

Abstract
In T4 bacteriophage, the DNA polymerase holoenzyme is responsible for accurate and processive DNA synthesis. The holoenzyme consists of the DNA polymerase gp43 and the clamp protein gp45. To form a productive holoenzyme complex, the clamp loader protein gp44/62 is required for the loading of gp45, along with MgATP, and also for the subsequent binding of polymerase to the loaded clamp. Recent evidence suggests that holoenzyme assembly in T4 replisome may take place via more than one pathway. In order to demonstrate unequivocally whether there are multiple pathways leading to the formation of a productive holoenzyme, single-molecule fluorescence microscopy has been used to study the potential clamp loading and holoenzyme assembly pathways on a single-molecule DNA substrate. The results obtained reveal four pathways that foster the formation of functional holoenzyme on DNA: (1) clamp loader-clamp complex binding to DNA followed by polymerase; (2) clamp loader binding to DNA followed by clamp then polymerase; (3) clamp binding to DNA followed by clamp loader then polymerase; and (4) polymerase binding to DNA followed by clamp loader-clamp complex. In all cases, MgATP is required. The possible physiological significance of the various assembly pathways is discussed in the context of replication initiation and lagging strand synthesis during various stages of T4 phage replication.
The T4 bacteriophage replication system is a model for DNA replication in general. The eight proteins that constitute the replisome are: DNA polymerase (gp43); the polymerase accessory proteins, clamp loader (gp44/62) and clamp (gp45); a single-strand DNA binding protein (gp32); a primase (gp61); a helicase (gp41); and a helicase assembly factor (gp59). A key question that has been the subject of several lines of inquiry is the assembly of the holoenzyme from the clamp, clamp loader and polymerase proteins in the presence of MgATP (1-15). To date two pathways have been documented: (1) the formation of a clamp loader-clamp complex associated with ATP hydrolysis followed by successive binding of the complex and polymerase to DNA with additional ATP hydrolysis; and (2) the binding of the clamp loader to DNA with concomitant ATP hydrolysis, followed by the successive binding of clamp and polymerase. Both pathways terminate with the departure of the clamp loader and active holoenzyme bound to the DNA.
In the work presented here, single molecule fluorescence microscopy has been used to clarify and extend further holoenzyme assembly. This method has the advantage of monitoring only what is happening on the DNA and directly observing proteins binding and dissociating from the DNA. Ensemble studies, in contrast, measure total changes in the reaction mixture. Thus single molecule methodology has the capability of providing an unambiguous characterization of the assembly process.
An unusual feature of the T4 clamp is its ring-open form in solution (16) that begs the question as to the advantage of this conformation and its influence on other potential clamp loading pathways. In this study, we have discovered that the open clamp may indeed bind directly to a DNA substrate and be processed to an active holoenzyme upon mandatory addition of clamp loader, followed by polymerase. Clamp loader fueled ATP hydrolysis is essential for this process. Additionally we find that holoenzyme can be reconstituted from a polymerase bound to DNA, followed by the addition of the clamp-clamp loader complex in the presence of MgATP. The existence of multiple holoenzyme assembly pathways may provide distinct advantages in the efficient assembly of the T4 replisome during various stages of replication.
Materials and Methods
Preparation of Proteins and DNA
Fluorescent dyes were purchased from Molecular Probes. Bacteriophage T4 proteins, exonuclease-deficient gp43 [gp43(exo-)], gp44/62, gp45, were purified as previously described (17, 18). All other chemicals were of analytical grade or better. The labeling of gp43(exo-) with Alexa Fluor 488 carboxylic acid, succinimidyl ester (A488) was carried out as before (19). The purification and labeling of V163C gp45 was carried out as previously reported with the following modifications (20). V163C gp45 was dialyzed in labeling buffer containing 20 mM tris(hydroxymethyl)aminomethane (Tris), pH 7.5, 150 mM NaCl, and 10% glycerol for 12 h and was incubated with a 5-fold excess of Alexa Fluor 555 C2 maleimide (A555) for 4 h. The labeled protein was then chromatographed on a Superose 12 column (Amersham Biosciences) equilibrated in labeling buffer to remove the unlabeled dye and frozen in aliquots at -80 °C. The Bio62/34/36mer primer-template forked DNA (21) (see Figure 1 for sequence) was prepared from a 34-mer primer annealed to a 3′ end biotinylated 62-mer template strand and a partially complementary 36-mer DNA strand (Integrated DNA Technologies, Inc.).
Figure 1.
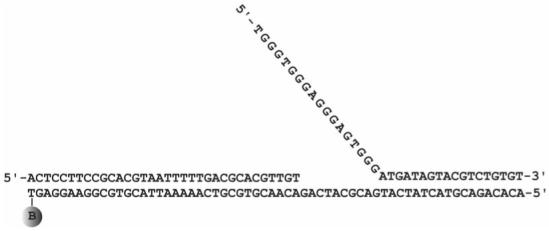
Structure of the forked DNA substrate used for the single molecule FRET study. The biotin tag used for attaching the DNA to the streptavidin coated glass slide is designated as B.
