. 0 and 3.0 Å have been utilized by many researchers to indicate correctly docked geometries of drug-like small molecules (MW <500).51-53 Considering that our penta- and hexasaccharides are significantly bigger (MW ∼2,000) and the resolution of antithrombin-heparin-thrombin crystal structure was only 2.5 Å,38 we employed a 2.5 Å RMSD criterion.
When the synthetic pentasaccharide H5CRYS was docked onto antithrombin, the GA search pre-terminated in all three independent runs, each with a high GOLD score of greater than 100. All 6 solutions were identical with an inter-solution RMSD of less than 0.6 Å, suggesting a strong preference for this orientation in the binding site (not shown). Comparison of these solutions with the H5CRYS structure present in the crystal suggests identical orientation and binding site. The maximal RMSD between the docked solutions and the crystal structure was also 0.6 Å. In addition, interaction at the atomic level for the docked and co-crystal complexes were found to be essentially identical, suggesting that the rigid-body docking procedure can reliably identify the binding geometry and the binding site of the most active pentasaccharide sequence.
Prediction of binding geometry of natural pentasaccharide sequence DEFGH with an “average GAG backbone” conformation to within 2.0 Å
X-ray fiber diffraction studies indicate that GAGs adopt a helical structure,54 which has now been confirmed through several crystal structures.29-35 For heparan sulfate-like GAGs (HL-GAGs), the helix has a 21 symmetry with inter-glycosidic torsion angles (ϕH [HUA-CUA-O-CGlcNp] and ψH [CUA-O-CGlcNp-HGlcNp]) constrained to generate the helix. These conclusions are borne out in a number of NMR and molecular dynamics studies on heparin20,47,55 and heparin oligosaccharides,46,48,49 which show that the ϕH/ψH angles vary within a relatively narrow range in solution. Thus, heparin appears to maintain a 2-fold helix irrespective of its sequence and substitution patterns. Additionally, crystal structures of pentasaccharide-antithrombin36-39 and hexasaccharide-FGF30 complexes demonstrate that only small changes in ϕH/ψH angles occur on protein binding, a result also observed with NMR studies of pentasaccharides bound to AT.46,49 Compiling these results indicates that ϕH and ψH values vary from 35° to 61° and 6° to 25°, respectively, for the UAp(1→4)GlcNp inter-glycosidic linkage, while ϕ varies between -68° and -9°H and ψH varies between -60° and -1° for GlcNp(1→4)UAp linkage. These variations represent a maximal change of only 19 to 59°, or less than ±30°, a relatively small deviation considering the large number of possibilities for a structurally diverse molecule. Thus, we reasoned that HL-GAGs, irrespective of their sequence and substitution pattern, could be simulated by an “average” structure, wherein the ϕH/ψH inter-glycosidic bond angles are held constant at the mean of known solution values. This implies that docking of any heparin/HS sequence with an “average backbone” model should reliably simulate the physiological GAG binding structure.
To test our hypothesis, the natural sequence-specific pentasaccharide H5 (Fig. 1) was modeled in which inter-glycosidic torsions were held at the ‘average backbone’ values, rather than set to those obtained from the crystal structure of H5CRYS. Docking was performed in which the inter-glycosidic torsions and intra-ring conformations were held constant, while substituents present at the 2-, 3-, and 6-positions were allowed conformational flexibility. All three docking experiments terminated rapidly indicating a strong preference for one type of binding geometry. Figure 4 shows a comparison of the 6 GOLD solutions with the geometry present in the crystal structure. All 6 solutions predict an essentially identical binding geometry, an observation typically not found in such statistical docking studies. Small differences are found for the orientation of the terminal -OSO3- and -OCH3 groups, especially in the G and H residues. All 6 solutions were within 2.5 Å RMSD (Fig. 4) of each other and comparison of these structures with H5CRYS present in the crystal structure indicates nearly identical orientation, conformational state and interactions (Fig. 4).
Figure 4. Comparison of GOLD predicted binding geometry of natural pentasaccharide H5 having ‘average backbone’ with that of H5CRYS determined in the crystal structure.
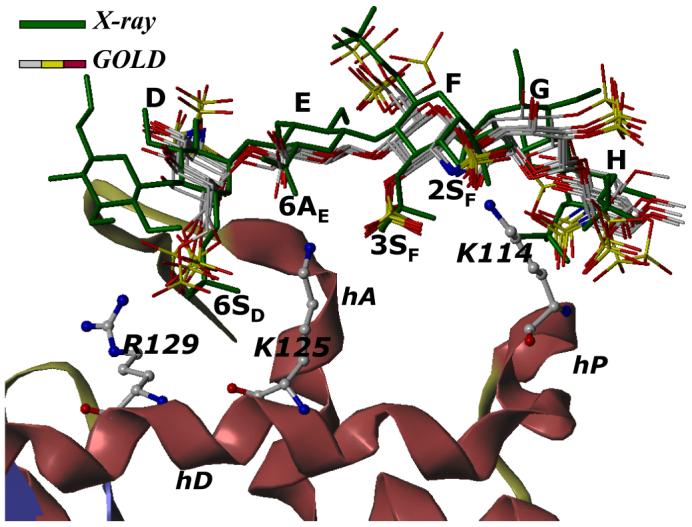
H5 with average inter-glycosidic bond parameters (‘the average backbone’), was docked onto antithrombin. Structurally H5 differs from H5CRYS (see Fig. 1). An overlay of 6 solutions from three independent docking runs shows high consistency in the predicted binding geometry, which matches the crystal structure geometry with an RMSD less than 2.5 Å. Structure in green is the crystal structure geometry, while those in atom-type color (red, yellow, grey and blue) are 6 docking solutions. Note the identical orientation of key groups, 2- and 3-OSO3- of residue F (2SF and 3SF), 6-COO- of residue E (6AE) and 6-OSO3- of residue D (6SD). Helices A, D and P of antithrombin (in ribbon diagram) are indicated as hA, hD and hP, while D, E, F, G, and H labels correspond to residues of the pentasaccharide. K114, K125 and R129 are shown in ball and stick representation. See text for details.
