

Abstract
PDZ domain scaffold proteins are capable of assembling macromolecular protein complexes in diverse cellular processes through PDZ-mediated binding to a short peptide fragment at the carboxyl tail of target proteins. How each PDZ domain specifically recognizes its target protein(s) remains a major conceptual question, as at least a few out of the several hundred PDZ domains in each eukaryotic genome share overlapping binding properties with any given target protein. Here, we show that the domain-swapped dimerization of zonula occludens-1 PDZ2 generates a distinct interface that functions together with the well-separated canonical carboxyl tail-binding pocket in each PDZ unit in binding to connexin43 (Cx43). We further demonstrate that the charge–charge interaction network formed by residues in the PDZ dimer interface and upstream residues of the Cx43 peptide not only provides the unprecedented interaction specificity for the complex but may also function as a phosphorylation-mediated regulatory switch for the dynamics of the Cx43 gap junctions. Finally, we provide evidence that such domain-swapped dimer assembly also occurs in other PDZ domain scaffold proteins. Therefore, our findings present a new paradigm for understanding how some PDZ domain proteins specifically bind to and regulate the functions of their target proteins.
Keywords: connexin43, domain swapping, gap junctions, PDZ domain, ZO-1
Introduction
PDZ domains are one of the most abundant protein–protein interaction modules in eukaryotic proteomes. By binding to a short peptide fragment located at the extreme carboxyl termini (C termini) of target proteins, PDZ domain proteins have critical functions in diverse cellular processes, including the development and maintenance of cell polarity, the establishment and sustaining of cell–cell adhesions, the orchestration of both release and the reception of transmitters in neurons as well as in other excitable cells and so on (Craven and Bredt, 1998; Zhang and Wang, 2003; Kim and Sheng, 2004). The molecular mechanisms governing the PDZ protein-mediated regulations of such broad cellular processes are certainly diverse. Nevertheless, two general PDZ protein-regulated cellular events have converged through ∼15 years of extensive studies since the discovery of PDZ domains (for selected reviews, see Craven and Bredt, 1998; Altschuler et al, 2003; Zhang and Wang, 2003; Kim and Sheng, 2004; Macara, 2004; Margolis and Borg, 2005; Suzuki and Ohno, 2006). First, PDZ proteins often exerts an effect as adaptors in linking their binding partners with cellular protein trafficking machineries, thereby regulating both the biogenesis and the cellular localization of PDZ-interacting targets. Second, PDZ proteins are known as scaffolds at specialized cellular regions capable of organizing supra-molecular signalling complexes and anchoring and/or clustering receptors/ion channels/cell adhesion molecules. The interaction mechanisms governing PDZ–ligand interactions are generally well understood, owing to extensive structural and biochemical studies in the past. The carboxyl tail from each PDZ targets is known to bind in a β-strand conformation to a well-defined ligand-binding groove of a cognate PDZ domain. In the vast majority of cases, only the last 3–5 residues of each target peptide are involved in binding to its cognate PDZ domains (Harris and Lim, 2001; Zhang and Wang, 2003). There are several hundred PDZ domains in each mammalian genome (for example, 335 non-redundant PDZ domains in humans by surveying the current version of the human genome database). It has been demonstrated that each individual PDZ domain can bind to multiple carboxyl peptides with comparable affinities. Conversely, each naturally occurring PDZ-binding peptide can interact with a panel of different PDZ domains (Songyang et al, 1997; Zhang et al, 2006). Given the fundamental and highly specific roles of PDZ proteins in diverse cellular processes, it is puzzling how living organisms can tolerate such promiscuities in PDZ–target interactions, although non-overlapping spatial distributions of PDZ proteins (or their targets) may partially explain the above dilemma.
Zonula occludens-1 (ZO-1) is the first identified member of the PDZ family proteins (Cho et al, 1992; Willott et al, 1993; Woods and Bryant, 1993). ZO-1, as with its related family members ZO-2 and ZO-3, contains three N-terminal PDZ domains followed by an SH3 domain and a guanylate kinase (GuK)-like domain. Accumulating evidence indicates that ZO proteins have critical functions in the formation and maintenance of intercellular junctions, including tight junctions (TJs), adheren junctions (AJs) and gap junctions (GJs) in diverse cellular settings (Itoh et al, 1999; Mitic et al, 1999; Giepmans, 2004; Hunter et al, 2005; McNeil et al, 2006; Umeda et al, 2006), presumably by functioning as scaffold proteins in organizing various junctional complexes. The complete loss of function of ZO-1 in mice by gene targeting is lethal due to early embryonic and extraembryonic development defects such as the disorganization of neural tubes and notochords and impaired angiogenesis (Katsuno et al, 2008). Interestingly, ZO-2−/− mice is also embryonic lethal due to decreased proliferation and increased apoptosis (Xu et al, 2008), but ZO-3−/− mice lack obvious phenotypes (Adachi et al, 2006; Xu et al, 2008). Therefore, at least in mice ZO-1/ZO-2/ZO-3 are not functionally redundant, although ZO-1 and ZO-2 were shown to be able to compensate for each other at the cellular level in some cell types (McNeil et al, 2006; Umeda et al, 2006). A number of ZO-interacting proteins were identified in the search for the molecular mechanisms governing the ZO protein-mediated development and maintenance of intercellular adhesive junctions. For example, most of the claudin family proteins contain conserved carboxyl tails that can bind to the first PDZ domain of ZO-1, ZO-2 and ZO-3, and this interaction with claudins is believed to integrate ZO proteins into the backbones of TJs (Itoh et al, 1999). JAM1 (junctional adhesion molecule-1) has been found to interact with PDZ3 of ZO-1 through its C terminus (Bazzoni et al, 2000). The SH3 and GuK domains of ZO-1 have been shown to interact with α-catenin, afadin and occludin (Itoh et al, 1997; Muller et al, 2005). Actin cytoskeletals have been found to bind directly to an ∼200-residue domain within the C-terminal half of ZO-1 (Itoh et al, 1997; Fanning et al, 2002).