Single Molecule FRET Measurements
Fields of well resolved fluorescent single molecules were observed using a Zeiss microscope with prism based total internal reflection optics and a 100x, 1.45 numerical aperture, oil immersion lens. Data were recorded on a Pentamax ICCD camera. The microscope filter sets were selected to observe fluorescence from three different sources: F1, emission from the fluorescence donor A488 (excitation at 488 nm/emission at 510-540 nm); F2, emission from FRET between A488, and A555 (excitation at 488 nm/emission at 595-645 nm); and F3, emission from the acceptor A555 (excitation at 514 nm/emission at 535-585 nm). All experiments were performed a minimum of three times at ambient temperature (∼25 °C).
Slides were prepared as previously described (22). In brief, glass microscope slides were extensively washed in a sonicating bath. Individual 30 min washes were done in methanol, 1 N NaOH, and 3 N HCl. Extensive rinsing in distilled H2O was done between washes. Slides were thoroughly dried under heated air and incubated 30 min in 0.2% 3-aminopropyltrimethoxysilane (Alfa Aesar; Ward Hill, MA) in hexane. Slides were then air dried, and a coverslip was attached via two double layers of double-sided transparent tape. One hundred microliters of approximately 10 mM Sulfo-NHS-LC-LC Biotin (Pierce; Rockford, IL) in 0.1 M sodium phosphate, pH 8.0, were passed between the coverslip and slide and allowed to react 30 min with the free amine groups on the silanized slide. This process was repeated three times. After biotinylation, slides were equilibrated in 0.1 M sodium phosphate, 3% (w/v) polyethylene glycol (molecular weight 3500), pH 8.0. Three 100 μL aliquots of a 100 nM solution of NeutrAvidin (Pierce; Rockford, IL) in the same buffer were added and allowed to incubate for 30 min. Unbound NeutrAvidin was removed by washing with buffer. (Three 100 μL is approximately 4-5 slide volumes.)
The forked DNA (Bio62/34/36mer) was attached to the surface of the slide as follows: a solution containing 100 nM forked DNA in 100 mM sodium phosphate, 3% (w/v) polyethylene glycol (molecular weight 3500), pH 8.0, was passed three times (300 μL total volume) through the space between the coverslip and avidin coated slide. After 30 min incubation, the unbound forked DNA was removed by washing with buffer. Forked DNA coated slides were then equilibrated in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9. These conditions were suitable for the observation of well resolved fluorescent spots in the microscope field.
The first protein to be added in a given experiment was passed between the coverslip and slide, containing bound forked DNA, three times using 100 μL volumes. Depending on the experiment, 100-500 nM fluorescently labeled or unlabeled protein in the Tris based buffer was used. Three consecutive 100 μL buffer washes were performed between protein additions to ensure that any unbound proteins were removed. Subsequent protein additions were performed with the slide mounted on the microscope stage without moving the slide. Control experiments in the absence of forked DNA confirmed that single molecules are observed only in the presence of forked DNA and therefore are not due to non-specific binding of the proteins to the slide. Catastrophic photobleaching experiments could not be used to confirm single molecules as multiple proteins are bound to the individual forked DNA substrates. However, slides prepared under identical conditions with the addition of a 5′-biotinylated 33 nucleotide ssDNA labeled with fluorescein at the 3′ end bleached as single molecules.
Single molecule images were collected using the program WinView32 version 2.5c (Roper Scientific; Trenton, NJ) with a 200 ms exposure time. A background subtraction was performed using the 2D Rolling Ball algorithm option with the rolling ball radius set at a value of 50 using NIH Image (http://rsb.info.nih.gov/nih-image/Default.html). The enhance contrast option was then applied with a saturated pixel level set at 0.5%. The final images were generated by cropping a representative section of the original field and sized with Adobe Photoshop (Adobe Systems, Inc.; San Jose, CA). The images presented in this report represent a small fraction of the hundreds of single molecules observed.
Results and Discussion
Validating the single-molecule FRET approach in studying T4 holoenzyme assembly
Ensemble experiments previously established a clamp loading pathway involving the initial formation of gp44/62-gp45 complex in the presence of MgATP (5, 7). The subsequent ATP hydrolysis by gp44/62 powers the additional opening of clamp gp45 and facilitates the loading of the opened clamp onto primer-template DNA. To demonstrate that the single-molecule system we developed is amenable for studying the holoenzyme assembly process, we first tested the pathway proposed on the basis of our previous ensemble holoenzyme assembly experiments. Specifically A555-gp45 and gp44/62 were mixed in the presence of 2.5 mM MgATP and introduced onto a microscope slide with single molecules of forked DNA (Bio62/34/36mer) attached via biotin-avidin interactions. The single molecule fluorescent spots due to the loaded fluorescent protein were viewed with fluorescence microscopy. As shown in Figure 2, the presence of A555-gp45 bound to DNA after addition of gp44/62-A555-gp45-MgATP is apparent with filter set F3. As expected, fluorescence is not observed with filter sets F1 and F2 as these filter sets are designed to monitor A488 emission (F1) and FRET between A488 and A555 (F2). Upon addition of A488-gp43 the binding of A488-gp43 is not readily evident with F1 as the fluorescence emission from the A488 is largely quenched by the A555. However, the binding of A488-gp43 could be verified by the FRET between the donor (A488) and the acceptor (A555) as seen with F2. The lack of fluorescence with F1 and the observation of FRET (F2) indicate close association between gp43 and gp45 on forked DNA.
Figure 2.
