

Abstract
Interestingly, some lymphoma cells, expressing high levels of transmembrane (tm)TNF-α, are resistant to secretory (s)TNF-α-induced necrosis but sensitive to tmTNF-α-mediated apoptosis. As tmTNF-α mediates “forward” as well as “reverse” signaling, we hypothesize that a balanced signaling between forward and reverse directions may play a critical role in determining the fate of cells bearing tmTNF-α. Using Raji cells as a model, we first added exogenous tmTNF-α on fixed, transfected NIH3T3 cells onto Raji cells to examine tmTNF-α forward signaling and its effects, showing that constitutive NF-κB activity and cellular inhibitor-of-apoptosis protein 1 transcription were down-regulated, paralleled with Raji cell death. As Raji cells express tmTNF-α, an inhibition of their tmTNF-α expression by antisense oligonucleotide caused down-regulation of NF-κB activity. Conversely, increasing tmTNF-α expression by suppressing expression of TNF-α-converting enzyme that cleaves tmTNF-α led to an enhanced activation of NF-κB, indicating that tmTNF-α, but not sTNF-α, contributes to constitutive NF-κB activation. We next transfected Raji cells with a mutant tmTNF-α lacking the intracellular domain to competitively suppress reverse signaling via tmTNF-α; as expected, constitutive NF-κB activity was decreased. In contrast, treating Raji cells with sTNFR2 to stimulate reverse signaling via tmTNF-α ehanced NF-κB activation. We conclude that tmTNF-α, when highly expressed on tumor cells and acting as a receptor, promotes NF-κB activation through reverse signaling, which is helpful to maintain tumor cell survival. On the contrary, tmTNF-α, when acting as a ligand, inhibits NF-κB activity through forward signaling, which is inclined to induce tumor cell death.
Keywords: tmTNF, sTNF, NF-κB, cancer
INTRODUCTION
TNF-α is a pleiotropic, proinflammatory cytokine with a wide range of biological effects [1]. TNF-α exists in two biologically active forms, transmembrane TNF-α (tmTNF-α) and secretory TNF-α (sTNF-α). tmTNF-α is expressed as a type II transmembrane protein, which can be cleaved by TNF-α-converting enzyme (TACE) to release the extracellular C-terminal portion with 157 aa, namely, sTNF-α [2]. tmTNF-α and sTNF-α possess bioactivities when structured as homotrimers and perform their functions by binding to two distinct TNFRs: TNFR1 and TNFR2 [3]. Although the biological activities mediated by sTNF-α, which include cell proliferation, apoptosis, and cytokine production in immune and inflammatory responses, are well-studied, the functional role of tmTNF-α is not completely understood.
Previous studies have shown that sTNF-α and tmTNF-α exhibit antitumor activity [4]. However, the mechanisms by which sTNF-α and tmTNF-α mediate cytotoxicity to tumor cells are different. tmTNF-α is not only cytotoxic to sTNF-α-sensitive target cells but is also cytotoxic to sTNF-α-resistant tumor cells [5, 6], thus exhibiting a broader, tumoricidal spectrum than sTNF-α. In addition, sTNF-α mediates signals predominantly via TNFR1, whereas tmTNF-α is the prime activating ligand for TNFR2. Although signals transmitted from tmTNF-α to target cells are likely to be mediated through both TNFRs, tmTNF-α seems to exert its cytotoxicity by a signaling pathway distinct from that of sTNF-α [7]. In vivo experiments have revealed that sTNF-α-induced tumor regression is associated with the infiltration of lymphocytes into the tumor, whereas tmTNF-α causes inhibition of tumor growth by promoting Fas expression in tumor cells [8].
It is known that tmTNF-α functions not only as an activating ligand to trigger forward signaling into target cells but also as a receptor to transmit reverse signals into tmTNF-α-bearing cells [9]. Ferran et al. [10] have demonstrated that stimulation of T cells with anti-TNF antibodies provides a costimulatory signal for CD3-mediated activation of transcription of IFN-γ and IL-4. In human monocytes/macrophages, reverse signaling through tmTNF-α confers resistance to LPS [11]. In addition, reverse signaling mediated by tmTNF-α induces E-selectin expression in activated human CD4+ T cells [12]. As E-selectin is a well-defined target of NF-κB, we have hypothesized that tmTNF-α-mediated reverse signaling may be able to activate NF-κB.
NF-κB is a transcriptional factor composed of homodimeric and heterodimeric complexes of related proteins from the Rel superfamily [13]. NF-κB, sequestered in the cytoplasm as a latent form stabilized by the inhibitory subunit IκB-α [14], becomes activated as a result of ubiquitination and degradation of IκB. Release of NF-κB from the NF-κB/IκB complex allows the active dimer to translocate into the nucleus, where it binds to the κB-responsive motif within the target gene promoter, thereby activating transcription of NF-κB-responsive genes [15]. In many cell types, TNF-α-induced apoptosis can be prevented by parallel, TNF-α-induced production of antiapoptotic proteins, such as cellular inhibitor-of-apoptosis protein (cIAP) and cellular Fas-association death domain-like IL-1β-converting enzyme-like inhibitory protein. It has been demonstrated that the production of these antiapoptotic proteins is mediated by NF-κB, and inhibiting NF-κB activation sensitizes cancer cells to TNF-α-induced apoptosis [16].
The present study has been prompted by our previous observation that tmTNF-α could kill sTNF-α-resistant Raji cells [6]. Using Raji cells as a model, we have investigated molecular mechanism(s) by which Raji cells are sensitive to tmTNF-α-induced apoptosis but resistant to sTNF-α-induced cell death. We found that exogenous tmTNF-α is able to kill Raji cells by inhibiting NF-κB activation via forward signaling, and the Raji cell’s own endogenous tmTNF-α appears to be responsible for constitutive activation of NF-κB via reverse signaling, rendering Raji cells resistant to sTNF-α-mediated cytotoxicity.