The interactions of connexins (especially connexin43 (Cx43), the most abundant connexin in mammals) with the second PDZ domain of ZO-1 received particular attention in recent years in the context of GJ formation and regulation (Giepmans and Moolenaar, 1998; Toyofuku et al, 1998; Giepmans, 2004; Hunter et al, 2005; Maass et al, 2007). Accumulating evidence indicates that in addition to having passive scaffolding functions in organizing GJ complexes, including connexins and cytoskeletals, ZO-1 also actively participates in the dynamic remodelling of GJs in a number of cellular systems, including cardiomyocytes, fibroblasts and neurons (Giepmans and Moolenaar, 1998; Hunter et al, 2005; Akoyev and Takemoto, 2007; van Zeijl et al, 2007). The formation of GJs requires the assembly of six connexins into a connexon/hemichannel, followed by the docking of a pair of connexons from the apposed membranes of adjacent cells. The carboxyl tail of Cx43 is required for it to bind to ZO-1 PDZ2 (Giepmans and Moolenaar, 1998; Toyofuku et al, 1998), and for ZO-1 to regulate GJ plaque size and dynamics in various cell models (Hunter et al, 2005; van Zeijl et al, 2007). Interestingly, both ZO-1 PDZ2 and ZO-2 PDZ2 have been shown to dimerize through inter-domain swapping of their respective first two β-strands, and this PDZ2-mediated dimerization has been suggested to increase the scaffolding capacity of ZO proteins (Fanning et al, 2007; Wu et al, 2007). However, it is not known whether the domain-swapped dimerization of ZO-1 PDZ2 has any implication in Cx43-mediated GJ formation.
The crystal structure of ZO-1 PDZ2 in complex with a carboxyl tail peptide of Cx43 solved here reveals that the domain swapping of ZO-1 PDZ2 generates a distinct Cx43-binding site distal to the canonical PDZ ligand-binding pocket. Importantly, the distal Cx43-binding sites in the PDZ2 dimer not only provide unprecedented interaction specificity between Cx43 and ZO-1 PDZ2 but also function as regulatory sites for the dynamics of the Cx43/ZO-1 GJs. Finally, we provide evidence that this domain-swapped dimerization assembly also exists in other PDZ scaffold proteins.
Results and discussion
Overall structure of ZO-1 PDZ2 in complex with the Cx43 peptide
As the first step towards understanding ZO-1-mediated GJ formation, we characterized the interaction between ZO-1 PDZ2 and a nine-residue Cx43 carboxyl tail peptide (NH2-RPRPDDLEI-COOH, referred to as the Cx43 peptide) in detail. A fluorescence-based assay showed that this nine-residue peptide specifically binds to ZO-1 PDZ2 with a Kd of ∼17 μM (Table I). Next, we used NMR spectroscopy to investigate the interaction between the Cx43 peptide and ZO-1 PDZ2. The wild-type ZO-1 PDZ2 displayed poor NMR spectra, which prevented us from doing a detailed analysis of the interaction (Supplementary Figure 1A). However, one of the ZO-1 PDZ2 mutants with Arg193 in the solvent exposed loop outside the ligand-binding pocket substituted with Ala showed excellent 1H–15N HSQC spectrum (Supplementary Figure 1B). The overall spectrum of the Arg193Ala mutant of ZO-1 PDZ2 is similar to that of the wild-type protein. Additionally, the Arg193Ala mutant and the wild-type PDZ2 bind to the Cx43 peptide with the same affinity (data not shown). We therefore used the Arg193Ala mutant of ZO-1 PDZ2 for further structural analysis, and this mutant will be referred to as ZO-1 PDZ2 hereafter unless otherwise specified. Sedimentation equilibrium experiments on ZO-1 PDZ2 at three different concentrations gave rise to a well-fitted MW of ∼21.8 kDa (Supplementary Figure 1C), indicating that the domain exists as a stable, symmetric dimer (the theoretical MW of the monomeric domain is 10.3 kDa and only one set of NMR peaks were observed for the domain). The titration of the 15N-labelled ZO-1 PDZ2 with the Cx43 peptide showed that a select set of peaks from the PDZ domain underwent dose-dependent chemical shift changes or peak broadenings (Figure 1A), further indicating that the Cx43 peptide specifically binds to ZO-1 PDZ2. However, the poor homogeneity of the spectrum of the ZO-1 PDZ2–Cx43 peptide complex made NMR-based high-resolution structure determination challenging. As an alternative approach, we resorted to X-ray crystallography to solve this complex structure (Table II).
Table 1.
The measured Kd (μM) of Cx43 peptides to different forms of ZO-1 PDZ2
| Proteins | Short peptide | Long peptide | Long S(−9)/E | Long S(−10)/E | Long pS(−9) |
|---|---|---|---|---|---|
| Wild type | 16.8±1.1 | 7.2±0.7 | 48.4±2.2 | 18.0±0.4 | >100a |
| K209A mutant | Undetectable | Undetectable | ND | ND | ND |
| R201A mutant | 124.5±6.7 | 136.5±4.3 | ND | ND | ND |
| K191A mutant | ND | 58.6±3.4 | ND | ND | ND |
| K246R mutant | 68.0±2.3 | 35.6±1.2 | ND | ND | ND |
| ND, not determined. | |||||
| aThe binding affinity between the pS(−9) Cx43 long peptide and ZO-1 PDZ2 could not be reliably measured using the same fluorescence polarization assay due to the poor titration curve fitting. The Kd of the peptide was estimated by a competition assay using increasing amount of the pS(−9) Cx43 long peptide to compete with the FITC-labelled Cx43 peptide for ZO-1 PDZ2. In this assay, the concentrations of the FITC-labelled Cx43 peptide and ZO-1 PDZ2 were fixed at a constant concentration. | |||||
Figure 1.

The overall structure of the ZO-1 PDZ2/Cx43 peptide complex. (A) Superposition plot of the 1H–15N HSQC spectra of ZO-1 PDZ2 with increasing molar ratios of the Cx43 peptide. The arrows indicate the peptide binding-induced peak shifts and the dotted boxes highlight the peptide binding-induced peak broadenings. (B) Stereo view of the final crystal structure model of the ZO-1 PDZ2/Cx43 peptide complex shown in the ribbon diagram. The Cx43 peptides are drawn in purple. The secondary structures are labelled following the scheme of the canonical PDZ domains. (C) The FO−FC map of the bound Cx43 peptide showing that the densities of all nine residues can be clearly assigned. The map is calculated by omitting the peptide from the final PDB file and contoured at 2.5σ.
Table 2.