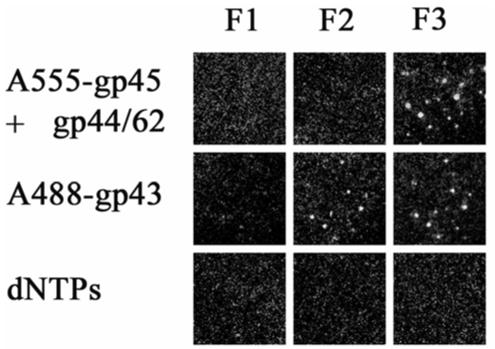
DNA polymerase holoenzyme formation on single molecules of forked DNA immobilized on a microscope slide. Each frame represents the fluorescence of single molecules of DNA with proteins bound in the order indicated at the side of each row. The microscope filters were set to observe fluorescence from three different sources as described in Materials and Methods: F1, fluorescence emission from donor; F2, FRET; and F3, fluorescence emission from acceptor. The proteins were in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9. For the addition of gp45 and gp44/62, 2.5 mM ATP was added. The loss of fluorescence upon the addition of dNTPs indicates the holoenzyme maintains strand displacement activity on single DNA molecules.
In order to verify that the holoenzyme is formed in an active conformation on single molecules of forked DNA, 100 μM dNTPs were added to the slides. Due to the strand displacement activity of the T4 holoenzyme, we expect the active holoenzyme will be able to carry out DNA synthesis and slide off the open end of nascent duplex product. Indeed we observed the loss of fluorescence with all three filter sets (Figure 2), which demonstrates the holoenzyme retains strand displacement activity, thus verifying the active conformation of the assembled holoenzyme complex on the single molecule of DNA substrate. (The rate of strand displacement cannot be determined with this assay.)
As a control, we carried out experiments in the same sequence, but in the absence of MgATP. A555-gp45-gp44/62 did not bind to single molecules of forked DNA as indicated by the absence of fluorescence spots with filter set 3 (data not shown). Subsequent addition of A488-gp43 showed that the labeled polymerase was able to bind to single molecules of forked DNA.
Nonspecific binding of gp45 to forked DNA does not support holoenzyme formation
The T4 gp45 belongs to a class of clamp protein that can encircle duplex DNA and increase the processivity of polymerase through direct interaction between the loaded clamp and the polymerase. However, T4 gp45 is unique in that it exists as a static open-ring structure in solution (11, 16). When A555 labeled clamp (A555-gp45) in the presence of 2.5 mM MgATP was added to single molecules of forked DNA immobilized on glass microscope slides, binding occurred as demonstrated with F3 of Figure 3. The clamp binds to single molecules of DNA tightly enough that the protein was not removed by several washes with buffer. This observation agrees with the notion of an open-ring gp45 structure in solution that can spontaneously encircle DNA and close onto DNA, presumably through electrostatic interactions between the positively charged residues located at the inner rim of gp45 and the negatively charged DNA. Upon addition of A488-gp43, fluorescent spots that overlap well with those detected previously with filter set F3 were observed with filter set F1 (Figure 3). A simultaneous disappearance of fluorescent spots also occurred with filter set F3. This suggests that gp43 has displaced the gp45 and that non-productive binding of gp45 occurs in the absence of gp44/62. The bound gp43 is an active polymerase, as the addition of dNTPs results in loss of gp43. To be certain that the binding of gp45 is not due to the relatively low salt concentration used, this experiment was repeated with the addition of 0.1 M NaCl with the same result (data not shown).
Figure 3.
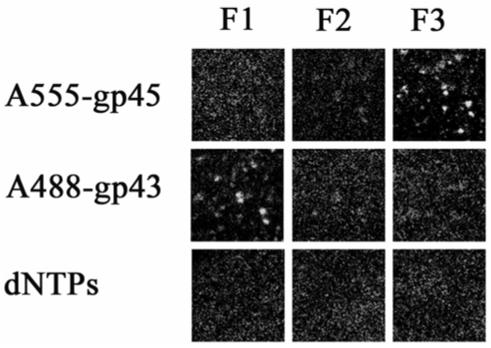
Non-productive binding of labeled gp45 (clamp) on single molecules of forked DNA in the absence of the clamp loading protein, gp44/62. In the absence of clamp loader, gp45 is displaced by the binding of gp43 (polymerase). Each frame represents the fluorescence of single molecules of DNA with proteins bound in the order indicated at the side of each row. The filter sets are as described in the legend to Figure 2. The proteins were in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9. For the addition of gp45, 2.5 mM ATP was included.
Although gp45 alone can bind to the duplex DNA, it may not bind in the conformation necessary to establish the required specific interactions with gp43 that is subsequently bound. The forked DNA substrate we used has a duplex region of 34 base pairs upstream of its primer end. This may represent a space too confined for the non-specific binding of gp45 and simultaneous binding of gp43 because the binding of gp45 to DNA requires ca. a 10 base pair space (23) and gp43 requires at least 10 base pairs (24). Due to the confined space, gp43 may dislodge gp45 from DNA, unless the gp44/62 is present as a chaperon to facilitate the formation of normal holoenzyme complex.