MATERIALS AND METHODS
Cell lines
Raji, a malignant B cell line derived from Burkitt lymphoma, was used as a target cancer cell line. The NIH3T3 cell line was stably transfected with pLW-TNF-α, a retrovirus plasmid inserted with the human wild-type (wt) TNF-α cDNA at the HpaI and XhoI cloning site of the pLXSN vector. TNF-α-transfected NIH3T3 cells overexpressing tmTNF-α on the cell surface were fixed in 1% paraformaldehyde and used as the source of exogenous tmTNF-α. To remove receptor-bound sTNF-α, we incubated cells with acid glycine buffer (Gly-NaCl, pH 3.0) for 15 min after fixation. All cell lines were cultured at 37°C in 5% CO2 in RPMI-1640 medium (Gibco, Grand Island, NY, USA), supplemented with 10% heat-inactivated (56°C, 30 min), pyrogen-free FCS (Sijiqing, Hangzhou, China), 1.0 mM sodium pyruvate, 2.0 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 × 10−5 M 2-ME.
Plasmids
Dr. M. Lienhard Schmitz (Deutsches Krebsforschungszentrum, Germany) provided the plasmid 3× NF-κB–luciferase reporter, containing the promoter for the κB-response element. The plasmid pcDNA3.0, containing the cytoplasmic segment (Δcs)-tmTNF-α that lacks a large portion of the cytoplasmic domain of TNF-α from positions −73 to −47, was constructed in our laboratory as described previously [17]. The pET-28a/sTNFR2 plasmid containing human cDNA that codes for the extracellular region of sTNFR2 (123–825) was generated by inserting a RT-PCR product into the pET-28a vector at the BamHI and HindIII sites. This RT-PCR product was amplified using a pair of primers: P1, 5′-CAGGATCCGTCGGACTGGAGCTCT-3′, and P2, 5′-CCCAAGCTTCAATCAGTCCAACTGGAAG-3′, for 30 cycles (94°C for 30 s, 66°C for 30 s, 72°C for 60 s).
Gene transfection, expression, and the reporter assays
Raji cells (1×106) were transiently cotransfected with a Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) solution containing 2 μg 3× NF-κB–luciferase reporter plasmid, plus a SV-40 promoter-driven β-galactosidase expression plasmid (Promega, Madison, WI, USA). The medium was changed to the complete culture medium after 4 h of transfection, and the incubation was continued for 24 h. These transfected Raji cells were challenged with sTNF-α (100 U/ml) or tmTNF-α (E:T ratio of 10:1) at different time-points before harvesting. Harvested cell extracts were prepared by addition of 100 μl cell lysis buffer. A portion of the extracts was tested for luciferase activity using the Luciferase assay system (Promega), according to the protocol provided, and then, the resulting luminescence was quantified with a Turner Luminometer Model 20. Another portion of extract was tested for β-galactosidase activity by addition of an equal volume of β-galactosidase assay buffer (200 mmol/L sodium phosphate buffer, pH 7.3, 2 mmol/L MgCl2, 100 mmol/L 2-ME, and 1.3 mg/mL o-nitrophenyl β-D-galactopyranoside), with incubation for 4 h at 37°C, followed by measurement of sample absorbance at 420 nm. Luciferase activities were normalized based on the paired β-galactosidase activities obtained.
For transfection of the Δcs-tmTNF-α-pcDNA3.0, 2 × 106 Raji cells were incubated with Lipofectamine 2000 containing 5 μg Δcs-tmTNF-α-pcDNA3.0 or as a control, empty vector pcDNA3.0. Following 6 h of transfection, 4 ml complete culture medium was added, and the incubation was continued for an additional 42 h. Subsequently, the cells were collected to test for tmTNF-α expression and NF-κB activation.
To obtain sTNFR2, the pET-28a/sTNFR2 plasmid was expressed in Escherichia coli stimulated with 1 mM isopropylthiogalactoside and purified by nickel ion 2±-nitrilotriacetic acid resin, up to 95% purity. Endotoxin was removed by using a Detoxi-Gel endotoxin-removing gel column (Pierce, Rockford, IL, USA), according to the manufacturer’s instructions. Residual endotoxin concentration was measured at <0.2 U/mg.
Confocal microscopy
Raji cells were harvested at different time-points after stimulation with tmTNF-α (at an E:T ratio of 10:1). The cells were fixed by incubation with 95% ethanol at 4°C for 2 h, washed three times with PBS, and then permeabilized by treatment with 0.1% Triton X-100/PBS for 10 min. After washing with PBS, they were incubated with a rabbit anti-NF-κB/p65 antibody (1:100) for 1 h. After further PBS washes, a FITC-labeled, anti-rabbit IgG was applied. The cells were also costained with propidium iodide (PI) for nuclear staining. A quantity of 1 × 104 cells in a volume of 50 μl PBS was mounted onto slides and observed under a confocal microscope FU5000 (Olympus, Tokyo, Japan).
RNA isolation and real-time RT-PCR
Total RNA was isolated using the Tripure isolation reagent (Roche, Indianapolis, IN, USA), according to the manusfacturer’s instructions. RNA (800 ng) was reversely transcribed to cDNA by using the GeneAmp RNA PCR kit (Perkin Elmer, Foster City, CA, USA). Real-time PCR was performed by using the Platinum SYBR Green Quantitative PCR SuperMix UDG kit (Invitrogen) in the Rotor gene3000 system (Corbett Research, Sydney, Australia). Each PCR mixture (in a total of 20 μl) contained 3 mM MgCl2, 200 μM each dNTP, 0.5 μM each primer, 1 μl cDNA, and 1.5 units Platinum Taq DNA polymerase. The following protocol was used: 94°C for 2 min and then 95°C 10 s, 55°C 20 s, and 72°C 20 s for 45 cycles. The following primers were chemically synthesized with a DNA synthesizer (Bioasia, China): cIAP1 (160 bp) [18], forward primer: 5′-AGCTGTTGTCAACTTCAGATACCACT-3′, reverse primer: 5′-TGTTTCACCAGGTCTCTATTAAAGCC-3′; β-actin (150 bp), forward primer: 5′-AGTTGCGTTACACCCTTTC-3′, reverse primer: 5′-CACCTTCACCGTTCCAGT-3′.
