

Abstract
The mechanisms that determine whether a tumor cell that has disseminated to a secondary site will resume growth immediately, die or enter a state of dormancy are poorly understood. Although tumor dormancy represents a common clinical finding, studying the mechanisms behind this stage of tumor progression has been challenging. Furthermore, it is thought that dormant tumor cells are refractory to chemotherapy due to their lack of proliferation. However, whether this is the only reason for their chemo-resistance remains to be proven. In this review we summarize recent findings that provide a mechanistic explanation about how stress signaling through the p38SAPK pathway and ER-stress signaling may coordinate the induction of growth arrest and drug-resistance in a model of squamous carcinoma dormancy. We further discuss how dormant tumor cells may enter this stage to adapt to strenuous conditions that do not favor immediate growth after dissemination. Finally, we propose that this response may recapitulate an evolutionarily conserved program of life-span extension through adaptation and tolerance to stress.
Keywords: p38, tumor dormancy, growth arrest, survival, stress signaling, metastasis, PERK
GROWTH CESSATION AND ADAPTATION TO STRESS AS A SELECTIVE ADVANTAGE FOR MULTI-CELLULAR ORGANISMS
Organisms across different kingdoms have evolved mechanisms to sense adverse environments and cope with stressful conditions (e.g., drought, cold, nutrient deprivation or heat shock) in order to survive. Usually these mechanisms result in a pause in development or growth and in the acquisition of stress-resistant phenotypes. These mechanisms provide species an adaptive advantage to resume development and/or growth after the stress is relieved, with the ultimate purpose of generating progeny. This stage is defined as dormancy in plants, diapause in nematodes or hibernation in mammals.1-3 A well-studied model of adaptation to microenvironment-imposed stress is the dauer larva stage in Caenorhabditis elegans. This alternative larval stage allows these animals to survive through periods of stress such as low nutrient availability.4 Genetic analysis has shown that Dauer formation (Daf) genes, such as TGFβ and IGF-1 orthologs, regulate this stage in response to conditions such as starvation.4 In addition, it was shown that resistance to osmotic stress in C. elegans can be regulated by an OSR-1->p38 pathway5 and that p38 can activate in this organism DAF-16, a dauer inducing gene.6 Interestingly, these pathways have been implicated in regulating growth (e.g., IGF)7,8 or suppression of malignancy,9 dormancy and stress resistance (e.g., p38)10-13 of tumor cells.
It has been proposed that as a tumor mass progresses, a micro-evolutionary process takes place where cells with growth advantageous properties will survive independently of other tumor cells and will be positively selected to grow and disseminate to distant organs.14 We would like to propose that within this process tumor cells acquire not only traits that select them to grow in a target organ after dissemination but also adaptation mechanisms that allow cells to accommodate to stressful conditions (Fig. 1A).12 Such a process might be the one observed during cancer dormancy that would ultimately allow tumor cells to pause and resume growth only when the conditions are propitious.10 This characteristic may represent a selective survival advantage.
Figure 1.