Clamp loaded by gp44/62-DNA complex is functional in forming holoenzyme with gp43
Recent studies (15) suggest gp44/62 is able to bind to primer-template DNA in the presence of ATP, and the gp44/62-DNA complex formed is functional in loading gp45 onto DNA. Consequently, we investigated this clamp loading sequence with the single-molecule experiments. In order to test whether this pathway supports the formation of active holoenzyme comprising gp43-gp45, unlabeled clamp loader complex (gp44/62) was added to single molecules of forked DNA immobilized on the surface of a microscope slide (Figure 4). After removing the free protein through washing with buffer, the addition of A555-gp45 resulted in fluorescent spots with filter set F3, suggesting the loading of clamp onto DNA by the preformed gp44/62-DNA complex. Upon addition of A488-gp43, fluorescent spots were observed with filter set F2, indicating that close association between gp43 and gp45 occurs on the forked DNA. Only a few dim fluorescence spots were observed with filter set F1 due to extensive quenching of the A488 by A555. Following the addition of 100 μM dNTPs, the loss of fluorescence was observed with all three filter sets, as expected for an active holoenzyme complex on DNA. Because gp44/62 loses activity upon dye labeling (data not shown), we were not able to visualize directly the binding of the clamp loader to DNA. Nevertheless, based on surface plasmon resonance binding studies (15), which share some similarity to the single-molecule system described here, we presume that gp44/62 pre-binds to DNA in the presence of ATP. Furthermore, we conclude that the subsequent binding of labeled clamp is most likely due to the clamp loading activity of gp44/62, rather than to nonspecific binding of A555-gp45 alone, because a functional holoenzyme was formed after addition of A488-gp43.
Figure 4.
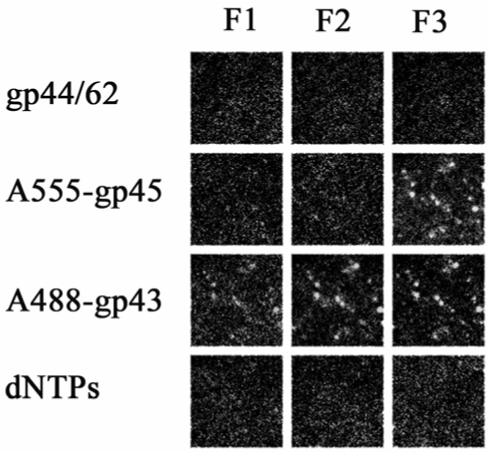
The gp45 (clamp), when loaded onto single molecules of forked DNA by the clamp loader (gp44/62) bound to DNA, forms an active holoenzyme upon addition of gp43. Each frame represents the fluorescence of single molecules of DNA with proteins bound in the order indicated at the side of each row. The filter sets are as described in the legend to Figure 2. The proteins were in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9. For the addition of gp45 and gp44/62, 2.5 mM ATP was included. The loss of fluorescence upon the addition of dNTPs indicates the holoenzyme maintains strand displacement activity on single DNA molecules.
Because ATP has been shown to be required for the formation of gp44/62-DNA complex (15), the same assembly sequence was tested in the absence of MgATP. Fluorescence spots were initially observed with filter set F3 after the individual additions of gp44/62 followed by A555-gp45 (data not shown). However, upon the addition of labeled polymerase, FRET was not observed between the A488-gp43 and A555-gp45. This observation, combined with the loss of fluorescence in F3, suggests that the A555-gp45 was nonspecifically associated with the single molecules of forked DNA and displaced by the polymerase.
Gp44/62 corrects the nonspecific binding of gp45 and allows functional holoenzyme formation
In a separate experiment, the order of addition of clamp loader (gp44/62) and clamp (gp45) was reversed (Figure 5). The binding of A555-gp45 to single molecules of forked DNA is indicated by the presence of fluorescent spots with filter set F3. No fluorescent spots were observed with filter set F1 or F2. From the previously described results, we know that A555-gp45 binds to forked DNA in a nonspecific way. No change in the fluorescence pattern was observed after addition of unlabeled gp44/62 with 2.5 mM MgATP, suggesting the A555-gp45 remains bound to DNA. However, in marked contrast to the previous experiment where gp44/62 is not included, the addition of A488-gp43 resulted in the same fluorescent spots with both filter sets F2 and F3, indicating the occurrence of FRET and the close association of A488-gp43 and A555-gp45. The addition of 100 μM dNTPs confirms that the holoenzyme assembled following this pathway has an active conformation. When the same experimental sequence was carried out in the absence of MgATP, FRET signals were not detected with filter set F2 upon final addition of A488-gp43. Fluorescence was observed with filter set F1, and a decrease in the number of fluorescent spots was observed with filter set F3 after the addition of A488-gp43, suggesting that the cofactor MgATP is required for gp44/62 to convert the nonspecifically loaded gp45 to a productive binding mode.
Figure 5.
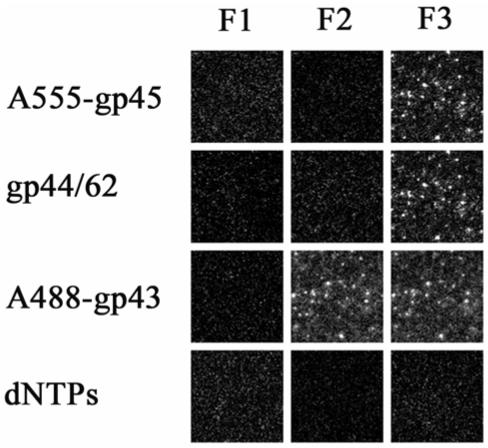
The gp44/62 (clamp loader) can correct non-productive binding of previously bound gp45 on individual molecules of forked DNA, resulting in an active holoenzyme upon addition of gp43. Each frame represents the fluorescence of single molecules of DNA with proteins bound in the order indicated at the side of each row. The filter sets are as described in the legend to Figure 2. The proteins were in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9. For the addition of gp45 and gp44/62, 2.5 mM ATP was included. The loss of fluorescence upon the addition of dNTPs indicates the holoenzyme maintains strand displacement activity on single DNA molecules.