Analysis of the GOLD solutions at the atomic level facilitates identification of groups important for specificity of interaction. For example, the average RMSDs for the sulfate groups at positions 2 and 3 of residue F were found to be 0.2±0.1 and 0.3±0.2 Å indicating a virtually invariant conformation in all solutions. Likewise, the average RMSDs for sulfates at 6 and 2 positions of residues D and H, respectively, were found to be 1.1±0.5 and 1.0±0.8 Å suggesting reduced tendency for alternative rotational states. In contrast, the average RMSDs of sulfates at the 6-, 2- and 6-positions of residues F, D and H were 2.4±1.2, 2.1±1.2 and 1.6±0.9 Å, respectively, indicating greater conformational variability. Our docking results suggest that four sulfate groups, at positions 6-, 2- and 3-, and 2- of residues D, F and H, respectively, have minimal conformational variability in the binding site. Thus, these sulfate groups organized in a specific three-dimensional orientation afford the specificity of interaction, a conclusion proposed earlier on the basis of a large number of structure-activity studies (see Fig. 1).2,14,15,26,61
Prediction of binding geometry with a flexible H5 fails to give reasonable solutions
The above docking procedure utilized constrained intra- and inter-glycosidic conformations to maintain solution-state structure in the protein-bound state. To test whether full conformational flexibility during docking would result in better predictability, docking of H5 was performed under conditions wherein the HL-GAG backbone and saccharide conformations were unrestrained. Striking differences were noted in these docking studies. None of the three docking experiments converged suggesting a lack of preference for a defined binding geometry (not shown). In addition, the final six solutions display completely random profiles. Further, the pyranose rings in the docked solutions were found to exhibit conformations not found in solution. To test whether partial flexibility is tolerated, H5 was docked with unrestrained inter-glycosidic torsions, but constrained intra-ring torsions matching geometries in their bound state. The RMSD matrix showed an inconsistent set of 6 solutions corresponding to several different binding geometries (not shown). Only 1 of these geometries was within 2.5 Å RMSD of H5CRYS. These experiments suggest that determining binding geometries with a fully flexible ligand, though highly desirable, is difficult. These results demonstrate that the imposition of an average backbone constraint significantly increases the chances of arriving at the true binding geometry by reducing the GA conformational search space.
The docking protocol sorts pentasaccharides based on specificity of interaction with antithrombin
The natural pentasaccharide sequence H5 is a rare sequence in heparin in which the EF unit (Fig. 1) with the presence of the 3-O-sulfate group is both unique and interesting. Studies with a large number of H5 derivatives have led to the understanding that other sulfate groups present in the pentasaccharide sequence, e.g., the N-sulfate of D and 6-O-sulfate of F, are not important.56 Interestingly, certain analogs with sulfation level higher than H5, e.g., H5+3SE and H5+3SG (Fig. 2), were less active than H5, in sharp contrast to the expectation that greater sulfation level, especially 3-O-sulfation, would induce higher antithrombin-affinity.56 At the same time, our studies suggested that 3-O-sulfation of residue H was not detrimental to biological activity.26,57
An obvious explanation for the changes in antithrombin affinity is the loss or gain of favorable or unfavorable interactions. Yet, this explanation suggests that individual interactions affect the changes independently. To gain a more fundamental understanding, we studied the docking of several H5 variants including H5-3SF, H5-6SD, and H5-2SH (Fig. 2), each lacking a sulfate at position 3 in F, position 6 in D, and position 2 in H, respectively, onto antithrombin. In addition, H5+3SE and H5+3SG, described above, and a heparin hexasaccharide with common repeat sequence GH (IdoAp2S-GlcNp2S6S) were also studied. Finally, a H5 sequence with IdoAp residue locked in the unfavorable 1C4 conformation (H5/G*) was studied to understand the importance of conformational flexibility in this residue.
These H5 analogs were prepared in silico, minimized at the average GAG ϕH /ψH values, and docked using the protocol described above for H5. Figure 5 shows the RMSD between the solutions obtained in three docking experiments for each of the H5 variants. Whereas for H5, all 6 solutions are essentially identical with RMSD less than 2.5 Å (Fig. 5A), none of the variants display this behavior. For example, H5-3SF, lacking the critical 3-O-sulfate group, displays only two RMSDs within 2.5 Å (Fig. 5B), while H5+3SE, possessing an additional 3-O-sulfate in residue E, displays only one (Fig. 5C). Likewise, the number of docked solutions that are within 2.5 Å RMSD of each other is 8, 7, and 4 for H5-2SH, H5-6SD, and (GH)3, respectively, out of the possible 15, while that for H5+3SG and H5/G* are 6 and 1, respectively (not shown). Thus, whereas 100% of solutions are found within 2.5 Å of each other for H5, only 53%, 47%, and 13% are found for H5-2SH, H5-6SD, and H5-3SF, respectively. For H5+3SE and H5+3SG, variants with additional 3-O-sulfation, this percentage is 13 and 40%, while for 1C4 conformationally locked H5 (H5/G*) it is only 6.7%.
Figure 5. Specificity of GAG sequence for antithrombin.