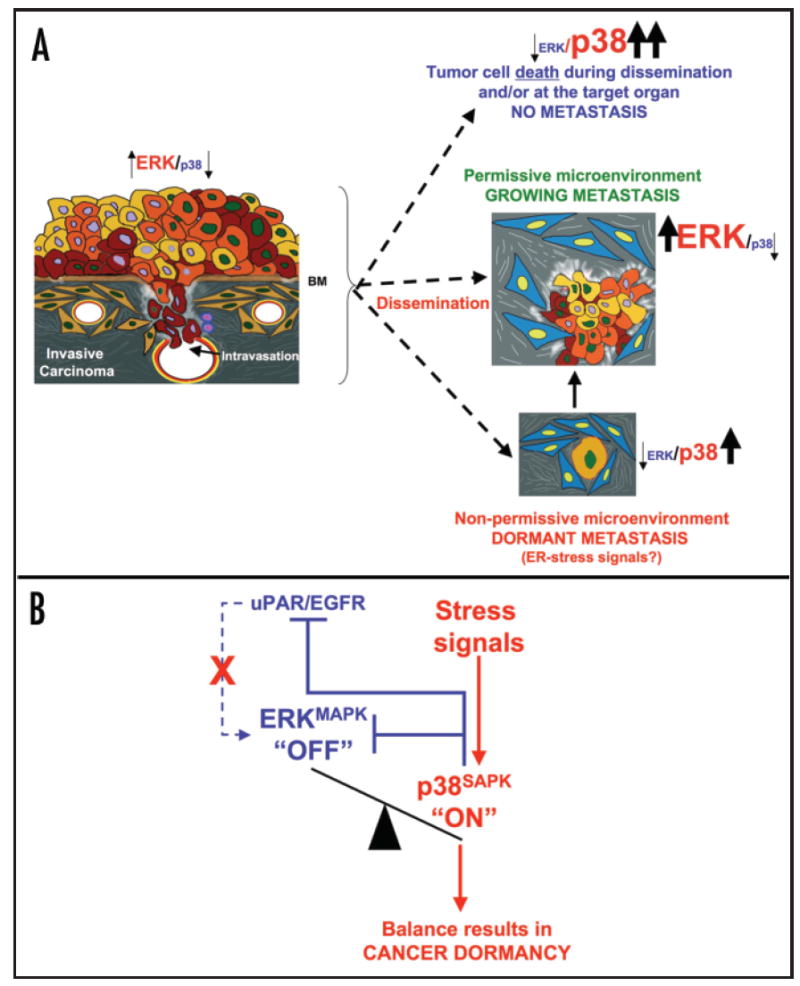
(A) Tumor cells that have acquired genetic or epigenetic changes leading to the acquisition of motile and invasive properties are able to degrade the basement membrane and invade the underlying stroma. Tumor cells degrade the matrix and the vascular walls (yellow) and disseminate through arterial or lymphatic routes. Following dissemination, tumor cells can proliferate, die, or enter a state of quiescence/dormancy, or after a period of dormancy they can resume growth. Depending on the environment, the proliferative signaling (ERK) may be outweighed by the stress-induced, growth suppressive or pro-apoptotic (p38) signaling, leading to either cell death or growth arrest depending on the intensity or duration of the stress signals. Alternatively, a growth permissive environment where high ERK signaling exceeds growth suppressive p38 signals favors tumor cell proliferation and metastasis formation. BM, basement membrane. (B) Stress signals (e.g., hypoxia, inappropriate extracellular matrix) can activate p38. Subsequently, p38 can inhibit proliferation by an immediate negative regulation on ERK and by inhibiting uPAR transcription, which is required for EGFR activation and further ERK signaling. This leads to a signaling balance that favors p38 over ERK activity that promotes G0/ G1 arrest and tumor dormancy.
DORMANCY AND ADAPTATION TO STRESS AS A SELECTIVE ADVANTAGE FOR METASTATIC CELLS
In cancer, dormancy can occur while the primary tumor is developing and the nascent tumor mass is unable to recruit blood vessels.15 Thus, the tumor mass, although containing proliferating cells, contains equal amount of apoptotic cells and therefore shows no net increase in size. In addition, a micrometastatic mass that is dormant due to lack of neovascularization resulting from a balance between apoptosis and mitosis may shed cells into circulation that are detectable for long periods.16,17 Interestingly, mounting evidence show that after dissemination single tumor cells can also enter a state of dormancy despite an active vasculature nurturing the target organ.13,18-20 In this review we will focus on recently discovered mechanisms that might propel disseminated single tumor cells into dormancy characterized by a growth arrest (Fig. 1A) and pathways that endow them with a survival mechanisms to resist stress conditions such as chemotherapy (Fig. 2A).12
Figure 2.