Holoenzyme can be assembled with gp43 binding to DNA first
The presence of A488-gp43 bound to individual molecules of forked DNA is demonstrated in Figure 6 as bright spots with filter set F1 after A488-gp43 is passed through the slide with immobilized DNA. No fluorescent spots are observed with filter sets F2 or F3, respectively. Addition of A555-gp45 and unlabeled clamp loader (gp44/62) in the presence of 2.5 mM MgATP results in formation of the holoenzyme on the same single molecules of forked DNA. The loss of fluorescence intensity with F1 and the formation of fluorescent spots with F2 demonstrate the close association between A488-gp43 and A555-gp45 on forked DNA that results in quenching of the fluorescence from A488-gp43 by the A555-gp45 and emission of A555-gp45 due to FRET between the two fluorophores. Fluorescent spots with F3 demonstrate that A555-gp45 is now bound to the forked DNA. Again, the addition of 100 μM NTPs results in the loss of fluorescence, indicating the enzyme is active.
Figure 6.
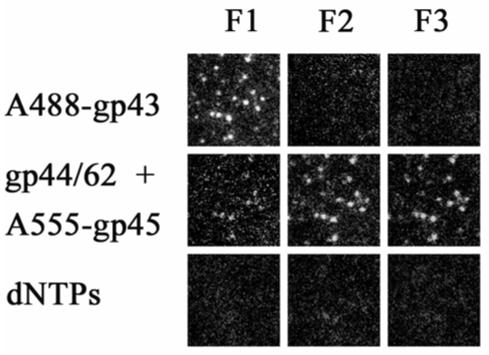
The binding of gp43 to forked DNA substrate, followed by gp45 and gp44/62, results in the formation of an active DNA polymerase holoenzyme. Each frame represents the fluorescence of single molecules of DNA with proteins bound in the order indicated at the side of each row. The filter sets are as described in the legend to Figure 2. The proteins were in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9. For the addition of gp45 and gp44/62, 2.5 mM ATP was included. The loss of fluorescence upon the addition of dNTPs indicates the holoenzyme maintains strand displacement activity on single DNA molecules.
In the absence of MgATP, holoenzyme formation was not observed with this assembly sequence. A488-gp43 was able to bind to single molecules of forked DNA as seen with filter set F1 (data not shown). Upon the addition of A555-gp45 and gp44/62 FRET was not observed with F2. The absence of fluorescent spots in F3 indicates that A555-gp45 was not bound. This again agrees with the notion that binding of gp43 to the DNA prohibits the concomitant nonspecific binding of gp45. Lastly dNTPs (100 μM) were added to verify that the labeled polymerase was bound in an active conformation. Disappearance of the fluorescent spots with F1 indicates that A488-gp43 was active.
Exchange of solution gp43 with clamp bound gp43 as observed by single molecule FRET
Previously through ensemble experiments, we demonstrated that the solution polymerase gp43 undergoes active exchange with gp43 tethered to clamp gp45 (25). The kinetics of this process was studied on a long stretch of DNA when polymerase is carrying out processive DNA synthesis. With the successful formation of labeled T4 holoenzyme on single-molecules of DNA substrate, the polymerase exchange process was studied at the single-molecule level. The experimental setup allows us to address the same question with a static holoenzyme, in contrast to the ensemble experiments.
Firstly unlabeled gp43 and A555-gp45 were assembled as holoenzyme on single molecules of forked DNA in the presence of gp44/62 and ATP. A488-gp43 (300 nM) was then added to determine if polymerase exchange between bound and free gp43 occurs. After a 10 min incubation the labeled gp43 was removed from the bulk solution by washing with 600 μL of buffer, and the image was then captured. This washing was necessary in order to reduce the fluorescence background due to unbound A488-gp43. An additional 300 nM aliquot of labeled polymerase was added, and a second 20 min incubation was performed. The labeled gp43 not bound to forked DNA was again removed and the image captured. Very little exchange of unlabeled gp43 with A488-gp43 was evident with filter set F2 after the 10 min incubation. Approximately 35% of the DNA contained bound A488-gp43 after the second incubation period. The fluorescent spots observed with filter set F3 due to the binding of A555-gp45 marked the location of single strands of forked DNA within the field of view. The addition of 100 μM dNTPs after the 30 min incubation period and subsequent loss of fluorescence indicated that both the unlabeled and labeled gp43 were bound in the correct conformation required for the polymerase reaction.
In a second set of experiments, the T4 replication holoenzyme consisting of A488-gp43, A555-gp45, and gp44/62 was assembled on single molecules of forked DNA. An aliquot of unlabeled gp43 (400 nM) was added to the slides, and the exchange of bound labeled gp43 for unlabeled gp43 in solution was monitored by the disappearance of fluorescent spots with filter set F2. The gp43 was shown to be enzymatically active after 1 hr incubation as all of the A488-gp43 fluorescence disappeared after the addition of dNTPs. The fraction of bound A488-gp43 molecules undergoing exchange with a lifetime τ were tabulated and plotted versus τ (Figure 7). The total number of single molecules that was observed to undergo exchange after 1 hr was 39 out of a total of 190. In the absence of unlabeled gp43 in the solution, the total number of A488-gp43 that was observed to dissociate from the single molecules of forked DNA was approximately 10% after 1 hour.