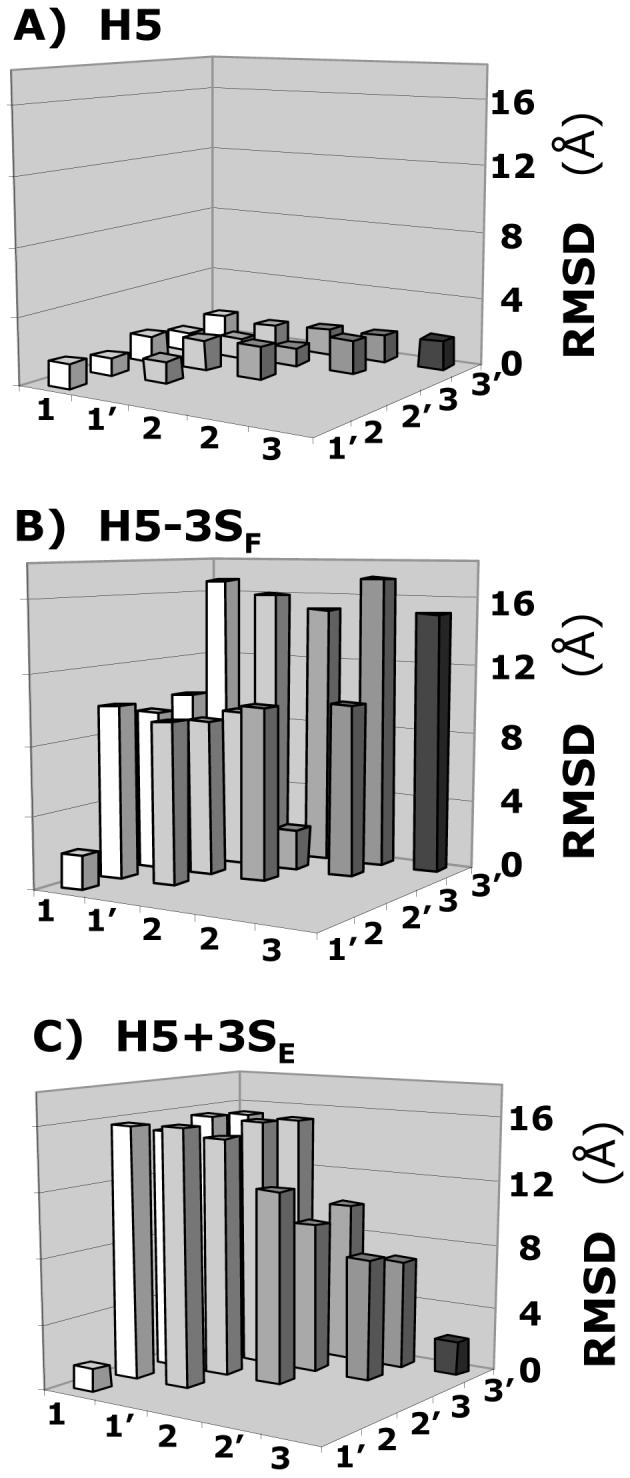
Variation in the RMSD between six solutions (1, 1′, 2, 2′, 3, and 3′) obtained from three independent docking experiments of heparin pentasaccharide H5 variants, containing either one additional sulfate group at 3-position of residue E or lacking a sulfate at the 3-position of residue F, binding to antithrombin. Whereas for H5 (A) the RMSD was less than 2.0 Å between each solution, for H5-3SF (B) and H5+3SE (C), a majority of RMSD values were much greater (5-17 Å). Other pentasaccharide variants were also studied. See text for details.
The above results indicate that self-consistency of docking geometries is sensitive to sulfate group distribution and pentasaccharide topology. Alternatively, the loss or gain of sulfate groups or 1C4 iduronate conformation gives rise to alternate binding modes significantly differing from each other and from that of the natural high-affinity sequence H5. A closer inspection of the binding geometries suggests that many of these variant sequences favor a completely different pattern of interactions. These altered preferences, especially a multitude of them, are likely to be non-productive, generating significant non-specific binding, thus resulting in a loss of binding affinity. Thus, these docking experiments indicate that the ‘self-consistency’ of the RMSD matrix of top-ranked solutions is a sensitive measure of GAG specificity.
GOLD scores correlate with antithrombin affinity
Implicit in our docking protocol is the assumption that GOLD scores correlate with protein binding affinity. To establish that this is indeed true, we studied the docking of pentasaccharide variants with antithrombin for which the binding affinity had been measured under standardized conditions.2,26,57-60 These include H5, H5+3SH, H5-GH, H5-H, H5-D, and H5-DE binding to wild-type antithrombin2,26,57 and H5 binding to R129H, K125M and K114Q antithrombin mutants.58-60 These 9 complexes represent a range of ∼12 kcal/mol in free energy of binding, a wide range spanning very strong to very weak interactions. Docking was performed in a manner identical to that for native pentasaccharide H5 and the top solution analyzed for binding energy correlation. Figure 6 shows a profile of modified GOLD score versus the observed free energy of binding under physiological conditions. For the 9 complexes studied, the modified GOLD score increased linearly from ∼95 to ∼150 giving a slope of 4.2 GOLD score per kcal/mol and an intercept of 78.9.
Figure 6. Correlation of GOLD score with antithrombin binding affinity.

Modified GOLD score (See Methods) determined following 100,000 iterations and multiple docking runs linearly increases with the observed antithrombin binding affinity under physiological conditions. Only those antithrombin complexes for which binding affinities were measured under essentially equivalent conditions are used for analysis. Labels refer to pentasaccharide or its variant, identified in Figures 1 and 2, interaction with wild-type antithrombin. H5/K114, H5/K125 and H5/R129 refer to natural pentasaccharide H5 interaction with mutant antithrombins.58-60 Solid line represents linear regression line with slope of 4.2 GOLD score per kcal/mol and an intercept of 78.9 GOLD score. See text for details.