ELISA
NF-κB activity was examined by an ELISA method described previously [19]. Briefly, 2 × 106-treated or untreated Raji cells were lysed in 50 μl lysis buffer (20 mM HEPES, pH 7.5, 0.35 M NaCl, 20% glycerol, 1% Nonidet P-40, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA) containing a protease inhibitor cocktail (Calbiochem, San Diego, CA, USA). After incubating on ice for 10 min, these lysates were centrifuged for 20 min at 13,000 rpm, and the supernatants were harvested for measurement. Two single-stranded oligonucleotide chains, 5′-AGTTGAGGGGACTTTCCCAGGC-C-(C)34-C-3-bio′, which is biotinylated at the 3′ end, and 5′-GCCTGGGAAAGTCCCCTCAACT-3′, were synthesized (Sangon, Shanghai, China). The two chains were mixed at a ratio of 1:1, denatured at 94°C for 10 min, and then allowed to anneal at room temperature to form the double-stranded probe, which bound to streptavidin-coated, 96-well plates at an end concentration of 2 pmol by its conjugated biotin. After washing these plates with PBS containing 0.1% Tween-20, 20 μl whole-cell lysate containing 5 μg protein mixed with 30 μl binding buffer (4 mM HEPES, pH 7.5, 100 mM KCl, 8% glycerol, 5 mM DTT, 0.2% BSA, 40 μg/ml salmon sperm DNA) was added and incubated for 1 h at room temperature. Then, the NF-κB activity was detected using a mAb against NF-κB p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:1000, followed by a secondary antibody of peroxidase-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology) at a dilution of 1:1000 and the substrate tetramethylbenzidine (Shanghai Lizhu-Dongfeng Biotechnology Corp., Shanghai, China). Subsequently, the OD was read at 450 nm in a microplate reader (Tecan, Austria), with 655 nm as the reference wavelength.
To measure secretory TNF-α, 1 × 106 Raji cells were first treated with antisense (AS) specific to TACE for 24 h, and then the supernatants were collected and tested for sTNF-α by ELISA (eBioscience, San Diego, CA, USA). For this, a mouse mAb specific for human TNF-α (clone mAb1) was used to coat tissue-culture plates. After incubation with samples and a recombinant human TNF-α standard, another biotin-conjugated mAb against TNF-α (clone mAb11) was added, followed by Avidin-HRP. Color developed following addition of the tetramethylbenzidine substrate, and OD was read at 450 nm under the same microplate reader, using 630 nm as the reference wavelength.
AS treatment
The AS used were designed to specifically target TNF-α and TACE. Their target sequences were 5′-CTGTGGTACTCGTGACTTTC-3′ [20] and 5′-CAGGAATAGGAGAGACTGCCT-3′, respectively. Also, two nonsense oligonucleotides (NS), 5′-CTTTTTGAGCCAGAAGAGGT-3′ and 5′-GATGAGAGAGTCTCCTAGTTG-3′, served as their respective controls. Raji cells were transfected with 10 μM AS or NS using the Lipofectamine 2000 reagent. Transfected cells were analyzed for tmTNF-α and TACE expression or tested for NF-κB activity and response to sTNF-α treatment.
Flow cytometry
After transfection with AS, cells were collected and washed with PBS, incubated with rabbit polyclonal antibodies against TNF-α or TACE (Santa Cruz Biotechnology) for 1 h at 4°C, and then incubated with secondary antibody against rabbit IgG labeled with FITC (Jackson Biotech, West Chester, PA, USA) for 1 h at 4°C. After washing with 0.5% BSA PBS, analyses for the Raji cells’ expression of tmTNF-α or TACE were performed on a FACSCalibur 440 E flow cytometer (Becton Dickinson, San Jose, CA, USA).
Cytotoxicity and proliferation assays
The biological activities of hsTNF-α (PeproTech Co., Rocky Hill, NJ, USA) and htmTNF-α, obtained through surface expression on NIH3T3 cells transfected by pLW-TNF-α, were detected by measuring their cytotoxicities against Raji cells. These target cells (5×104) were seeded into 96-well microtiter plates and incubated overnight at 37°C in 5% CO2. The following day, 100 U/ml sTNF-α and 5 × 105 fixed-transfected or nontransfected NIH3T3 cells per well (at an E:T ratio of 10:1) were added and incubated for 24 h. Cell viability was then measured by staining for 4 h with 30 mM glucose-PBS containing 0.5 mg/ml MTT (Sigma Chemical Co., St. Louis, MO, USA), followed by cell lysis with 0.1 ml DMSO. The photometric measurement was performed at 570 nm on a microplate reader (Titertek Multiskan Scanner, Flow Laboratories, McLean, VA, USA). TNF-α-induced cytotoxicity or proliferation was calculated by using the following formulas: Cell death rate (%) = (1–ODsample/ODcontrol) × 100%, and cell proliferation rate (%) = (ODsample/ODcontrol–1) × 100%.
Statistical analysis
All data were statistically analyzed by a software Statistical Package for the Social Sciences. Differential significance was analyzed using the Student’s t-test. Data are presented as mean ± sd. Differences were considered to be statistically significant at P < 0.05.
RESULTS
Raji cells responded differently to tmTNF-α and sTNF-α
To compare the effects of tmTNF-α and sTNF-α on Raji cells, we treated these cells separately with 100 U/ml sTNF-α and exogenous tmTNF-α expressed on the surface of 1% paraformaldehyde-fixed NIH3T3 cells that were stably transfected with TNF-α at an E:T ratio of 10:1. As shown in Figure 1, tmTNF-α killed ∼60% of Raji cells after 24 h of incubation, as compared with Raji cells that were incubated with nontransfected NIH3T3 cells (P<0.01). This cytotoxic effect of tmTNF-α on Raji cells could be blocked effectively by anti-TNF-α antibody. In contrast, sTNF-α failed to kill Raji cells but stimulated cell proliferation instead.
Fig. 1.

The cytotoxic and proliferative effects of tmTNF-α and sTNF-α on Raji cells, respectively. For sTNF-α activity, Raji cells (5×104) were incubated with 100 U/ml sTNF-α or medium alone (control). For exogenous tmTNF-α activity, the same number of Raji cells was incubated with 5 × 105-fixed NIH3T3 cells transfected with tmTNF-α or with an empty vector (vehicle). The treatment was for 24 h. The specificity of TNF-α activity was corroborated by 1 h preincubation of the fixed transfectants with an anti-human TNF-α mAb. Cell viability was measured by MTT assay. Data represent mean percentage (bars, ±sd) of three replicates from four independent experiments; *, P < 0.01.