Figure 7.
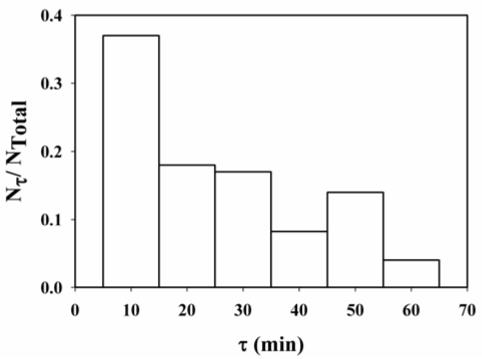
Plot of the fraction of bound A488-gp43 molecules with a lifetime τ, Nτ/NTotal, versus τ. Nτ is the number of molecules with a lifetime τ, and NTotal is the total number of molecules undergoing exchange, 38. The concentration of unlabelled gp43 in 20 mM Tris, 5 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9 was 300 nM. After 1 hour, 20% of the bound A488-gp43 underwent exchange. In the absence of unlabeled gp43, less than 10% of the single gp43 molecules was observed to dissociate from the forked DNA in the same time period. The clamp protein (gp45) was also present on the DNA.
These results indicate that gp43 in solution exchanges with gp43 in the holoenzyme bound to the DNA. The quantitation of the exchange process suggests that the half-time for ensemble exchange is >30 min (k < 4 × 10-4 s-1). The exchange rate determined with the single-molecule experiment is much slower than suggested by the ensemble experiments (25). The slower exchange kinetics observed here may be intrinsic to the static holoenzyme complex assembled on the DNA fork substrate, in contrast to the mobile holoenzyme carrying out processive DNA syntheses assayed in the ensemble experiments. This result combined with our previous observations emphasizes the dynamic nature of this polymerase exchange process.
Multiple holoenzyme assembly pathways may be functional in T4 bacteriophage DNA replication
The single-molecule FRET studies have demonstrated that the T4 bacteriophage holoenzyme can be assembled through four major different pathways. These pathways are summarized by the cartoon in Figure 8.
Figure 8.
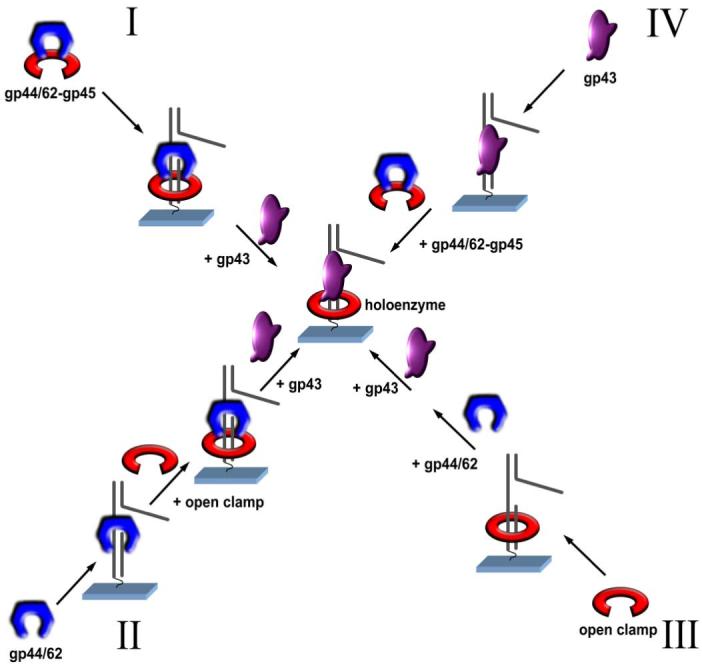
Multiple pathways for assembling the T4 DNA polymerase holoenzyme on a forked DNA substrate. The gp43, gp44/62 and gp45 are colored as magenta, blue and red respectively. In pathway I, gp44/62-gp45 complex formed in the presence of ATP binds to DNA, followed by binding of gp43 to form holoenzyme; in pathway II, gp44/62 binds to DNA in the presence of ATP and recruits an open clamp from solution to form gp44/62-gp45 complex on DNA, followed by binding of gp43 to form holoenzyme; in pathway III, open clamp gp45 encircles DNA spontaneously, followed by binding of gp44/62 and gp43 to form holoenzyme; in pathway IV, gp43 binds to DNA first followed by addition of gp44/62-gp45 to form holoenzyme. MgATP is required for all assembly pathways.
In bacteriophage T4, multiple pathways for initiating phage T4 DNA replication have been documented (26). In the early stage of T4 phage infection, the leading strand DNA synthesis is initiated from a RNA transcript that forms an R loop structure. Subsequent DNA replication initiation in T4 is primed by DNA recombination intermediates. The partially unreplicated 3′ termini of the replicative intermediate invades the homologous region of another chromosome DNA, thus forming a unique D-loop structure. The holoenzyme is presumably assembled on the DNA/DNA or DNA/RNA loop structure and initiates the processive leading strand DNA synthesis. On the other hand, lagging strand DNA synthesis requires the holoenzyme to be assembled repetitively on a short RNA pentamer. The lagging strand DNA possesses a unique structure, different than either the D-loop or R-loop structure used by the holoenzyme assembly machinery for initiating processive leading strand synthesis. Therefore, we hypothesize that the assembly pathways of the holoenzyme may be biased by the specific DNA structures and protein interactions at the different sites of phage replisome.