Statistics of X-ray crystallographic data collection and model refinement
| Data collection | |
| Space group | C2221 |
| Unit cell parameters | |
| a, b, c (Å) | 41.04, 68.14, 149.69 |
| Resolution range (Å) | 35.16–2.40 (2.53–2.40) |
| No. of total reflections | 41 480 |
| No. of unique reflections | 8265 |
| I/σ | 20.9 (4.5) |
| Completeness (%) | 96.9 (95.9) |
| Rmerge (%)a | 5.1 (34.2) |
| Structure refinement | |
| Resolution (Å) | 30–2.4 |
| Rcryst/Rfree (%)b | 21.5/25.4 |
| r.m.s.d bonds (Å)/angles (deg) | 0.009/1.22 |
| No. of reflection working set/test set | 7860/388 |
| No. of atoms | |
| Protein atoms | 1252 |
| Peptide atoms | 156 |
| Water molecules | 77 |
| Average B-factor (Å2) | |
| Main chain | 47.31/45.9 |
| Side chain | 48.3/47.4 |
| Peptide | 48.3/46.7 |
| Water | 49.4 |
| Ramachandran plot | |
| Most favoured regions (%) | 92.9 |
| Additionally allowed (%) | 7.1 |
| Generously allowed (%) | 0.0 |
| aRmerge=∑∣Ii−Im∣/∑Ii, where Ii is the intensity of the measured reflection and Im is the mean intensity of all symmetry-related reflections. | |
| bRcryst=∑∣∣Fobs∣−∣Fcalc∣∣/∑∣Fobs∣, where Fobs and Fcalc are observed and calculated structure factors. | |
| Rfree=∑T∣∣Fobs∣−∣Fcalc∣∣/∑T∣Fobs∣, where T is a test data set of about 5% of the total reflections randomly chosen and set aside prior to refinement. | |
| Numbers in parentheses represent the value for the highest resolution shell. | |
The crystal structure of ZO-1 PDZ2 in complex with the Cx43 peptide was solved using the molecular replacement method. In the final refined model at 2.4-Å resolution, each asymmetric unit contains two PDZ domains that adopt a symmetric swapped dimer conformation, and two Cx43 peptides are bound to each PDZ dimer (Figure 1B). The electron densities of all nine residues of the Cx43 peptide can be clearly assigned; thus, the conformation of the entire peptide is well defined (Figure 1C). The last three residues of the Cx43 peptide bind to the αB/βB-groove in each PDZ unit, and the other six residues intimately interact with one of the two symmetric pockets formed by the PDZ domain-swapped assembly (Figure 1B, and see below for more details). This domain-swapped dimer assembly in the ZO-1 PDZ2–Cx43 peptide complex was also observed in the ligand-free forms of ZO-1 and ZO-2 PDZ2 (Fanning et al, 2007; Wu et al, 2007). Three distinct, intermolecular interaction regions between the two PDZ domains contribute to the extensive dimer interfaces: the inter-domain βA/βF pair, the extended antiparallel inter-domain βBC/βBC assembly, and the two pairs of inter-domain salt bridges formed by the side chains of Lys209 and Glu238 (Supplementary Figure 2B). A total of 26 inter-domain backbone hydrogen bonds, together with numerous side chain interactions, are expected to keep this domain-swapped dimer at a very stable conformation. It is unlikely that the PDZ domains in the ZO-1 PDZ2 dimer can readily dissociate from each other to form heterodimers with ZO-2 PDZ2 or ZO-3 PDZ2. We confirmed this prediction by mixing 15N-labelled ZO-1 PDZ2 with unlabelled ZO-2 PDZ2 at a 1:1 stoichiometric ratio. No detectable ZO-1 PDZ2–ZO-2 PDZ2 heterodimer could be observed after the prolonged incubation of this mixture (up to 1 week at 30°C), as the 1H–15N HSQC spectrum of ZO-1 PDZ2 did not change at all after the incubation (Supplementary Figure 1D).
Domain-swapped dimer assembly is required for ZO-1 PDZ2 to bind to Cx43
Amino-acid sequence analysis has revealed that ZO-1 PDZ2, in contrast to the known monomeric PDZ domains, lacks any connecting residues between the βB and βC strands (Figure 3A). In all likelihood, the lack of connecting residues between the βB and βC strands prevents the βC strand from folding back to form an intramolecular antiparallel β-sheet with the βB strand, and forces the formation of the domain-swapped βBC/βBC assembly. To test this hypothesis, we inserted four flexible residues (GGGA) into the middle of the βBC strand of ZO-1 PDZ2 and investigated the folding property of the mutant in detail. Interestingly, the insertion mutant of ZO-1 PDZ2 was well folded, as indicated by its nicely dispersed and homogenous 1H–15N HSQC spectrum (Figure 2A). Furthermore, the insertion of ‘GGGA' in the middle of βBC strand converted ZO-1 PDZ2 into a stable monomer, as shown by the sedimentation equilibrium analysis (Figure 2B). We completed the backbone chemical shift assignments of the wild-type PDZ2 dimer and the GGGA-inserted PDZ2 monomer, plotted the amide backbone chemical shift differences of the two forms as a function of the residue number and further mapped shift differences onto the 3D structure of the ZO-1 PDZ2 dimer (Figure 2C). The largest chemical shift differences were restricted to the residues within the GGGA-inserted region and the dimer interface. For instance, the insertion led to large chemical shift changes to Lys209 and Glu238, as the conversion of the ZO-1 PDZ2 dimer to a monomer breaks the inter-molecular salt bridges formed between these two residues (Supplementary Figure 2B). The data shown in Figure 2A and C also indicate that the overall PDZ fold in the GGGA insertion mutant is retained. We determined the secondary structure of the GGGA insertion mutant based on the 13Cα and 13Cβ chemical shift values of each residue (Supplementary Figure 3). The data showed that the GGGA insertion mutant adopts the same secondary structure as that of the wild-type protein, except for the GGGA insertion region. The CD spectra of the GGGA insertion mutant and the wild-type protein are also highly similar (data not shown), further indicating that the secondary structures of the two proteins are essentially the same.
Figure 3.

The domain-swapped dimer interface of ZO-1 PDZ2 provides a distinct specificity-determining binding site for Cx43. (A) Structure-based sequence alignment of ZO-1 PDZ2 from different species. In this alignment, the conserved hydrophobic residues are shown in orange, negatively charged residues in magenta, positively charged residues in blue and the rest of the highly conserved residues in cyan. The residues that are directly involved in the binding to the Cx43 peptide are boxed and highlighted with red stars. (B) Stereo view showing the detailed interactions of the Cx43 peptide with the residues from the swapped dimer of ZO-1 PDZ2. The hydrogen bonds involved in the binding are shown as the dotted lines. (C) Surface representation showing the binding interface between the ZO-1 PDZ2 and the Cx43 peptide. In this presentation, the hydrophobic amino-acid residues in PDZ2 surface model are drawn in yellow, the positively charged residues in blue, the negatively charged residues in red and the uncharged polar residues in grey. The Cx43 peptide is shown in the stick model. (D) The combined stick-dot model and the ribbon representation showing the charge–charge interaction network formed in the ZO-1 PDZ2–Cx43 peptide complex.
Figure 2.