Different effects of tmTNF-α and sTNF-α on NF-κB activation in Raji cells
NF-κB is known to be an important regulator at the crossroad for deciding cell death versus survival and proliferation. To evaluate the possibility that tmTNF-α and sTNF-α induce distinct cellular responses via regulation of NF-κB, we tested NF-κB activity in Raji cells treated with these two forms of TNF-α. The NF-κB activity was measured using a luciferase assay in Raji cells transfected with a plasmid containing the κB response element in the promoter. As seen in Figure 2A, Raji cells transfected with this reporter plasmid expressed high levels of luciferase activity, suggesting that these malignant cells have constitutive activation of NF-κB. Treatment with sTNF-α did not further increase this NF-κB activity. Interestingly, we found that treatment with tmTNF-α significantly down-regulated the constitutive activation of NF-κB, and luciferase activity declined as early as 30 min after exposure to tmTNF-α. When exposure time was prolonged to 4 h, the constitutive activation of NF-κB in Raji cells was suppressed completely (Fig. 2B). In consonance with this observation, confocal microscopy revealed that the persistent translocation of NF-κB from the cytoplasm to the nucleus gradually decreased within the stimulatory time until it became nearly completely blocked by 4 h after tmTNF-α treatment (Fig. 2D).
Fig. 2.

Inhibitory effect of tmTNF-α on NF-κB activation and NF-κB-mediated transcription of cIAP-1 in Raji cells, which (A) when transiently cotransfected with a 3× NF-κB–luciferase reporter construct and a β-galactosidase expression vector, were stimulated for 30 min with sTNF-α (100 U/ml) or with fixed NIH3T3 cells transfected with tmTNF-α (at an E:T ratio of 10:1). Cells treated with nontransfection and left untreated served as controls. Luciferase activity was measured and normalized against the β-galactosidase activity. Data represent the mean of three independent experiments. Time course of inhibitory effect of tmTNF-α on NF-κB activation and cIAP transcription was performed by incubation of the Raji cells transfected by 3× NF-κB–luciferase reporter with fixed NIH3T3 cells (at an E:T ratio of 10:1) transfected with tmTNF-α or empty plasmid for the indicated time-points. Luciferase activity was measured, and data represent the mean of three independent experiments (B). The transcription level of cIAP-1 was analyzed by real-time RT-PCR. The results shown are representative of three independent experiments (C). The translocation of NF-κB in Raji cells was detected by confocal microscopy (D). An anti-NF-κB/p65 polyclonal antibody, a FITC-labeled, anti-IgG antibody, and PI nuclear counterstain were used to stain cells. NF-κB (green stain), translocated from the cytoplasm to the nucleus (red stain), showed as yellow fluorescence in the merged images, which were representative of three slides scanned; *, P < 0.01.
The gene of cIAP-1, an antiapoptotic molecule, is one of NF-κB-targeted genes. It is also constitutively transcribed into mRNA in Raji cells. The treatment with tmTNF-α led to obvious down-regulation of cIAP-1 transcription at 60 min, one-half hour later than inhibition of NF-κB activation. This suppressive effect on cIAP-1 transcription was more evident when protracted incubation time to 240 min (Fig. 2C). The inhibition of cIAP-1 transcription owing to down-regulation of NF-κB by tmTNF-α may be responsible for the sensitivity of Raji cells to tmTNF-α cytotoxicity.
Inhibition of NF-κB activation sensitized Raji cells to sTNF-α-induced cytotoxicity
As the sensitization of Raji cells to tmTNF-α-induced apoptosis is a result of its inhibition of constitutive activation of NF-κB, we investigated whether inhibition of NF-κB by direct use of the NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) could sensitize Raji cells to sTNF-α-mediated cytotoxicity. Indeed, treatment of Raji cells with PDTC inhibited NF-κB activation in a dose-dependent manner. PDTC alone, in the higher concentrations above 50 μM, significantly inhibited NF-κB activation and induced cell death (Fig. 3, A and B). However, at the concentration of 5 μM, PDTC did not affect cell viability, yet it still suppressed NF-κB activity by 50%. Interestingly, at this low dose, PDTC conferred sensitivity to the cytotoxic effect of sTNF-α (Fig. 3, C and D). These results indicate that the constitutive NF-κB activation contributes to the resistance of Raji cells to sTNF-α-induced cell death.
Fig. 3.

Sensitization of Raji cells to sTNF-α-induced cytotoxicity by the NF-κB inhibitor PDTC. (A and B) Raji cells (5×104) were treated with different concentrations of PDTC as indicated for 1 h. (C and D) Raji cells (5×104) pretreated with 5 μM PDTC for 1 h were incubated with 100 U/ml sTNF-α for 24 h. Untreated cells and cells treated with sTNF-α or PDTC alone served as controls. Cell viability was measured by MTT, and NF-κB activity was determined by ELISA. Data represent the mean of three independent experiments; *, P < 0.01.
Constitutive NF-κB activation was associated with a high level of tmTNF-α in Raji cells
As NF-κB is constitutively activated in Raji cells, we evaluated whether this NF-κB activation was associated with endogenous tmTNF-α expression. Through flow cytometry analysis, we found that there was a high level of tmTNF-α expressed by Raji cells (Fig. 4A). To further determine the contribution of this endogenous tmTNF-α to NF-κB activation, Raji cells were transfected with TNF-α AS to silence the TNF-α expression. As shown in Figure 4A, only transfection of TNF-α AS, but not the control NS, inhibited over 50% of endogenous tmTNF-α expression. Consequently, the constitutive NF-κB activation was down-regulated as compared with the nontransfection and transfection with NS (P<0.01; Fig. 4B). Interestingly, the down-regulation of endogenous tmTNF-α by specific AS sensitized Raji cells to exogenous sTNF-α cytotoxicity (Fig. 4C).
Fig. 4.

Constitutive NF-κB activation and resistance to sTNF-α cytotoxicity mediated by overexpression of endogenous TNF-α. Raji cells (2×106) were transfected with 10 μM TNF-α antisense or with 10 μM mismatched NS for 24 h. (A) tmTNF-α expression on the surface of Raji cells was detected by flow cytometry using a polyclonal antibody specific to TNF-α. The results as shown by mean fluorescence intensity (MFI) are representative of at least three independent experiments. (B) The transfectants and the nontransfected cells were lysed, and the NF-κB activity was measured by ELISA. (C) Cells (5×104) with transfection of TNF-AS or NS or with nontransfection were incubated with 100 U/ml sTNF-α for 24 h. The viability of cells was determined by MTT. Data represent the mean of three independent experiments; *, P < 0.01.