The reasonable linear correlation between modified GOLD score and ΔG°OBS validates the docking protocol. Yet, the slope of 4.2 GOLD score units per kcal/mol reflects a less-than-optimal sensitivity to structural change, especially considering that multiple docking geometries were not considered in the analysis. In general, predicting protein-binding affinities of GAGs has been difficult because of the exposed nature of the binding site and the unquantified interaction parameters of sulfate groups. In addition, knowledge-based or empirical scoring functions do not take into account entropic contributions to ΔG°, which arise from the release of Na+ and H2O species from antithrombin surface following saccharide binding and are the dominant factors in the stabilization of the complex. Finally, the scoring function cannot compute the energy required for conformational flips of iduronate residues as well as the energy consumed to induce an allosteric, two-step binding interaction necessary for antithrombin.26,57 In light of these unknowns, the correlation observed in Figure 6 is reasonably good, especially considering that the affinity varies from the high μM to the low nM.
Identification of high-affinity, high specificity sequences through a combinatorial virtual screening approach
Although a large number of proteins interact with HL-GAGs in our body, little information is available on sequences that recognize these proteins. This is also the status for other GAGs, such as dermatan sulfate, chondroitin sulfate and keratan sulfate. In addition, knowledge regarding the specificity of these interactions is completely lacking. We reasoned that our protocol, which combines two functions - affinity in the form of GOLD score and specificity in the form of consistency of docking - represents a powerful tool to deduce high-affinity GAG sequences that bind proteins with high specificity.
To test this hypothesis, we generated a combinatorial library of heparin hexasaccharides for docking onto antithrombin. The library was generated from a set of 19 disaccharides (Fig. 3), which included all monosaccharides found in nature, except for the rare free glucosamines.22 The library considered the conformational flexibility of IdoAp residues in an explicit manner through inclusion of both 1C4 and 2SO conformations. Thus, the combinatorial library consisted of 6,859 unique heparin hexasaccharides built with the ‘average backbone’ geometry in an automated manner.
A biphasic docking protocol was used for screening this library. The first phase evaluated the binding of each hexasaccharide onto antithrombin using 10,000 iterations (7-8x speed-up), instead of 100,000 (no speed-up), followed by GOLD score screening. The histogram of GOLD score frequency obtained from this analysis suggests an approximately Gaussian distribution of sequences centered between 40 and 55 GOLD score units with the highest at 115 (Fig. 7). A majority of hexasaccharides, 6,352 sequences or 92% of the total, have a GOLD score of 75 or less suggesting that these sequences minimally or do not bind antithrombin. Nearly 6.4% or 439 sequences have a GOLD score between 75 and 100. This score corresponds to μM to mM antithrombin affinity, most probably arising from non-specific, non-productive interactions.
Figure 7. Histogram of number of HS hexasaccharide sequences for every 5 unit change in GOLD score.

Modified GOLD score was calculated for all 6,859 hexasaccharides docked onto antithrombin following the first phase of combinatorial library screening. Inset shows an expanded portion of the histogram in the range 95-120 GOLD score. See text for details.
Less than 1% of the sequences or 28 sequences, each with a GOLD score greater than 100 were identified as ‘hits’ or high-affinity sequences. Of these 28 sequences, 23 were strikingly similar in possessing three sulfate groups, 6SD, 3SF, and 2SF. In addition, these 23 sequences had glucuronic acid residue in the ‘E’ position. This similarity is particularly significant considering that 513 sequences of the 6,859 hexasaccharides, i.e., ∼7%, contain these structural features. In other words, the ‘DEF’ motif has been enriched from ∼7% to ∼82% through the affinity filter.
The 28 ‘hits’ identified in the affinity screen were then subjected to specificity analysis through a ‘consistency test’ with 100,000 iterations over triplicate docking runs. This step involved greater evolution of docking geometries, whereby significantly more conformational space around the binding site is searched multiple times. Of the 28 high-affinity sequences, 10 docked to consistently give a single geometry with an RMSD of less than 2.5 Å among the top six solutions (Fig. 8). The other 18 sequences did not dock consistently 100% of the time. The self-consistent binding geometry for each of the 10 sequences indicates a high specificity interaction. Of the 10 sequences, 9 bind antithrombin in manner identical to the natural pentasaccharide H5, while one hexasaccharide interacts with a significantly different geometry and orientation (Fig. 8).
Figure 8. Finding a needle in a haystack.

A) An overlay of final 10 hexasaccharide sequences obtained after second phase of combinatorial library screening. Structure in green is the H5CRYS, while those in atom-type color are 9 sequences with nearly identical binding orientation and geometry. Sequence in purple color was found to bind antithrombin reproducibly with high specificity and affinity but dramatically different orientation. Labels 2SF, 3SF, 6AE and 6SD represent sulfate or carboxylate groups at the 2- and 3-position of residue F, 6-position of residue E and 6-positon of residue D. B) Symbolic representation of the high-affinity, high-specificity hexasaccharide structures shown above. The hexasaccharide library sequence runs {UAp(1→4)GlcNp(1→4)}3, where UA is either IdoAp (shaded hexagon) or GlcAp (shaded square). Sulfated substitution at 2-, 3- or 6-positions of either UAp or GlcNp is indicated with a line (—), while acetate substitution at the 2-positon of GlcNp is indicated with a line-dot (—·). Iduronic acid residues in 2SO conformation are shown as fully shaded hexagons, while those in 1C4 conformation are shown as half-filled hexagons. See text for details. See Supplementary Information for a MOL2 file of all the combinatorial sequences.
A closer look at these 9 sequences shows that 8 contain the →4) GlcNp6S (1→4) GlcAp (1→4) GlcNp2S3S (1→ structure that matches key structural features within DEF necessary for high-affinity interaction with antithrombin (Fig. 1).2,26,57 The last sequence in this category also has this structure, except for the absence of 6SD. Structural differences within DEF in these eight sequences include the presence of either an acetyl or a sulfate group at the 2 position of residue D and the presence or absence of a sulfate at 6-position of residue F. Our virtual screening shows that an IdoAp residue, either with or without a sulfate in 2-position, is consistently present at the G location in these 9 sequences (Fig. 8). Alternatively, GlcAp residue is forbidden in this position. Further, the IdoAp residue overwhelmingly prefers 2SO conformation over 1C4 (∼89%) suggesting that the self-consistency filter is greatly sensitive to the local topology of the GAG helix. Finally, location H with GlcNp residue tolerates considerable variations, e.g., sulfation and acetylation in the 6- and 2-positions assuming that the high-affinity DEF sequence is present.