Domain-swapped dimerization is required for ZO-1 PDZ2 to bind to Cx43. (A) 1H–15N HSQC spectrum of the GGGA insertion mutant of ZO-1 PDZ2. The well-dispersed spectrum indicates that the mutant is well folded. (B) Sedimentation equilibrium analysis showing that the GGGA insertion mutant ZO-1 PDZ2 is a stable monomer. The insert shows a schematic model illustrating the GGGA insertion-induced conversion of the ZO-1 PDZ2 dimer into a monomer. (C) Plot of backbone amide chemical shift differences as a function of the residue number of ZO-1 PDZ2 between the wild-type dimer and the GGGA insertion monomer. The two residues (Lys209 and Glu238) forming inter-domain salt bridges in the wild-type ZO-1 PDZ2 are highlighted using red stars. The secondary structures of PDZ2 are also indicated at the top of the figure. The ribbon diagram shows the shift changes mapped onto the 3D structure of the ZO-1 PDZ2 dimer. In this representation, the combined 1H and 15N chemical shift changes are defined as: 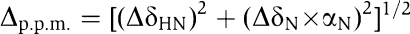 where ΔδHN and ΔδN represent chemical shift differences of amide proton and nitrogen chemical shifts of the each residue of ZO-1 PDZ2. The scaling factor (αN) used to normalize the 1H and 15N chemical shift is 0.17. (D) Fluorescence-based measurement of the binding affinities of the wild-type ZO-1 PDZ2 domain and the GGGA insertion mutant towards the Cx43 peptide. (E) Comparison of the cellular localizations of the GFP-tagged full-length wild-type ZO-1 and the ZO-1 mutant with the GGGA insertion in its PDZ2 in HeLa cells. The overexpressed wild-type ZO-1 forms plaques at the contact regions between transfected cells, whereas the ZO-1 mutant transfected cells lack such intercellular ZO-1 punta.
where ΔδHN and ΔδN represent chemical shift differences of amide proton and nitrogen chemical shifts of the each residue of ZO-1 PDZ2. The scaling factor (αN) used to normalize the 1H and 15N chemical shift is 0.17. (D) Fluorescence-based measurement of the binding affinities of the wild-type ZO-1 PDZ2 domain and the GGGA insertion mutant towards the Cx43 peptide. (E) Comparison of the cellular localizations of the GFP-tagged full-length wild-type ZO-1 and the ZO-1 mutant with the GGGA insertion in its PDZ2 in HeLa cells. The overexpressed wild-type ZO-1 forms plaques at the contact regions between transfected cells, whereas the ZO-1 mutant transfected cells lack such intercellular ZO-1 punta.
The GGGA insertion mutant provided us with a unique opportunity to address the role of the domain-swapped dimer assembly of ZO-1 PDZ2 in its target binding. In contrast to the specific interaction between the wild-type ZO-1 PDZ dimer and the Cx43 peptide, the GGGA insertion mutant showed no detectable interaction with the Cx43 peptide as assayed by both fluorescence- and NMR-based techniques (Figure 2D, and data not shown). To evaluate the role of PDZ2-mediated dimerization in ZO-1's cellular localization, we transfected the GFP-tagged full-length wild-type ZO-1 and the ZO-1 mutant with the GGGA insertion in its PDZ2 into HeLa cells. The wild-type ZO-1 overexpressed in HeLa cells formed dense intercellular junctional puncta. In sharp contrast, the ZO-1 mutant containing the GGGA insertion in its PDZ2 completely lacked intercellular ZO-1 puncta (Figure 2E). Therefore, we conclude that the domain-swapped dimer assembly of ZO-1 PDZ2 is absolutely required for its binding to Cx43 and likely to other connexins as well.
Would the βBC-strand-swapped dimer assembly seen in PDZ2 of ZO proteins also occur in other PDZ proteins? To answer this question, we developed a computer algorithm to search the human genome for PDZ domains that may lack βB/βC-connecting residues. Several PDZ domains in addition to PDZ2 of ZO-1, ZO-2 and ZO-3 showed up in our search result, and one of them was the second PDZ domain of DLG5, another cell polarity/junction regulatory protein (Nechiporuk et al, 2007). Amino-acid sequence alignment analysis revealed that DLG5 PDZ2 bears a similar feature seen in the βB and βC region of ZO-1 PDZ2 (that is, lack of any connecting residues between the two β-strands; Supplementary Figure 2A). We confirmed that DLG5 PDZ2 also exists as a dimer in solution, as revealed by a DSG-mediated chemical cross-linking assay (Supplementary Figure 2C) and an analytical ultra-centrifugation analysis (data not shown). Therefore, it is likely that the domain-swapped dimer formation seen in ZO-1 PDZ2 also occurs in some other PDZ proteins.
The domain-swapped dimer interface of ZO-1 PDZ2 determines Cx43-binding specificity
The interaction between the last three residues of the Cx43 peptide and the canonical binding pocket of ZO-1 PDZ2 is typical for type II PDZ domain–ligand complexes (Zhang and Wang, 2003): the hydrophobic side chain of Ile(0) of the Cx43 peptide inserts into the hydrophobic pocket situated at the end of the αB/βB groove, the carboxyl group of Ile(0) forms three hydrogen bonds with the backbone amides of Tyr198, Gly199, Leu200 (the so-called ‘GLGF motif' in PDZ domains) and a water-mediated salt bridge with the side chain of Lys191, and the side chain of Leu(−2) packs with the side chain of Leu202 at the αB1 position of the PDZ domain (Figure 3B and C). The domain-swapped assembly leads to the direct apposition of the two αB/βB grooves of the PDZ dimer (Figure 1B). Consistent with this arrangement, the Cx43 peptide in each PDZ monomer unit makes a sharp turn at the completely conserved Pro(−5) to avoid clashing into each other in the complex (Figures 1B and 3B).