However, TNF-α AS not only inhibited tmTNF-α expression but also suppressed sTNF-α production. To resolve whether sTNF-α may also be responsible for NF-κB activation, we performed an assay to decrease sTNF-α but increase tmTNF-α expression by using a specific AS against TACE that inhibits the cleavage of tmTNF-α into sTNF-α. We found that transfection of Raji cells with TACE AS significantly reduced the expression of TACE by ∼70% (Fig. 5A). Consequently, tmTNF-α expression was increased by fourfold (Fig. 5B), and sTNF-α release was decreased by 46.78% (Fig. 5C). In Figure 5D, we show that NF-κB activity was up-regulated by treatment with TACE AS. These results suggest that tmTNF-α, but not sTNF-α, is associated with the constitutive activation of NF-κB in Raji cells.
Fig. 5.

Enhancement of NF-κB activity by suppressing the cleavage of tmTNF-α into sTNF-α. Raji cells (6×106) were transfected with 10 μM TACE-AS or NS as control for 24 h. Expression of TACE (A) and tmTNF-α (B) on the cell surface was detected by flow cytometry using polyclonal antibodies specific for TACE or TNF-α. The results are representative of at least three independent experiments. Release of sTNF-α in the supernatants was determined by ELISA (C), and NF-κB activity was measured by ELISA (D). Data represent the mean of three independent experiments; *, P < 0.01.
tmTNF-α-mediated reverse signaling for NF-κB activation
It is known that tmTNF-α functions not only as an activating ligand that binds TNFR to deliver forward signals but also as a receptor that receives and transmits reverse signals. To clarify whether tmTNF-α mediates reverse signals for constitutive activation of NF-κB, the plasmid containing a Δcs-tmTNF-α mutant that lacks the cytoplasmic segment was transfected into Raji cells. We anticipated that overexpression of the mutant tmTNF-α could block endogenous wt-tmTNF-α-mediated reverse signaling. As expected, transfection of Δcs-tmTNF-α diminished the constitutive activation of NF-κB as compared with the cells without transfection or with transfection of the control vector (P<0.01; Fig. 6A).
Fig. 6.
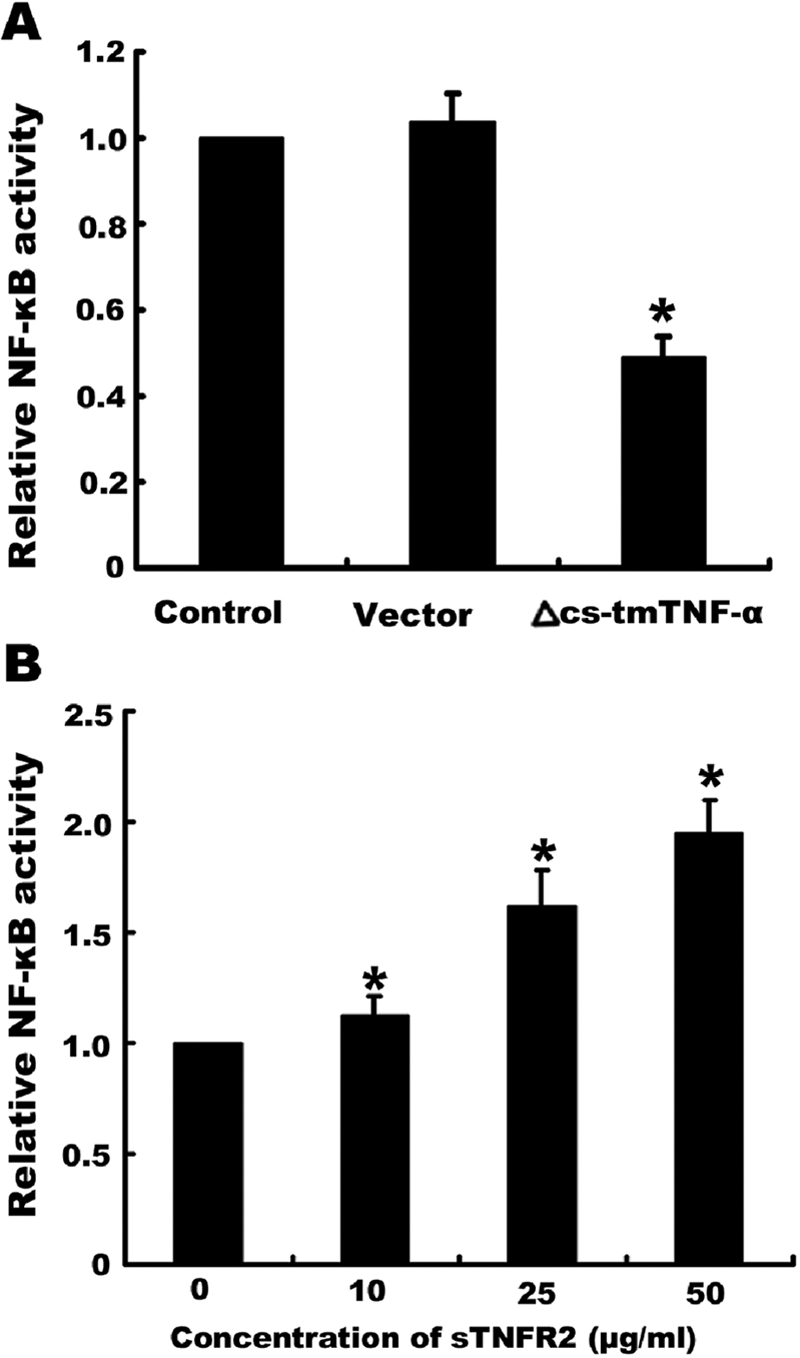
The effect on NF-κB activation of tmTNF-α-mediated reverse signaling by transfection of a mutant tmTNF-α plasmid or by treatment with sTNFR2. (A) Raji cells (1×106) were transfected with pcDNA3.0 containing Δcs-tmTNF-α cDNA or the control empty vector. After a 48-h transfection, cells were harvested and lysed in lysis buffer. NF-κB activity was detected by ELISA. (B) Raji cells (1.5×106) were incubated with different concentrations of sTNFR2, as indicated, for 30 min. The cells were then lysed, and NF-κB activity was detected by ELISA. Data represent the mean of three independent experiments; *, P < 0.01.