The GOLD scores for the 9 hexasaccharide sequences ranged from 109.5 to 127.5 suggesting each of these sequences to be potent antithrombin ligands. Yet, despite having all the key structural features these are not as potent as sequence-specific pentasaccharide H5, which shows a GOLD score of 140. Comparison of the docking geometry of H5 with that of the hexasaccharide sequences reveals a lateral displacement of 0.3-0.5 Å in all solutions irrespective of substitution pattern. This effect is most noted on G and H residues resulting in weaker interactions at the reducing end of the sequence. Although the small difference in binding geometry between pentasaccharide H5 and hexasaccharides remains unconfirmed, previous biochemical experiments have suggested a lateral movement as chain length increases.61
Identification of an unusual high-affinity, high-specificity sequence
Our combinatorial virtual screening approach results in one hexasaccharide sequence that binds antithrombin in an unusual, unexpected manner. This sequence, IdoAp2S (1→4) GlcNp2Ac (1→4) IdoAp2S (1→4) GlcNp2Ac (1→4) IdoAp (1→4) GlcNp2S6S (Fig. 8), does not have the critical DEF scaffold, is much less sulfated than H5, and has all three IdoAp residues in 1C4 conformation. The sequence binds antithrombin reproducibly in triplicate docking experiments with a GOLD score of 109.0 indicating a high-affinity, high specificity interaction. The binding geometry is unique but distinct from H5 and other sequences identified above (Fig. 8), specifically the sequence occupies part of both the pentasaccharide-binding site and the extended-heparin binding site in antithrombin.2 Significant binding energy originates from hydrogen bonding with amino acids Arg13, Arg46, Arg47, Lys125 and Arg129, in which the sulfate groups at the 2-position of the GlcNp and IdoAp residues make multi-valent interactions. At the same time, greater than 50% of its binding energy, as predicted by the modified GOLD score, originates from van der Waals interactions. Thus, despite a lower than normal sulfation level, a fortuitous combination of sequence and conformational features introduces high antithrombin affinity in this sequence.
It is difficult to predict whether this sequence will be physiologically active. The observation that less sulfated GAG sequence can demonstrate high antithrombin specificity in silico is exciting. Assuming that specific interactions of H5 are responsible for the allosteric conformational change in antithrombin, the dramatically different binding geometry suggests reduced likelihood of an agonist activity in this unusual sequence. At the same time, the high affinity of the sequence suggests an antagonistic activity may be expected.
Conclusion and Significance
Our interest in heparin/HS stems from our work on rational design of antithrombin-based anticoagulants.2,42-44,62 Despite being the anticoagulant of choice for the past 8 decades, heparin-based therapy is still beset with several adverse effects, principal being an enhanced risk toward bleeding. A large body of structure-activity studies spanning more than two decades resulted in several potent pentasaccharides, of which fondaparinux was introduced in the clinic in late 2001.14,15,63 Yet, these pentasaccharides, and also several low-molecular-weight heparins introduced in mid-1990s,62 are essentially ‘heparin’ and structurally represent a small advance.
Non-heparin anticoagulants with high specificity for antithrombin may afford a solution to current adverse effects of heparin therapy. Yet, designing non-heparin, organic activator(s) of antithrombin is challenging, especially due to the non-availability of techniques that address specificity of binding.43 Previous two simulations of heparin pentasaccharide binding to antithrombin were successful in identifying only a few of the critical interactions.40,41 A major reason for this is the difficult simulation of GAG interactions originating from the primary sequence complexity of GAG polymers, the inadequate parameterization of sulfate groups, and the shallow, exposed and highly flexible nature of GAG binding pockets.
Most docking approaches to date focus primarily on the affinity of interaction, and minimally on the specificity of interaction.64 Predicting affinity accurately of protein-ligand complexes is in its infancy, especially due to the poorly defined contribution of water molecules in the process. Further, affinity alone is not a powerful tool in predicting potent GAG sequences because of poor sensitivity to structural change, as the current study (see Fig. 6) and work elsewhere demonstrate.42 Instead of affinity, our genetic algorithm-based approach places emphasis on specificity of interaction. This approach affords screening of a large amount of conformational space that ensures elimination of a number of false positives. Thus, specificity is perhaps a more critical filter that offers significantly high reliability.
Our work represents the first approach on combinatorial library screening for heparin/HS GAGs. The phenomenal structural diversity of these GAGs is a challenge to medicinal chemists and represents a frontier extremely difficult to traverse through traditional structure-activity studies.63 More specifically, it is impossible to ever isolate all the sequences so as to fully delineate their interaction with proteins. This work demonstrates that library screening is feasible for heparin/HS oligosaccharides, especially if a high-resolution crystal structure of the protein is available. Further, the approach is geared toward predicting high specificity sequences. Thus, it is expected to be especially useful for heparin binding proteins, e.g., heparin co-factor II, protein C inhibitor and growth factors, for which specificity features remain poorly defined. In addition, for many other proteins, including glycoprotein D of herpes simplex virus-1 and protease nexin 1, information regarding heparin-binding site may also become feasible.