The most salient feature of the ZO-1 PDZ2–Cx43 peptide complex structure is the extended ligand-binding pocket located in the interface of the PDZ dimer (Figure 3B and C). An extensive, continuous charge–charge interaction network was formed between Asp(−3), Asp(−4) and Arg(−6) of the Cx43 peptide and charged residues located in the PDZ dimer interface (Arg201, Lys209 and Glu238). The charge–charge interaction between Glu(−1) of Cx43 and Arg201 and Glu210 further extends the above charge interaction network into the canonical ligand-binding region (Figure 3D). The charge–charge interactions in the dimer interface are expected to enhance the PDZ's affinity for Cx43, but perhaps more importantly to provide ZO-1 PDZ2 with the exquisite specificity in binding to Cx43. Consistent with this notion, the substitution of Lys209 at the centre of this charge–charge interaction network with an Ala completely abolished ZO-1 PDZ2's binding to Cx43 (Table I). This notion is further supported by our earlier data, which show that the GGGA insertion mutant lacking the Lys209–Glu238 salt bridge as well as the entire PDZ dimer interface does not bind to Cx43 at all. In comparison, the disruption of a peripheral salt bridge in the canonical peptide-binding pocket of PDZ2 (substituting Arg201 with Ala) had a modest impact (∼8-fold decrease; Table I) on the PDZ's binding to Cx43, further substantiating the critical role of the charge–charge interaction network at the dimer interface for the domain to bind to its targets. Finally, amino-acid sequence analysis showed that the residues involved in the formation of the charge–charge network are strictly conserved for both ZO-1 PDZ2 (Figure 3A) and Cx43 (Figure 4A) throughout evolution. We note that Cx43 and its related connexins are the only known binding partners of ZO-1 PDZ2 reported to date. A phage display-based peptide library search for ZO-1 PDZ2 ligands also failed to identify any additional positive binders (Zhang et al, 2006). We believe that it is the unique charge–charge interactions enabled by the domain-swapped dimerization that provides such exquisite target binding specificity of ZO-1 PDZ2. Consistent with this notion, a search for carboxyl peptide sequences with the pattern of ‘R/K-X-R/K-P-D/E-D/E-ψ-D/E-ψ*' (where ‘X' represents any amino acids and ‘ψ' stands for hydrophobic amino acids) in the genomes of human, rat and mouse failed to identify any positive matches with the exceptions of connexins.
Figure 4.

The interaction between Cx43 and ZO-1 PDZ2 could be regulated by the phosphorylation of Cx43. (A) Amino-acid sequence alignment of the C-terminal tail of Cx43 from different species. The highly conserved Pro(−5) is highlighted with a red star. Three well-conserved SSR repeats are boxed. Ser(−9) and Ser(−10) of Cx43 are highlighted with red triangles. (B) Overlay plot of a selected region of the 1H–15N HSQC spectrum of free ZO-1 PDZ2 and the protein in the presence of saturating amounts of short and long Cx43 peptides. (C) Superposition plots of the 1H–15N HSQC spectrum of free ZO-1 PDZ2 and with excess amount of the short, long, long S(−9)/E, long S(−10)/E, and phosphor-S(−9) Cx43 peptides, For clarity, only three well-resolved peaks are shown.
It has been reported that ZO-2 PDZ2 can also bind to the Cx43 C-terminal tail, although the affinity and functional role of this interaction have not been determined (Singh et al, 2005). We found that ZO-2 PDZ2 binds to the Cx43 peptide with a much weaker affinity (Kd∼391 μM), although the residues that are critical for ZO-1 PDZ2 binding to Cx43 are essentially conserved in ZO-2 PDZ2 except for the residue situated at one helical turn above the αB1 residue (Arg367 in ZO-2 PDZ2 and Lys246 in ZO-1 PDZ2; Supplementary Figure 4). At first, we thought that the larger and more polar nature of the Arg side chain in ZO-2 PDZ2 may be responsible for its weaker binding to Cx43, as the corresponding Lys in ZO-1 PDZ2 directly participates in the interaction with the side chain of Leu(−2) from the Cx43 peptide (Figure 3B). The substitution of this Lys in ZO-1 PDZ2 with an Arg indeed decreased its binding to the Cx43 peptide, although at a modest level (Kd∼68 μM; Supplementary Figure 4C). The above data also suggest that additional amino-acid differences, possibly in the swapped βBC/βBC region, are also responsible for the differential Cx43-binding properties of these two PDZ domains. Nevertheless, our data imply that ZO-2 may not be involved in the Cx43-mediated GJ formation due to its much weaker binding to Cx43.
The Cx43/ZO-1 complex can be further regulated by the phosphorylations of Ser(−9) and Ser(−10) in Cx43
As the electron densities of all nine residues of the Cx43 peptide can be clearly defined in the ZO-1 PDZ2–Cx43 peptide complex, we wondered if amino-acid residues further upstream of Arg(−8) of Cx43 could also be involved in binding to ZO-1 PDZ2. To test this possibility, we extended the Cx43 peptide by three residues (‘ASSRPRPDDLEI', referred to as the long Cx43 peptide) to include the completely conserved Ser(−9) and Ser(−10) (Figure 4A). The 1H–15N HSQC spectrum of ZO-1 PDZ2 saturated with the long Cx43 peptide is clearly different from that of ZO-1 PDZ2 saturated with the nine-residue Cx43 peptide (Figure 4B), indicating that the completely conserved Ser(−9) and Ser (−10) of Cx43 are indeed involved in binding to ZO-1 PDZ2. Consistent with this NMR data, quantitative fluorescence-based binding affinity measurement showed that the long Cx43 has >1-fold higher binding affinity than the short Cx43 peptide in binding to ZO-1 PDZ2 (Table I). Extending the Cx43 peptide to residues further upstream did not increase its binding to ZO-1 PDZ2 any more (data not shown). As Ser(−9) and Ser(−10) have been reported to be substrates of several kinases, including PKC and Akt (Keiichiro et al, 2002; Solan and Lampe, 2005; Park et al, 2007), it would be informative to elucidate the role(s) of these two Ser residues in the binding of Cx43 to ZO-1, preferably by obtaining atomic-level pictures of the interaction. The complex structure of ZO-1 PDZ2/Cx43 peptide also points to the likelihood that the phosphorylation of Ser(−9) and/or Ser(−10) would interfere with the charge–charge interaction network formed by Cx43 and the residues in the dimer interface of ZO-1 PDZ2 (Figure 3B–D). We tried extensively to obtain the crystals of the ZO-1 PDZ2/long Cx43 peptide, but failed to achieve this goal. As an alternative approach, we resorted to biochemical methods to evaluate the roles of the two Ser residues of Cx43 in binding to ZO-1 PDZ2. We substituted Ser(−9) and Ser(−10) in the long Cx43 peptide with glutamate individually to mimic their phosphorylation, and measured the binding affinities of the mutant peptides towards ZO-1 PDZ2. Interestingly, the Ser(−9)Glu and the Ser(−10)Glu Cx43 peptides displayed ∼7- and ∼2.5-fold lower affinities, respectively, compared with the wild-type peptide (Table I). Next, we directly evaluated the impact of Ser(−9) phosphorylation on the Cx43 peptide's binding to ZO-1 PDZ2 using a phospho-Cx43 peptide at Ser(−9). The phospho-Cx43 peptide was found to bind to ZO-1 PDZ2 with an affinity even weaker than that of the Ser(−9)Glu peptide (Table I). We also compared the bindings of all above Cx43 peptides to ZO-1 PDZ2 using NMR spectroscopy. Consistent with the results of the fluorescence-based assay, the long peptide-binding induced most significant chemical shift changes to the ZO-1 PDZ2, and the phosphorylation of Ser(−9) or the removal of Ser(−10) and Ser(−9) from the Cx43 peptide (the nine-residue peptide) significantly reduced the amplitude of the peptide binding-induced chemical shift changes of the PDZ domain (Figure 4C). Taken together, the above biochemical data indicate that the phosphorylation of the two Ser residues (Ser(−9) in particular) can indeed weaken the binding between Cx43 and ZO-1 PDZ2. Considering the polyvalent properties of both Cx43 in connexons and ZO-1 PDZ2 (ZO-1 can be further multimerized by inter-molecular interactions between its SH3 and GuK domains in addition to its PDZ2-mediated dimerization; McGee et al, 2001; Tavares et al, 2001), the impact of Ser(−9) and/or Ser(−10) phosphorylation of Cx43 on its ZO-1 binding (that is, decrease in the binding) is very large. Therefore, the phosphorylation/dephosphorylation of Ser(−9) and/or Ser(−10) on Cx43 can be readily used as an on/off switch for the Cx43–ZO-1 complex formation.