Although transfection of Δcs-tmTNF-α could make it compete with endogenous wt-tmTNF-α for TNFR binding to block the wt-tmTNF-α-mediated reverse signaling, it is possible that the mutant expressed by the transfectant also augments the ligands that bind TNFR on cells, increasing forward signaling, which when mediated by tmTNF-α, was shown in Figure 2 to inhibit the constitutive activation of NF-κB. To exclude the possibility that forward signaling mediated an effect on NF-κB, soluble TNFR2 was added into the culture (since Raji cells mainly express TNFR2). This treatment with sTNFR2 could block forward signaling by inhibiting the interaction between TNF-α and TNFR on the cell surface and at the same time, triggering reverse signaling through binding to tmTNF-α. As was expected, sTNFR2 induced an obvious increase in NF-κB activity in a dose-dependent manner (Fig. 6B), suggesting that the constitutive activation of NF-κB is most likely mediated by reverse signaling via tmTNF-α.
Preferential forward or reverse signaling resulted in opposite effects on NF-κB activation in Raji cells
Raji cells express tmTNF-α and TNFR. In this cell model, forward and reverse signaling via tm-TNF-α may become integrated when both signals reach a balance. The likely outcome is that the reverse signaling via tmTNF-α is dominant, as shown above. To further confirm this, we incubated unfixed Raji cells (as responder) with the same amounts of fixed Raji cells (as stimulator), on which tmTNF-α or the TNFRs were previously blocked by specific antibodies, respectively, therefore interfering with the balance between bidirectional signaling of tmTNF-α in unfixed cells. As 1% paraformaldehyde-fixed Raji cells could not be lysed by lysis buffer, and the total protein was, thereby, unable to be isolated (data not shown), NF-κB activity was only detectable in the extract from unfixed cells that were not required for separation from the mixture. As shown in Figure 7, both types of TNFRs on fixed Raji cells were blocked by specific antibodies, leaving only tmTNF-α that bound TNFR on unfixed cells, which delivered forward signaling. The tmTNF-α-mediated reverse signaling was over-ruled. Consequently, we observed that constitutive NF-κB activation was suppressed (P<0.01). Conversely, when tmTNF-α on fixed Raji cells was neutralized by antibody, leaving the TNFRs that bound tmTNF-α on the unfixed cells, the tmTNF-α-mediated reverse signaling in these cells was strengthened, resulting in increased NF-κB activity (P<0.01).
Fig. 7.

Experimental design (A) and results (B) of opposite effects of tmTNF-α-mediated, bidirectional signaling on NF-κB activity. Raji cells were fixed with 1% polyformaldehyde at room temperature for 15 min and then pretreated with antibodies against TNFR1 and TNFR2 or against TNF-α for 1 h. After washes with PBS, the cells were mixed with the same amount of unfixed Raji cells for 30 min. In the mixture, only unfixed cells could be lysed in lysis buffer; thereby, their NF-κB activity was detectable by ELISA. The data are represented by the mean of three independent experiments; *, P < 0.01. RS, reverse signaling; FS, forward signaling.
DISCUSSION
In this study, we have demonstrated that there are opposite effects produced by tmTNF-α and sTNF-α on the malignant B cell Raji cell line. Although tmTNF-α as a type 2 membrane protein is identical with sTNF-α in its extracellular part, when tmTNF-α was added into the culture, it induced Raji cells to die via forward signaling, whereas sTNF-α stimulated Raji cells to proliferate. However, when tmTNF-α was expressed internally by Raji cells, it protected these cells from sTNF-α-induced cell death. Importantly, the distinct effects of exogenous and endogenous tmTNF-α on Raji cells have been shown to correlate with the activation of the NF-κB pathway. Although exogenous tmTNF-α induced a rapid down-regulation of NF-κB activity in Raji cells followed by cell death, endogenous tmTNF-α on Raji cells apparently caused constitutive activation of NF-κB and as a result, promoted cell survival.
It is known that NF-κB plays an important role in regulating cell proliferation, survival, and apoptosis. Activation of NF-κB is induced by TNF-α through signals mediated independently by two distinct types of TNFRs [21,22,23]. As a result of constitutively and highly active NF-κB found in Raji cells, sTNF-α treatment did not further stimulate NF-κB activation. Surprisingly, we found that treatment with exogenous tmTNF-α inhibited the translocation of NF-κB from the cytoplasm into the nucleus and inactivated constitutive NF-κB activation in Raji cells. The down-regulation of NF-κB activity by exogenous tmTNF-α appears to be specific but not a result of cell death, as the decrease in NF-κB activity occurred rapidly (30 min, Fig. 2B), and cell death was detected after 24 h of exposure to tmTNF-α. Although the mechanism(s) for inactivation of NF-κB by exogenous tmTNF-α are obscure, down-regulated NF-κB activity is followed by consequent suppression of transcription of its targeted genes, including apoptotic inhibitors, cIAP-1, which may play a critical role in tmTNF-α-induced cell death. This notion is supported by the results from studies about PDTC, which had been shown to inhibit NF-κB activation by stabilizing IκB-α and has now been widely used as an inhibitor of the NF-κB signaling pathway [24]. In our hands, as shown in Figure 3, at higher doses, PDTC completely abolished NF-κB activation in Raji cells and induced cell death, and at lower doses, although PDTC still significantly inactivated NF-κB, it did not affect viability of Raji cells but sensitized them to sTNF-α-mediated cytotoxicity. These observations indicate that constitutive NF-κB activation is necessary for the resistance of Raji cells to sTNF-α-induced cell death.