Our dual filter strategy - affinity and specificity - utilized fairly stringent criteria for selection. While the affinity filter selected the upper 20% of sequences, the specificity filter was set to select only those sequences that satisfy self-consistency 100% of the time. In combination, a high overall enrichment was realized (∼660-fold). If one considers only those hexasaccharide sequences that possess IdoAp (1→4) GlcNp (1→4) GlcAp (1→4) GlcNp (1→4) IdoAp (1→4) GlcNp structure, which is the base sequence present in the final 9 hexasaccharides, even then the enrichment remains substantial (∼77-fold). It was possible to use high filtering stringency because antithrombin-heparin interaction is biochemically a well-studied system. However, the stringent criteria may eliminate potentially useful information, especially for less well-understood systems.
Our combinatorial screening approach relies on a fundamental hypothesis. The combinatorial library was generated based on the ‘average backbone’ hypothesis in which uniformity of inter-glycosidic torsion angles, irrespective of sequence and intra-ring conformational variability, is assumed.46-49 For heparin/HS, this hypothesis appears to hold true for all known cases, although only a fraction of possible homogeneous sequences have been studied in solution. For GAGs other than HS, such as dermatan sulfate and chondroitin sulfate that involve mixed inter-glycosidic linkages, the approach needs rigorous testing.
Another approximation that our work relies upon is the rigid framework of the protein binding site during docking, despite the highly flexible nature of arginine and lysine side chains. The heparin-binding site in antithrombin is an engineering marvel. It possesses an exquisite asymmetrical arrangement of its positively charged residues, which are involved in the formation of intricate networks of charges.58-60,65,66 Such networks, possibly of salt bridge-type or hydrogen-bonding-type, likely engineer some ‘rigidity’ in the binding site, thus supporting our rigid binding site approximation. For other proteins such an approximation may not work. In such cases, a more rigorous virtual screening approach would be to combinatorially search the best combination of arginine and lysine side chain torsions, instead of the torsions frozen from the crystal structure. Such an exhaustive exercise is expected to be computationally time and cost expensive.
Although our approach was designed to identify individual GAG sequences with high specificity, it facilitates the extraction of a ‘pharmacophore’, key interactions that drive binding specificity. A map of RMSD of individual atoms, backbone as well as functional groups, can be readily created from the combinatorial library screening experiments to identify those locations with minimal variation, e.g., 3SF, 2SF, and 6SD in DEF, which define the pharmacophore. In contrast, domains with higher RMSD, and therefore possessing significant movement, define locations in which structural modification can be introduced to design new ligands. Thus, this approach is also likely to be useful for designing therapeutically useful molecules.
In conclusion, the work describes identification of high-affinity high specificity heparin/HS sequences that bind antithrombin utilizing a combinatorial virtual library screening approach. The approach relies on a dual filter strategy involving affinity and specificity filters and is based on average heparin/HS backbone hypothesis. The approach uses genetic algorithm based docking and scoring protocol. The dual filter strategy, if found useful for other heparin/HS systems, is likely to be extremely useful in the design of pharmaceutically useful agents.
Supplementary Material
Acknowledgements
We thank Professors Richard B. Westkaemper and Glen E. Kellogg of Virginia Commonwealth University for two GOLD licenses and critical comments on the manuscript, respectively. We also thank Cambridge Crystallographic Data Centre (CCDC) User Support for helpful technical support. This work was supported by the National Heart, Lung and Blood Institute (RO1 HL069975 and R41 HL081972) and the American Heart Association National Center (EIA 0640053N).
Footnotes
- AT
- antithrombin
- DEFGH
- sequence specific heparin pentasaccharide
- DS
- dermatan sulfate
- GAG
- glycosaminoglycan
- GlcAp
- glucuronic acid residue
- GlcNp
- glucosamine residue
- HCII
- heparin cofactor II
- HS
- heparan sulfate
- HL-GAG
- heparan sulfate-like glycosaminoglycans
- IdoAp
- iduronic acid residue
- PDB
- Brookhaven Protein Data Bank
- RMSD
- root mean square difference
- serpin
- serine protease inhibitor
- SPL
- SYBYL Programming Language
- UAp
- uronic acid residue
References
- 1.Pike RN, Buckle AM, Le Bonniec BF, Church FC. Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. 2005;272:4842–4851. doi: 10.1111/j.1742-4658.2005.04880.x. [DOI] [PubMed] [Google Scholar]
- 2.Desai UR. Antithrombin activation and designing novel heparin mimics. In: Garg HG, Linhardt RJ, Hales CA, editors. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier; NY: 2005. pp. 483–513. [Google Scholar]
- 3.Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Rops AL, van der Vlag J, Lensen JF, Wijnhoven TJ, van den Heuvel LP, van Kuppevelt TH, Berden JH. Heparan sulfate proteoglycans in glomerular inflammation. Kidney Int. 2004;65:768–785. doi: 10.1111/j.1523-1755.2004.00451.x. [DOI] [PubMed] [Google Scholar]
- 5.Kelton JG. The pathophysiology of heparin-induced thrombocytopenia: biological basis for treatment. Chest. 2005;127:9S–20S. doi: 10.1378/chest.127.2_suppl.9S. [DOI] [PubMed] [Google Scholar]
- 6.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nature Rev. Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Thorpe SC. Cell surface heparan sulfate and its roles in assisting viral infections. Med. Res. Rev. 2002;22:1–25. doi: 10.1002/med.1026. [DOI] [PubMed] [Google Scholar]