The Cx43 GJ dynamic regulation requires the functional interaction of the Cx43 tail with ZO-1 PDZ2
We next investigated the role of the Cx43 carboxyl tail-mediated binding to ZO-1 PDZ2 in GJ formation in HeLa cells. The HeLa cell line was chosen as the model system because HeLa cells lack the endogenous Cx43 but contain endogenous ZO-1. Consistent with the data reported earlier (Hunter et al, 2005), the exogenously expressed, N-terminal GFP-tagged full-length Cx43 localizes on the cell–cell contact regions as well as in cytosols. The membrane-localized Cx43 forms small, highly dense GJ plaques between two adjacent cells (Figure 5A1) when cells were grown to full confluence, and such Cx43 plaques colocalize very well with the endogenous ZO-1 plaques (Figure 5A2 and A4). We noted that the ZO-1 plaques are constantly formed regardless of the expression of GFP-Cx43. We next investigated whether the phosphorylation of Ser(−9) and Ser(−10) would affect Cx43 GJ formation using an S(−9)E/S(−10)E phosphorylation mimetic mutant of Cx43. In sharp contrast to the wild-type Cx43, the S(−9)E/S(−10)E Cx43 mutant appeared as much more diffused plaques with expanded sizes along plasma membranes (Figure 5B1). The distribution pattern of the S(−9)E/S(−10)E Cx43 mutant is very similar to that of Cx43 with the deletion of its carboxyl ZO-1 PDZ2-binding tail (Hunter et al, 2005). It is important to note that the expanded S(−9)E/S(−10)E Cx43 clusters no longer overlap with the dense ZO-1 plaques (Figure 5B2 and B4). As a control, GFP expressed in HeLa cells was diffused and had no impact on ZO-1 plaque formation and distribution (Figure 5C1–C4). The above data strongly indicate that the specific interaction between ZO-1 PDZ2 and Cx43 is indispensable for Cx43 GJ formation. Our data further imply that the ZO-1-mediated Cx43 GJ formation can be dynamically regulated by the phosphorylation of Ser373 and/or Ser372 of Cx43 (equivalent to Ser(−9) and Ser(−10) of the long Cx43 peptide).
Figure 5.

The specific interaction between ZO-1 PDZ2 and Cx43 is required for the formation of Cx43 GJs. (A1–C4) Comparison of the cellular localizations of the wild-type Cx43 and the S(−10)E, S(−9)E mutant of Cx43 in HeLa cells. The endogenous ZO-1 was stained with an anti-ZO-1 antibody and various forms of Cx43 were visualized by fluorescence signals from GFP. DAP staining (A3, B3, and C3) was used to show nuclei of cells. Colocalization of the wild-type Cx43 signal with the endogenous ZO-1 plaques between two adjacent transfected cells is indicated by arrowheads (A1–A4). Empty arrows indicate ZO-1 plaques between two adjacent cells that were transfected by GFP vector control or the GFP-Cx43 S(−10)E, S(−9)E mutant. The arrows highlight the endogenous ZO-1 plaques between two adjacent, untransfected cells. (D) A structural model showing six Cx43 pack together to form a connexon through their transmembrane helices. The Cx43 connexon model is built based on the connexon structure (PDB id: 1TXH). The cytoplasmic side of the connnexon points to readers and the distance between the two helix tails from two adjacent Cx43 is labelled. (E) The ribbon diagram representation showing the distance between two Cx43 peptides in the ZO-1 PDZ2–Cx43 peptide complex structure. In this drawing, the distance between the Cα atoms of Arg(−8) in Cx43 peptides is labelled. (F) A cartoon model showing the synergistic interactions between multimerized ZO-1 and polyvalent Cx43 connexons. In this mode, the hexameric Cx43 connexon can bind to the domain-swapped dimer of ZO-1 PDZ2, thereby greatly enhancing both the affinity and specificity of the binding of Cx43 connexon to ZO-1. Multimerization of ZO-1 through its SH3-GuK module can further enhance the interaction avidity and specificity.
Taking all structural, biochemical and cell biology data presented in this work together, we constructed a model depicting the ZO-1-mediated Cx43 GJ complex assembly (Figure 5D–F). The distance between the tails of the two neighbouring transmembrane helices of Cx43 is ∼36 Å (Figure 5D). Considering the long (∼140 residues) and flexible connecting sequence between the transmembrane helix and the PDZ-binding tail of Cx43, the existence of two Cx43-binding sites in the ZO-1 PDZ2 dimer, which are ∼30 Å apart (Figure 5E), should be able to promote and stabilize the assembly of six Cx43 molecules with one hemichannel as well as to cluster multiple hemichannels distributed on plasma membranes for the formation of functional GJs. The potential multimerization of ZO-1 through its SH3 and GuK domains should further increase its Cx43 GJ formation capacity.
In summary, the complex structure of ZO-1 PDZ2–Cx43 peptide provides a new paradigm for specific PDZ–target interactions as well as the dynamic regulation of these interactions. In the case of ZO-1 PDZ2, domain-swapped dimer assembly generates a highly specific Cx43-interacting site distinct to the canonical ligand-binding pocket of the PDZ domain. The extensive charge–charge interaction network in the second binding site formed between residues from both ZO-1 PDZ2 and Cx43 not only enhances the affinity and specificity of the ZO-1–Cx43 interaction but also builds a phosphorylation-dependent interaction switch into the ZO-1–Cx43 complex. Finally, the domain-swapped dimerization of PDZ domains represents a ‘clever' mechanism for achieving the highly specific PDZ–target interactions required by living cells.