Constitutive activation of NF-κB has been detected in various cancer cells, including leukemia. A high level of NF-κB activation is associated with cell survival, resistance to apoptosis, and tumor progression [25, 26]. Although the mechanism for constitutive activation of NF-κB to occur is not clear, several studies have demonstrated that enhanced degradation of IκB-α and aberrant activation of IκB kinase can lead to constitutive NF-κB activation in some cases of neoplastic diseases [27, 28]. Here, our own study has suggested a novel mechanism for the constitutive activation of NF-κB in Raji cells, in which the expression of tmTNF-α is found to be responsible. First, our results showed that inhibition of endogenous tmTNF-α expression by specific TNF-α AS inactivated NF-κB. Importantly, similar to treatment with PDTC, the down-regulation of NF-κB by TNF-α AS was able to sensitize Raji cells to sTNF-α-mediated cytotoxicity. In contrast, increased tmTNF-α expression by treating Raji cells with specific AS against TACE, which reduces release of sTNF-α, resulted in enhanced NF-κB activation. These results indicate that endogenously expressed tmTNF-α, but not released sTNF-α, contributes to the constitutive activation of NF-κB.
An increasing body of evidence suggests that the membrane-integrated ligands act as receptors receiving and transmitting positive- and negative-feedback signals to ligand-bearing cells [17]. As Raji cells express tmTNF-α and TNFR, the interaction between tmTNF-α as a ligand and TNFR can occur on the same cell (autotropic) as well as on the neighboring cell (juxtatropic) to initiate forward signaling. Simultaneously, this interaction can also transmit reverse signals via tmTNF-α when acting as a receptor. Thus, NF-κB may become activated by forward signaling or reverse signaling as a result of tmTNF-α/TNFR interactions. Our results demonstrated that the constitutive NF-κB activation in Raji cells is induced by tmTNF-α-mediated reverse signaling. Suppression of NF-κB by transfection of mutant TNF-α lacking the intracellular domian likely resulted from blockage of the endogenous tmTNF-α-mediated reverse signaling, as a result of competitive binding by the mutant protein to the TNFR. However, it is possible that transfection of the mutant TNF-α may also have increased the forward signaling that is capable of inhibiting NF-κB activation as shown in Figure 2. To exclude the effect of forward signaling on NF-κB, we treated Raji cells with sTNFR2 that inhibits forward signaling by blocking the interaction between tmTNF-α and TNFR on Raji cells but triggers reverse signaling by binding to tmTNF-α. Our results have shown that addition of this sTNFR2 into the cell culture significantly enhanced constitutive NF-κB activation in Raji cells (Fig. 6B). In agreement with our findings, studies by others [29] have demonstrated that in tmTNF-α-transfected Hela cells expressing TNF-α–TNFR complexes on the cell surface, NF-κB and p38 MAPK are constitutively activated. Interestingly, a recent study demonstrated that two proteases, signal peptide peptidase-like protein 2a (SPPL2a) and SPPL2b, promoted the release of the TNF-α intracellular domain, which in turn, triggers expression of IL-12 but not IL-6 and TNF-α [30]. As all of the three cytokines are well-known targets of NF-κB, this observation suggests that the intracellular domain of tmTNF-α-stimulated IL-12 expression is regulated by other cellular signaling rather than the NF-κB pathway. Our results showed that endogenous tmTNF-α via interacting with TNFR mediates reverse signaling to activate NF-κB, implying that tmTNF-α-mediated NF-κB activation may depend on its membrane-anchored cytoplasmic segment but not the released intracellular part of tmTNF-α.
Based on these observations, it would be reasonable to propose that in a Raji cell, its TNFR binds to tmTNF-α on a neighboring Raji cell, thus transmitting forward signaling to negatively regulate NF-κB activation, and its tmTNF-α binds to TNFR on the neighboring cell, triggering reverse signaling to positively regulate NF-κB activation. By an as-yet unknown mechanism, the reverse signaling via tmTNF-α is dominant in Raji cells, and therefore, NF-κB is constitutively activated in this cancer cell line. However, the NF-κB activation is limited by forward signaling via tmTNF-α. When both types of TNFRs on fixed Raji cells were blocked, leaving only tmTNF-α to bind to TNFRs on unfixed cells, we observed an enhanced forward signaling via tm-TNF-α, resulting in a decrease in constitutive NF-κB activation in unfixed Raji cells. In sharp contrast, when tmTNF-α on fixed Raji cells was neutralized, leaving only TNFRs to bind to tmTNF-α on unfixed cells, we noticed an augmented reverse signaling via tmTNF-α, leading to an increase in constitutive NF-κB activation (Fig. 7).
In conclusion, the results presented here have demonstrated the bidirectional tmTNF-α pathways for its opposite effects on tumor cells, survival, or death. When tmTNF-α functions as a ligand, it induces cytotoxicity to tumor cells through the down-regulation of NF-κB activity via forward signaling. On the contrary, when tmTNF-α acts as a receptor and is highly expressed, it induces constitutive activation of NF-κB via reverse signaling that protects tumor cells from death. Whereas the forward signaling mediated by the TNF-α/TNFR interaction has been well-studied, the reverse signaling induced by tmTNF-α remains largely unclear. In this context, our current observations certainly warrant further investigations into addressing issues of the reverse signaling pathway and the molecular events associated with tmTNF-α-induced activation of NF-κB in tumor cells. A detailed study of this pathway may reveal how we could better control the deviation of the bidirectional tmTNF-α pathways with the goal to render tumor cells once again susceptible to apoptotic agents.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (30471586), National Key Basic Research Program of China from the Ministry of Science and Technology of the People’s Republic of China (2004AA215162), National Institutes of Health (R01 CA123490 to M. Z.), and the Leukemia and Lymphoma Society (6249-05 and 6033-08 to M. Z.). We thank Kathleen Kite-Powell for editing the manuscript.