Given the recent finding that gp43 and gp59 form a tight complex with the forked DNA structure (19) and the fast binding of gp43 to DNA, we speculate that the assembly of holoenzyme may proceed via a pathway in which gp43 locates the 3′ end of DNA through specific interactions with fork-bound gp59 and triggers the clamp loading process. This notion is supported by the finding that pre-binding of gp43 to DNA allows the loading of clamp and the subsequent formation of holoenzyme. Since the binding of gp43 to the primer-template junction excludes the simultaneous binding of gp44/62 to the same DNA site, we speculate that gp44/62 may be guided to the site through interaction with gp43.
Previous studies has suggested that during lagging strand DNA synthesis, clamp loader may have the function of displacing primase from the newly synthesized RNA primer through interaction with single-strand DNA binding protein (27, 28, 29). Subsequently the clamp can be loaded onto DNA and replicative polymerase recruited to form active holoenzyme. For bacteriophage T4, it has been hypothesized that the lagging strand holoenzyme assembly involves the initial binding the gp44/62-gp45 complex to the RNA pentamer synthesized by primase gp61. However, given the small size of the RNA pentamer annealed to the lagging strand DNA template, it is difficult to picture how the gp44/62-gp45 complex, whose binding site covers approximately 16 DNA base pairs, can be accommodated on such a short RNA pentamer even without considering the simultaneous binding of gp61. The binding of gp44/62 followed by gp45 may effectively relieve the steric clash generated between primase gp61 and gp44/62-gp45, and may represent a functional pathway for lagging strand holoenzyme formation. Our experimental results provide evidence for such an alternative pathway for lagging strand holoenzyme assembly.
Abbreviations used
- A488
Alexa Fluor 488
- A555
Alexa Fluor 555
- FRET
fluorescence resonance energy transfer
- dNTPs
equimolar mixture of adenine, cytosine, guanine and thymine nucleotide triphosphates
- Tris
tris(hydroxymethyl) aminomethane
Footnotes
This research was supported by grants from the National Institutes of Health, GM65128, (G. G. H.) and GM13306 (S. J. B.).
References
- 1.Alley SC, Trakselis MA, Mayer MU, Ishmael FT, Jones AD, Benkovic SJ. Building a replisome solution structure by elucidation of protein-protein interactions in the bacteriophage T4 DNA polymerase holoenzyme. J. Biol. Chem. 2001;276:39340–39349. doi: 10.1074/jbc.M104956200. [DOI] [PubMed] [Google Scholar]
- 2.Kuriyan J, O’Donnell M. Sliding clamps of DNA polymerases. J Mol Biol. 1993;234:915–925. doi: 10.1006/jmbi.1993.1644. [DOI] [PubMed] [Google Scholar]
- 3.Alley SC, Abel-Santos E, Benkovic SJ. Tracking sliding clamp opening and closing during bacteriophage T4 DNA polymerase holoenzyme assembly. Biochemistry. 2000;39:3076–3090. doi: 10.1021/bi992377r. [DOI] [PubMed] [Google Scholar]
- 4.Trakselis MA, Alley SC, Abel-Santos E, Benkovic SJ. Creating a dynamic picture of the sliding clamp during T4 DNA polymerase holoenzyme assembly by using fluorescence resonance energy transfer. Proc. Natl Acad. Sci. U.S.A. 2001;98:8368–8375. doi: 10.1073/pnas.111006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaboord BF, Benkovic SJ. Dual role of the 44/62 protein as a matchmaker protein and DNA polymerase chaperone during assembly of the bacteriophage T4 holoenzyme complex. Biochemistry. 1996;35:1084–1092. doi: 10.1021/bi9520747. [DOI] [PubMed] [Google Scholar]
- 6.Piperno JR, Alberts BM. An ATP stimulation of T4 DNA polymerase mediated via T4 gene 44/62 and 45 proteins. The requirement for ATP hydrolysis. J. Biol. Chem. 1978;253:5174–5179. [PubMed] [Google Scholar]
- 7.Jarvis TC, Newport JW, von Hippel PH. Stimulation of the processivity of the DNA polymerase of bacteriophage T4 by the polymerase accessory proteins. The role of ATP hydrolysis. J. Biol. Chem. 1991;266:1830–1840. [PubMed] [Google Scholar]
- 8.Berdis AJ, Benkovic SJ. Role of adenosine 5′-triphosphate hydrolysis in the assembly of the bacteriophage T4 DNA replication holoenzyme complex. Biochemistry. 1996;35:9253–9265. doi: 10.1021/bi952569w. [DOI] [PubMed] [Google Scholar]
- 9.Young MC, Weitzel SE, von Hippel PH. The kinetic mechanism of formation of the bacteriophage T4 DNA polymerase sliding clamp. J. Mol. Biol. 1996;264:440–452. doi: 10.1006/jmbi.1996.0652. [DOI] [PubMed] [Google Scholar]
- 10.Sexton DJ, Kaboord BF, Berdis AJ, Carver TE, Benkovic SJ. Dissecting the order of bacteriophage T4 DNA polymerase holoenzyme assembly. Biochemistry. 1998;37:7749–7756. doi: 10.1021/bi980088h. [DOI] [PubMed] [Google Scholar]