- 8.Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem. Rev. 2005;105:2745–2764. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- 9.Gettins PG. Serpin structure, mechanism, and function. Chem. Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 10.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Herault JP, Herbert JM, Bjork I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb. Haemost. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 11.Nakato H, Kimata K. Heparan sulfate fine structure and specificity of proteoglycan functions. Biochim. Biophys. Acta. 2002;1573:312–318. doi: 10.1016/s0304-4165(02)00398-7. [DOI] [PubMed] [Google Scholar]
- 12.Maccarana M, Lindahl U. Mode of interaction between platelet factor 4 and heparin. Glycobiology. 1993;3:271–277. doi: 10.1093/glycob/3.3.271. [DOI] [PubMed] [Google Scholar]
- 13.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Atha DH, Stephens AW, Rimon A, Rosenberg RD. Sequence variation in heparin octasaccharides with high affinity from antithrombin III. Biochemistry. 1984;23:5801–5812. doi: 10.1021/bi00319a020. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl U, Thunberg L, Backström G, Riesenfeld J, Nordling K, Björk I. Extension and structural variability of the antithrombin-binding sequence in heparin. J. Biol. Chem. 1984;259:12368–12376. [PubMed] [Google Scholar]
- 16.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Shriver Z, Pope RM, Thorp SC, Duncan MB, Copeland RJ, Raska CS, Yoshida K, Eisenberg RJ, Cohen G, Linhardt RJ, Sasisekharan R. Characterization of a heparan sulfate octasaccharide that binds to herpes simplex virus type 1 glycoprotein D. J. Biol. Chem. 2002;277:33456–33467. doi: 10.1074/jbc.M202034200. [DOI] [PubMed] [Google Scholar]
- 18.Keiser N, Venkataraman G, Shriver Z, Sasisekharan R. Direct isolation and sequencing of specific protein-binding glycosaminoglycans. Nature Med. 2001;7:123–128. doi: 10.1038/83263. [DOI] [PubMed] [Google Scholar]
- 19.Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat. Prod. Rep. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 20.Mulloy B, Forster MJ. Conformation and dynamics of heparin and heparan sulfate. Glycobiology. 2000;10:1147–1156. doi: 10.1093/glycob/10.11.1147. [DOI] [PubMed] [Google Scholar]
- 21.Ferro DR, Provasoli A, Ragazzi M, Casu B, Torri G, Bossennec V, Perly B, Sinay P, Petitou M, Choay J. Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 1990;195:157–167. doi: 10.1016/0008-6215(90)84164-p. [DOI] [PubMed] [Google Scholar]
- 22.Esko JD, Selleck SB. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 23.Cardin AD, Weintraub HJR. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 24.Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J. Biol. Chem. 1993;268:19228–19231. [PubMed] [Google Scholar]
- 25.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. BioEssays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 26.Desai UR, Petitou M, Björk I, Olson ST. Mechanism of heparin activation of antithrombin. Role of individual residues of the pentasaccharide activating sequence in the recognition of native and activated states of antithrombin. J. Biol. Chem. 1998;273:7478–7487. doi: 10.1074/jbc.273.13.7478. [DOI] [PubMed] [Google Scholar]
- 27.Thompson LD, Pantoliano MW, Springer BA. Energetic characterization of the basic fibroblast growth factor-heparin interaction: Identification of the heparin binding domain. Biochemistry. 1994;33:3831–3840. doi: 10.1021/bi00179a006. [DOI] [PubMed] [Google Scholar]
- 28.Olson ST, Halvorson HR, Björk I. Quantitative characterization of the thrombin-heparin interaction. Discrimination between specific and nonspecific binding models. J. Biol. Chem. 1991;266:6342–6352. [PubMed] [Google Scholar]
- 29.Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J. Biol. Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- 30.Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 31.Pellegrini L, Burke DF, von Delft F, Mulloy B, Blundell TL. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature. 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- 32.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 33.Capila I, Hernaiz MJ, Mo YD, Mealy TR, Campos B, Dedman JR, Linhardt RJ, Seaton BA. Annexin V-heparin oligosaccharide complex suggests heparan sulfate--mediated assembly on cell surfaces. Structure. 2001;9:57–64. doi: 10.1016/s0969-2126(00)00549-9. [DOI] [PubMed] [Google Scholar]
- 34.Moon AF, Edavettal SC, Krahn JM, Munoz EM, Negishi M, Linhardt RJ, Liu J, Pedersen LC. Structural analysis of the sulfotransferase (3-O-sulfotransferase isoform 3) involved in the biosynthesis of an entry receptor for herpes simplex virus 1. J. Biol. Chem. 2004;279:45185–45193. doi: 10.1074/jbc.M405013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw JP, Johnson Z, Borlat F, Zwahlen C, Kungl A, Roulin K, Harrenga A, Wells TNC, Proudfoot AEI. The X-ray structure of RANTES:heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure (Cambridge) 2004;12:2081–2093. doi: 10.1016/j.str.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Jin L, Abrahams J-P, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc. Nat. Acad. Sci. USA. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson DJD, Huntington JA. Crystal structure of antithrombin in a heparin-bound intermediate state. Biochemistry. 2003;42:8712–8719. doi: 10.1021/bi034524y. [DOI] [PubMed] [Google Scholar]
- 38.Li W, Johnson DJ, Esmon CT, Huntington JA. Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Biol. 2004;11:857–862. doi: 10.1038/nsmb811. [DOI] [PubMed] [Google Scholar]