References
- Aggarwal B B. Signaling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Moss M L, Jin S L, Milla M E, Bickett D M, Burkhart W, Carter H L, Chen W J, Clay W C, Didsbury J R, Hassler D. Cloning of a disintegrin metalloproteinase that processes precursor tumor-necrosis factor-α. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Tang P, Hung M-C, Klostergaard J. Human pro-tumor necrosis factor is a homotrimer. Biochemistry. 1996;35:8216–8225. doi: 10.1021/bi952182t. [DOI] [PubMed] [Google Scholar]
- Kriegler M, Perez C, DeFay K, Albert I, Lu S D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53:45–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- Peck R, Brockhaus M, Frey J R. Cell surface tumor necrosis factor (TNF) accounts for monocyte- and lymphocyte-mediated killing of TNF-resistant target cells. Cell Immunol. 1989;122:1–10. doi: 10.1016/0008-8749(89)90143-3. [DOI] [PubMed] [Google Scholar]
- Shi W Z, Li F G. Comparison of cytotoxicity by transmembrane and secreted TNF-alpha. Chin J Microbiol Immunol. 1998;18:499–513. [Google Scholar]
- Shi W, Li L, Shi X, Zheng F, Zeng J, Jiang X, Gong F, Zhou M, Li Z. Inhibition of nuclear factor-κB activation is essential for membrane-associated TNF-α-induced apoptosis in HL-60 cells. Immunol Cell Biol. 2006;84:366–373. doi: 10.1111/j.1440-1711.2006.01436.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Li L, Shi W, Jiang X, Xu Y, Gong F, Zhou M, Edwards C K, Li Z. Mechanism of action differences in the antitumor effects of transmembrane and secretory tumor necrosis factor-α in vitro and in vivo. Cancer Immunol Immunother. 2006;55:1470–1479. doi: 10.1007/s00262-006-0150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissner G, Kohlhuber F, Grell M, Ueffing M, Scheurich P, Hieke A, Multhoff G, Bornkamm G W, Holler E. Critical involvement of transmembrane tumor necrosis factor-α in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin. Blood. 1995;86:4184–4193. [PubMed] [Google Scholar]
- Ferran C, Dautry F, Merite S, Sheehan K, Schreiber R, Grau G, Bach J F, Chatenoud L. Anti-tumor necrosis factor modulates anti-CD3-triggered T cell cytokine gene expression in vivo. J Clin Invest. 1994;93:2189–2196. doi: 10.1172/JCI117215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissner G, Kirchner S, Lindner H, Kolch W, Janosch P, Grell M, Scheurich P, Andreesen R, Holler E. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol. 2000;164:6193–6198. doi: 10.4049/jimmunol.164.12.6193. [DOI] [PubMed] [Google Scholar]
- Harashima S, Horiuchi T, Hatta N, Morita C, Higuchi M, Sawabe T, Tsukamoto H, Tahira T, Hayashi K, Fujita S, Niho Y. Outside-to-inside signal through the membrane TNF-α induces E-selectin (CD62E) expression on activated human CD4+ T cells. J Immunol. 2001;166:130–136. doi: 10.4049/jimmunol.166.1.130. [DOI] [PubMed] [Google Scholar]
- Baeuerle P A, Henkel T. Function and activation of NF-κ B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin A S. The NF-κ B and I κ B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baeuerle P A, Baltimore D. NF-κ B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Kitahara J, Sakamoto H, Tsujimoto M, Nakagawa Y. Involvement of NF-κB in the protection of cell death by tumor necrosis factor in L929 derived TNF resistant C12 cells. Biol Pharm Bull. 2000;23:397–401. doi: 10.1248/bpb.23.397. [DOI] [PubMed] [Google Scholar]
- Xin L, Wang J, Zhang H, Shi W, Yu M, Li Q, Jiang X, Gong F, Gardner K, Li Q Q, Li Z. Dual regulation of soluble tumor necrosis factor-α induced activation of human monocytic cells via modulating transmembrane TNF-α-mediated “reverse signaling”. Int J Mol Med. 2006;18:885–892. [PubMed] [Google Scholar]
- Hasegawa T, Suzuki K, Sakamoto C, Ohta K, Nishiki S, Hino M, Tatsumi N, Kitagawa S. Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood. 2003;101:1164–1171. doi: 10.1182/blood-2002-05-1505. [DOI] [PubMed] [Google Scholar]
- Jin S, Lu D, Ye S, Ye H, Zhu L, Feng Z, Liu S, Wang D, Hu Q. A simplified probe preparation for ELISA-based NF-κB activity assay. J Biochem Biophys Methods. 2005;65:20–29. doi: 10.1016/j.jbbm.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Lloyd B H, Giles R V, Spiller D G, Grzybowski J, Tidd D M, Sibson D R. Determination of optimal sites of antisense oligonucleotide cleavage within TNFα mRNA. Nucleic Acids Res. 2001;29:3664–3673. doi: 10.1093/nar/29.17.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmann K, Schutze S, Kampen E, Himmler A, Machleidt T, Kronke M. Human 55-kDa receptor for tumor necrosis factor coupled to signal transduction cascades. J Biol Chem. 1992;267:17997–18001. [PubMed] [Google Scholar]
- Rothe M, Wong S C, Henzel W J, Goeddel D V. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel D V. The TNF receptor 1-associated protein TRADD signals cell death and NF-κ B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Schreck R, Meier B, Mannel D N, Droge W, Baeuerle P A. Dithiocarbamates as potent inhibitors of nuclear factor κ B activation in intact cells. J Exp Med. 1992;175:1181–1194. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M L, Neering S J, Upchurch D, Grimes B, Howard D S, Rizzieri D A, Luger S M, Jordan C T. Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- Giri D K, Aggarwal B B. Constitutive activation of NF-κB causes resistance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells. Autocrine role of tumor necrosis factor and reactive oxygen intermediates. J Biol Chem. 1998;273:14008–14014. doi: 10.1074/jbc.273.22.14008. [DOI] [PubMed] [Google Scholar]
- Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dorken B, Scheidereit C. Molecular mechanisms of constitutive NF-κB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene. 1999;18:943–953. doi: 10.1038/sj.onc.1202351. [DOI] [PubMed] [Google Scholar]
- Shattuck-Brandt R L, Richmond A. Enhanced degradation of I-κB α contributes to endogenous activation of NF-κB in Hs294T melanoma cells. Cancer Res. 1997;57:3032–3039. [PubMed] [Google Scholar]
- Haas E, Grell M, Wajant H, Scheurich P. Continuous autotropic signaling by membrane-expressed tumor necrosis factor. J Biol Chem. 1999;274:18107–18112. doi: 10.1074/jbc.274.25.18107. [DOI] [PubMed] [Google Scholar]
- Friedmann E, Hauben E, Maylandt K, Schleeger S, Vreugde S, Lichtenthaler S F, Kuhn P H, Stauffer D, Rovelli G, Martoglio B. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFα in activated dendritic cells to trigger IL-12 production. Nat Cell Biol. 2006;8:843–848. doi: 10.1038/ncb1440. [DOI] [PubMed] [Google Scholar]