- 11.Alley SC, Shier VK, Abel-Santos E, Sexton DJ, Soumillion P, Benkovic SJ. Sliding clamp of the bacteriophage T4 polymerase has open and closed subunit interfaces in solution. Biochemistry. 1999;38:7696–7709. doi: 10.1021/bi9827971. [DOI] [PubMed] [Google Scholar]
- 12.Pietroni P, Young MC, Latham GJ, von Hippel PH. Dissection of the ATP-driven reaction cycle of the bacteriophage T4 DNA replication processivity clamp loading system. J. Mol. Biol. 2001;309:869–891. doi: 10.1006/jmbi.2001.4687. [DOI] [PubMed] [Google Scholar]
- 13.Berdis AJ, Benkovic SJ. Mechanism of bacteriophage T4 DNA holoenzyme assembly: The 44/62 protein acts as a molecular motor. Biochemistry. 1997;36:2733–2743. doi: 10.1021/bi962139l. [DOI] [PubMed] [Google Scholar]
- 14.Trakselis MA, Berdis AJ, Benkovic SJ. Examination of the role of the clamp-loader and ATP hydrolysis in the formation of the bacteriophage T4 polymerase holoenzyme. J. Mol. Biol. 2003;326:435–451. doi: 10.1016/s0022-2836(02)01330-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhuang Z, Berdis AJ, Benkovic SJ. An alternative clamp loading pathway via T4 clamp loader gp44/62-DNA complex. Biochemistry. 2006 doi: 10.1021/bi0601205. submitted for publication. [DOI] [PubMed] [Google Scholar]
- 16.Millar D, Trakselis MA, Benkovic SJ. On the solution structure of the T4 sliding clamp (gp45) Biochemistry. 2004;43:12723–12727. doi: 10.1021/bi048349c. [DOI] [PubMed] [Google Scholar]
- 17.Frey MW, Nossal NG, Capson TL, Benkovic SJ. Construction and characterization of a bacteriophage T4 DNA polymerase deficient in 3′-to-5′ exonuclease activity. Proc. Natl Acad. Sci. U.S.A. 1993;90:2579–2583. doi: 10.1073/pnas.90.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nossal NG. DNA replication with bacteriophage T4 proteins. Purification of the proteins encoded by T4 genes 41, 45, 44, and 62 using a complementation assay. J. Biol. Chem. 1979;254:6026–6031. [PubMed] [Google Scholar]
- 19.Xi J, Zhuang Z, Zhang Z, Selzer T, Spiering MM, Hammes GG, Benkovic SJ. Interaction between the T4 Helicase Loading Protein (gp59) and the DNA Polymerase (gp43): A Locking Mechanism to Delay Replication During Replisome Assembly. Biochemistry. 2005;44:2305–2318. doi: 10.1021/bi0479508. [DOI] [PubMed] [Google Scholar]
- 20.Soumillion P, Sexton DJ, Benkovic SJ. Clamp subunit dissociation dictates bacteriophage T4 DNA polymerase holoenzyme disassembly. Biochemistry. 1998;37:1819–1827. doi: 10.1021/bi972526a. [DOI] [PubMed] [Google Scholar]
- 21.Kaboord BF, Benkovic SJ. Accessory proteins function as matchmakers in the assembly of the T4 DNA polymerase holoenzyme. Curr. Biol. 1995;5:149–157. doi: 10.1016/s0960-9822(95)00036-4. [DOI] [PubMed] [Google Scholar]
- 22.Rajagopalan PT, Zhang Z, McCourt L, Dwyer M, Benkovic SJ, Hammes GG. Interaction of dihydrofolate reductase with methotrexate: ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13481–13486. doi: 10.1073/pnas.172501499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moarefi I, Jeruzalmi D, Turner J, O’Donnell M, Kuriyan J. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 2000;296:1215–1223. doi: 10.1006/jmbi.1999.3511. [DOI] [PubMed] [Google Scholar]
- 24.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Zhuang Z, Roccasecca RM, Trakselis MA, Benkovic SJ. The dynamic processivity of the T4 DNA polymerase during replication. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8289–8294. doi: 10.1073/pnas.0402625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosig G, Colowick N, Gruidl ME, Chang A, Harvey AJ. Multiple initiation mechanisms adapt phage T4 DNA replication to physiological changes during T4′s development. FEMS Microbiol. Rev. 1995;17:83–98. doi: 10.1111/j.1574-6976.1995.tb00190.x. [DOI] [PubMed] [Google Scholar]
- 27.Tsurimoto T, Stillman B. Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase alpha and delta during initiation of leading and lagging strand synthesis. J. Biol. Chem. 1991;266:1961–1968. [PubMed] [Google Scholar]
- 28.Maga G, Stucki M, Spadari S, Hubscher U. DNA polymerase switching. I. Replication factor C displaces DNA polymerase alpha prior to PCNA loading. J. Mol. Biol. 2000;295:791–801. doi: 10.1006/jmbi.1999.3394. [DOI] [PubMed] [Google Scholar]
- 29.Yuzhakov A, Kelman Z, O’Donnell M. Trading places on DNA: A three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell. 1999;96:153–163. doi: 10.1016/s0092-8674(00)80968-x. [DOI] [PubMed] [Google Scholar]