Materials and methods
Protein expression and purification
A DNA fragment encoding human ZO-1 PDZ2 (residues 182–273) was amplified by PCR using the full-length human ZO-1 cDNA (from Alan Fanning at UNC-CH) as the template and cloned into an in-house modified version of pET32a vector. The full-length Cx43 gene was amplified by PCR from a rat brain cDNA library and cloned into the pEGFP.C3 vector. All point mutations of ZO-1 PDZ2, the full-length ZO-1 and the full-length Cx43 used in this study were created using the standard PCR-based mutagenesis method and confirmed by DNA sequencing. Recombinant proteins were expressed in BL21 (DE3) Escherichia coli cells at 16°C. His6-tagged ZO-1 PDZ2 proteins expressed in bacterial cells were purified by Ni2+-NTA agarose (Qiagen) affinity chromatography followed by size-exclusion chromatography. The N-terminal His6 tag of each recombinant protein was cleaved by protease 3C and removed by size-exclusion chromatography. Uniformly 15N or 1 5N, 13C-labelled PDZ proteins were prepared by growing bacteria in M9 minimal medium using 15NH4Cl as the sole nitrogen source or 15NH4Cl and 13C6-glucose (Cambridge Isotope Laboratories Inc.) as the sole nitrogen and carbon sources, respectively.
Crystallography
Crystals of ZO-1 PDZ2 in complex with the Cx43 peptide were obtained by the hanging drop vapour diffusion technique at 16°C. Freshly purified PDZ2 was concentrated to 4 mM before saturating amount of the Cx43 peptide (up to 4 molar ratio of the peptide to PDZ2) was added. The Cx43 peptide contained the last nine amino-acid residues of Cx43 and was commercially synthesized. The PDZ2/Cx43 peptide mixture was set up in hanging drops with equal volume of 0.2 M CaCl2, 0.1 M HEPES, pH 7.5 and 14% (w/v) polyethylene glycol 400. No further cryo-protectant was used. A 2.4-Å resolution X-ray data set was collected at 100 K by a Rigaku R-AXIS IV++ imaging-plate system with a Rigaku MicroMax-007 copper rotating-anode generator. The diffraction data were processed using the MOSFLM (Leslie, 1992) and scaled by the SCALA modules in the CCP4 suite (Storoni et al, 2004).
Molecular replacement was employed to solve the phase problem by using the PDZ domain structures of synaptojanin-2 binding protein (PDB id:2JIN) and PICK1 (PDB id: 2GZV) as the search models with PHASER (Storoni et al, 2004). The phase improvement and the initial model building were performed by RESOLVE (Terwilliger, 2000). The initial model was rebuilt manually and then refined using REFMAC (Murshudov et al, 1997) against the 2.4-Å resolution data set. Further manual model building and adjustment were completed using COOT (Emsley and Cowtan, 2004). The stereo-chemical quality of the final model was validated by PROCHECK (Laskowski et al, 1993). The final refinement statistics are listed in Table II. The structure figures were prepared using the program PyMOL (http://pymol.sourceforge.net/).
NMR spectroscopy
All protein samples for NMR titration experiments were concentrated to ∼0.2 mM in 50 mM Tris buffer containing 100 mM NaCl, 1 mM DTT and 1 mM EDTA at pH 6.5. NMR spectra were acquired at 30°C on Varian Inova 500 or 750 MHz spectrometers. Backbone resonance assignments were achieved by a combination of standard heteronuclear correlation experiments, including HNCO, HNCACB, CBCA(CO)NH using 15N/13C-labelled protein samples at a concentration of ∼1 mM (Bax and Grzesiek, 1993).
Fluorescence polarization assay
Fluorescence anisotropy binding assays were performed on a PerkinElmer LS-55 fluorimeter equipped with an automated polarizer at 25°C. Various forms of FITC-labelled Cx43 peptides were commercially synthesized, and the purities of the peptide were >95%. Fluorescence titration was performed with additions of increasing amount of ZO-1 PDZ2 proteins and to a constant concentration of FITC-labelled peptide (∼1 μM). The titration curves were fitted with the MicroCal Origin software package.
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed on a Beckman XL-I analytical ultracentrifuge equipped with an eight-cell rotor at 25°C. The partial specific volume of protein samples and the buffer density were calculated using the program SEDNTERP (http://www.rasmb.bbri.org/). The final sedimentation equilibrium data were analysed using the XL-A/XL-I data analysis software provided by the manufacturer.
Chemical cross-linking
Purified DLG5 PDZ2 was diluted to the indicated concentrations and centrifuged at 14 000 r.p.m. for 5 min to remove possible aggregation. The protein alone or in the mixture with 0.2 or 2 mM of disuccinimidyl glutarate (DSG) was incubated at room temperature for 30 min before the cross-linking reaction was quenched by the addition of 1 M Tris–HCl (pH 7.5). Samples were then analysed by SDS–PAGE.
Cell culture, immunostaining and imaging
HeLa cells were cultured in MEM medium (Invitrogen, Grand Island, NY) supplemented with fetal bovine serum. For immunostaining, HeLa cells were cultured on coverslips coated with 0.2% gelatin. The wild-type/mutant GFP-Cx43/GFP-ZO-1 was introduced into HeLa cells by lipofectamine transfection method. The cells were fixed at 36–48 h after transfection by 4% paraformaldehyde and 4% sucrose in PBS, then permeabilized by 0.2% Triton X-100 in PBS for 10 min at room temperature. After blocking with 10% normal donkey serum (NDS) in PBS, the cells were incubated with anti-ZO-1 mAb (from B Peng, Department of Biology, HKUST) in 3% NDS for 1 h at room temperature, followed by 1 h incubation with Red-X-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA). The cells were imaged with a Nikon Eclipse TE2000 (Nikon, Tokyo, Japan) inverted fluorescence microscope.
Coordinates
The atomic coordinates of the ZO-1 PDZ/Cx43 peptide complex have been deposited in the Protein Data Bank under the accession code 3CYY.
Supplementary Material
Supplementary Figure and Legends
Acknowledgments
We thank Miss Ling-Nga Chan for the help in cell biology experiments, Dr Alan Fanning for the full-length ZO-1 cDNA used in this study, Dr H Benjamin Peng for the anti-ZO-1 antibody and Mr Anthony Zhang for the critical reading of the paper. This study was supported by grants from the Research Grants Council of Hong Kong to MZ (HKUST6419/05M, 6442/06M, 663407, CA07/08.SC01, AoE/B-15/01-II and AoE/M-04/04). The NMR spectrometer used in this study was purchased with funds donated to the Biotechnology Research Institute by the Hong Kong Jockey Club. MZ was a recipient of the Croucher Foundation Senior Research Fellow Award.
Conflict of interest The authors declare that they have no competing financial interests.