- 39.Dementiev A, Petitou M, Herbert JM, Gettins PG. The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nat. Struct. Mol. Biol. 2004;11:863–866. doi: 10.1038/nsmb810. [DOI] [PubMed] [Google Scholar]
- 40.Grootenhuis PDJ, van Boeckel CAA. Constructing a molecular model of the interaction between antithrombin III and a potent heparin analogue. J. Am. Chem. Soc. 1991;113:2743–2747. [Google Scholar]
- 41.Bitomsky W, Wade RC. Docking of glycosaminoglycans to heparin-binding proteins: Validation for aFGF, bFGF, and antithrombin and application to IL-8. J. Am. Chem. Soc. 1999;121:3004–3013. [Google Scholar]
- 42.Gunnarsson GT, Desai UR. Designing small, non-sugar activators of antithrombin using hydropathic interaction analyses. J. Med. Chem. 2002;45:1233–1243. doi: 10.1021/jm020012q. [DOI] [PubMed] [Google Scholar]
- 43.Gunnarsson GT, Desai UR. Interaction of sulfated flavanoids with antithrombin: Lessons on the design of organic activators. J. Med. Chem. 2002;45:4460–4470. doi: 10.1021/jm020132y. [DOI] [PubMed] [Google Scholar]
- 44.Gunnarsson GT, Desai UR. Hydropathic interaction analyses of small organic activators binding to antithrombin. Bioorg. Med. Chem. 2004;12:633–640. doi: 10.1016/j.bmc.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 45.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 46.Ragazzi M, Ferro DR, Perly B, Sinay P, Petitou M, Choay J. Conformation of the pentasaccharide corresponding to the binding site of heparin for antithrombin III. Carbohydr. Res. 1990;195:169–185. doi: 10.1016/0008-6215(90)84165-q. [DOI] [PubMed] [Google Scholar]
- 47.Mulloy B, Forster MJ, Jones C, Drake AF, Johnson EA, Davies DB. The effect of variation of substitution on the solution conformation of heparin: a spectroscopic and molecular modelling study. Carbohydr. Res. 1994;255:1–26. doi: 10.1016/s0008-6215(00)90968-6. [DOI] [PubMed] [Google Scholar]
- 48.Mikhailov D, Linhardt RJ, Mayo KH. NMR solution conformation of heparin-derived hexasaccharide. Biochem. J. 1997;328:51–61. doi: 10.1042/bj3280051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hricovini M, Guerrini M, Bisio A, Torri G, Petitou M, Casu B. Conformation of heparin pentasaccharide bound to antithrombin III. Biochem. J. 2001;359:265–272. doi: 10.1042/0264-6021:3590265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Personal communication fromBattle G. User Support of Cambridge Crystallographic Data Centre, UK. Also see http://www.ccdc.cam.ac.uk/products/life_sciences/gold/faqs/scientific_faq.php#bindingaffinity.
- 51.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 52.Erickson JA, Jalaie M, Robertson DH, Lewis RA, Vieth M. Lessons in molecular recognition: the effects of ligand and protein flexibility on molecular docking accuracy. J. Med. Chem. 2004;47:45–55. doi: 10.1021/jm030209y. [DOI] [PubMed] [Google Scholar]
- 53.Kroemer RT, Vulpetti A, McDonald JJ, Rohrer DC, Trosset JY, Giordanetto F, Cotesta S, McMartin C, Kihlen M, Stouten PF. Assessment of docking poses: interactions-based accuracy classification (IBAC) versus crystal structure deviations. J. Chem. Inf. Comput. Sci. 2004;44:871–881. doi: 10.1021/ci049970m. [DOI] [PubMed] [Google Scholar]
- 54.Atkins ED, Nieduszynski IA. Crystalline structure of heparin. Adv. Exp. Med. Biol. 1975;52:19–37. doi: 10.1007/978-1-4684-0946-8_2. [DOI] [PubMed] [Google Scholar]
- 55.Mulloy B, Forster MJ, Jones C, Davies DB. NMR and molecular-modeling studies of the solution conformation of heparin. Biochem. J. 1993;293:849–858. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Boeckel CAA, Petitou M. The unique antithrombin III binding domain of heparin: A lead to new synthetic antithrombotics. Angew. Chem. Intl. Ed. Engl. 1993;32:1671–1818. [Google Scholar]
- 57.Desai UR, Petitou M, Björk I, Olson ST. Mechanism of heparin activation of antithrombin: Evidence for an induced-fit model of allosteric activation involving two interaction subsites. Biochemistry. 1998;37:13033–13041. doi: 10.1021/bi981426h. [DOI] [PubMed] [Google Scholar]
- 58.Desai UR, Swanson RS, Bock SC, Björk I, Olson ST. The role of arginine 129 in heparin binding and activation of antithrombin. J. Biol. Chem. 2000;275:18976–18984. doi: 10.1074/jbc.M001340200. [DOI] [PubMed] [Google Scholar]
- 59.Arocas V, Bock SC, Raja S, Olson ST, Björk I. Lysine 114 of antithrombin is of crucial importance for the affinity and kinetics of heparin pentasaccharide binding. J. Biol. Chem. 2001;276:43809–43817. doi: 10.1074/jbc.M105294200. [DOI] [PubMed] [Google Scholar]
- 60.Schedin-Weiss S, Desai UR, Bock SC, Gettins PGW, Olson ST, Björk I. The importance of lysine 125 for heparin binding and activation of antithrombin. Biochemistry. 2002;41:4779–4788. doi: 10.1021/bi012163l. [DOI] [PubMed] [Google Scholar]
- 61.Belzar KJ, Dafforn TR, Petitou M, Carrell RW, Huntington JA. The effect of a reducing-end extension on pentasaccharide binding by antithrombin. J. Biol. Chem. 2000;275:8733–8741. doi: 10.1074/jbc.275.12.8733. [DOI] [PubMed] [Google Scholar]
- 62.Desai UR. New antithrombin-based anticoagulants. Med. Res. Rev. 2004;24:151–181. doi: 10.1002/med.10058. [DOI] [PubMed] [Google Scholar]
- 63.van Boeckel CAA, Petitou M. The unique antithrombin III binding domain of heparin : A lead to new synthetic antithrombotics. Angew. Chem. Int. Ed. 1993;32:1671–1818. [Google Scholar]
- 64.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 65.Schedin-Weiss S, Arocas V, Bock SC, Olson ST, Bjork I. Specificity of the basic side chains of Lys114, Lys125, and Arg129 of antithrombin in heparin binding. Biochemistry. 2002;41:12369–12376. doi: 10.1021/bi020406j. [DOI] [PubMed] [Google Scholar]
- 66.Monien BH, Krishnasamy C, Olson ST, Desai UR. Importance of tryptophan 49 of antithrombin in heparin binding and conformational activation. Biochemistry. 2005;44:11660–11668. doi: 10.1021/bi050741i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.