References
- Adachi M, Inoko A, Hata M, Furuse K, Umeda K, Itoh M, Tsukita S (2006) Normal establishment of epithelial tight junctions in mice and cultured cells lacking expression of ZO-3, a tight-junction MAGUK protein. Mol Cell Biol 26: 9003–9015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akoyev V, Takemoto DJ (2007) ZO-1 is required for protein kinase C gamma-driven disassembly of connexin 43. Cell Signal 19: 958–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler Y, Hodson C, Milgram SL (2003) The apical compartment: trafficking pathways, regulators and scaffolding proteins. Curr Opin Cell Biol 15: 423–429 [DOI] [PubMed] [Google Scholar]
- Bax A, Grzesiek S (1993) Methodological advances in protein NMR. Acc Chem Res 26: 131–138 [Google Scholar]
- Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E (2000) Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem 275: 20520–20526 [DOI] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB (1992) The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 9: 929–942 [DOI] [PubMed] [Google Scholar]
- Craven SE, Bredt DS (1998) PDZ proteins organize synaptic signaling pathways. Cell 93: 495–498 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fanning AS, Lye MF, Anderson JM, Lavie A (2007) Domain swapping within PDZ2 is responsible for dimerization of ZO proteins. J Biol Chem 282: 37710–37716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning AS, Ma TY, Anderson JM (2002) Isolation and functional characterization of the actin-binding region in the tight junction protein ZO-1. FASEB J 16: 1835–1837, 02-0121fje [DOI] [PubMed] [Google Scholar]
- Giepmans BN (2004) Gap junctions and connexin-interacting proteins. Cardiovasc Res 62: 233–245 [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Moolenaar WH (1998) The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol 8: 931–934 [DOI] [PubMed] [Google Scholar]
- Harris BZ, Lim WA (2001) Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci 114: 3219–3231 [DOI] [PubMed] [Google Scholar]
- Hunter AW, Barker RJ, Zhu C, Gourdie RG (2005) Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell 16: 5686–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 147: 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Nagafuchi A, Moroi S, Tsukita S (1997) Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol 138: 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno T, Umeda K, Matsui T, Hata M, Tamura A, Itoh M, Takeuchi K, Fujimori T, Nabeshima YI, Noda T, Tsukita S, Tsukita S (2008) Deficiency of ZO-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell 19: 2465–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiichiro Y, Takuya O, Motofusa A, Norihiro I, Tatsuo T (2002) Identification and functional analysis of novel phosphorylation sites in Cx43 in rat primary granulosa cells. FEBS Lett 531: 132–136 [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5: 771–781 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067 [DOI] [PubMed] [Google Scholar]
- Leslie AGW (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography, no. 26
- Maass K, Shibayama J, Chase SE, Willecke K, Delmar M (2007) C-Terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ Res 101: 1283–1291 [DOI] [PubMed] [Google Scholar]
- Macara IG (2004) Par proteins: partners in polarization. Curr Biol 14: R160–R162 [PubMed] [Google Scholar]
- Margolis B, Borg JP (2005) Apicobasal polarity complexes. J Cell Sci 118: 5157–5159 [DOI] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell 8: 1291–1301 [DOI] [PubMed] [Google Scholar]
- McNeil E, Capaldo CT, Macara IG (2006) Zonula occludens-1 function in the assembly of tight junctions in Madin–Darby canine kidney epithelial cells. Mol Biol Cell 17: 1922–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitic LL, Schneeberger EE, Fanning AS, Anderson JM (1999) Connexin-occludin chimeras containing the ZO-binding domain of occludin localize at MDCK tight junctions and NRK cell contacts. J Cell Biol 146: 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller SL, Portwich M, Schmidt A, Utepbergenov DI, Huber O, Blasig IE, Krause G (2005) The tight junction protein occludin and the adherens junction protein alpha-catenin share a common interaction mechanism with ZO-1. J Biol Chem 280: 3747–3756 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nechiporuk T, Fernandez TE, Vasioukhin V (2007) Failure of epithelial tube maintenance causes hydrocephalus and renal cysts in Dlg5−/− mice. Dev Cell 13: 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DJ, Wallick CJ, Martyn KD, Lau AF, Jin C, Warn-Cramer BJ (2007) Akt phosphorylates connexin43 on Ser373, a ‘mode-1' binding site for 14-3-3. Cell Commun Adhes 14: 211–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Solan JL, Taffet SM, Javier R, Lampe PD (2005) Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem 280: 30416–30421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD (2005) Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta 1711: 154–163 [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275: 73–77 [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr 60: 432–438 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ohno S (2006) The PAR-aPKC system: lessons in polarity. J Cell Sci 119: 979–987 [DOI] [PubMed] [Google Scholar]
- Tavares GA, Panepucci EH, Brunger AT (2001) Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol Cell 8: 1313–1325 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr 56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M (1998) Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 273: 12725–12731 [DOI] [PubMed] [Google Scholar]
- Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S (2006) ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126: 741–754 [DOI] [PubMed] [Google Scholar]
- van Zeijl L, Ponsioen B, Giepmans BN, Ariaens A, Postma FR, Varnai P, Balla T, Divecha N, Jalink K, Moolenaar WH (2007) Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J Cell Biol 177: 881–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott E, Balda M, Fanning A, Jameson B, Itallie C, Anderson J (1993) The tight junction protein ZO-1 is homologous to the Drosophila discs––large tumor suppressor protein of septate junctions. Proc Natl Acad Sci 90: 7834–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods DF, Bryant PJ (1993) ZO-1, DlgA and PSD-95/SAP90: homologous proteins in tight, septate and synaptic cell junctions. Mech Dev 44: 85–89 [DOI] [PubMed] [Google Scholar]
- Wu J, Yang Y, Zhang J, Ji P, Du W, Jiang P, Xie D, Huang H, Wu M, Zhang G, Wu J, Shi Y (2007) Domain-swapped dimerization of the second PDZ domain of ZO2 may provide a structural basis for the polymerization of claudins. J Biol Chem 282: 35988–35999 [DOI] [PubMed] [Google Scholar]
- Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W (2008) Early embryonic lethality of mice lacking ZO-2, but not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol 28: 1669–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang W (2003) Organization of signaling complexes by PDZ-domain scaffold proteins. Acc Chem Res 36: 530–538 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yeh S, Appleton BA, Held HA, Kausalya PJ, Phua DCY, Lee Wong W, Lasky LA, Wiesmann C, Hunziker W, Sidhu SS (2006) Convergent and divergent ligand specificity among PDZ domains of the LAP and zonula occludens (ZO) families. J Biol Chem 281: 22299–22311 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure and Legends