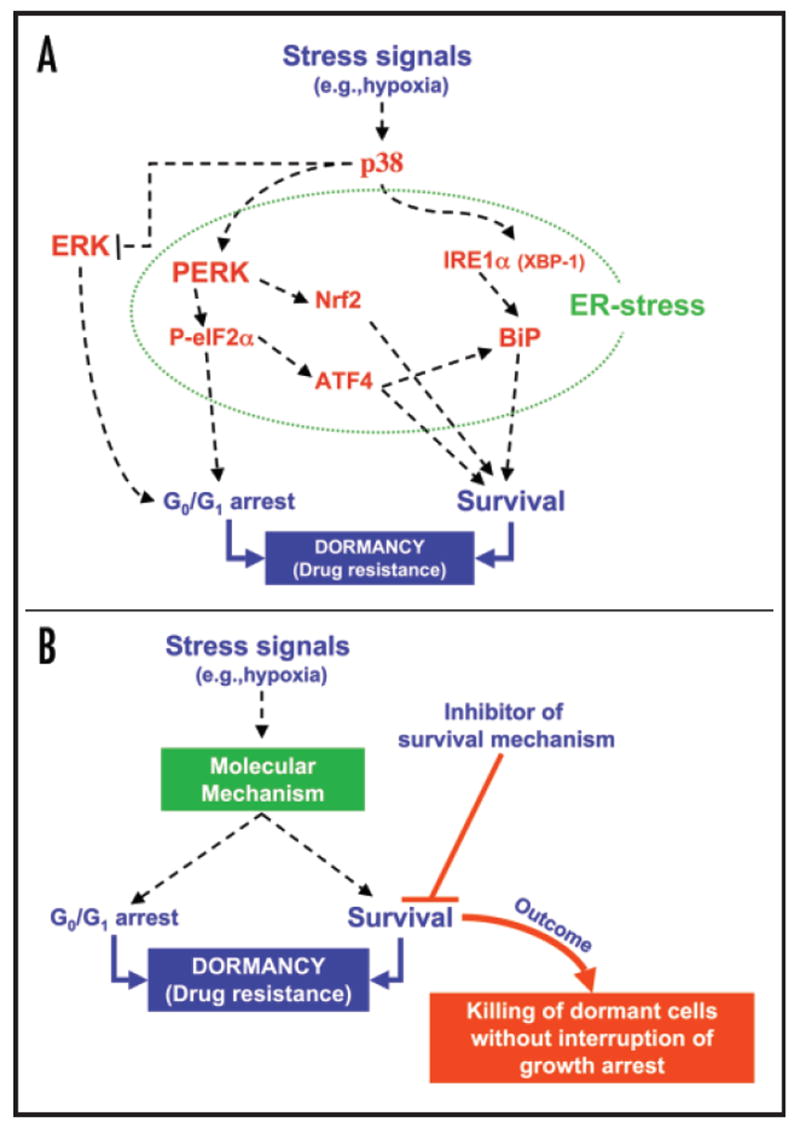
(A) Stress signals that result in p38 activation can suppress ERK signaling. Further, active p38 induces an ER-stress response that coordinates growth arrest and survival through the activation of PERK and IRE-1 and inducing the expression of BiP, ATF4 and Nrf2. These pathways fulfill the two key requirements of dormancy, growth arrest and robust survival. Green dotted line surrounds the signaling molecules that participate in the ER stress response. (B) Two potential therapies emerge from understanding dormancy: one is the induction of dormancy of growing and otherwise therapy resistant lesions (not shown); the second would involve targeting residual local or disseminated disease by inhibiting the survival signals that promote drug resistance in dormant tumor cells without interrupting their growth arrest. This would lead to longer disease free periods or eradicating the disease.
Metastatic disease rather than the primary tumor itself is one of the major causes of morbidity and mortality among cancer patients. Although metastatic dissemination may occur even at early stages of tumor development, only a subset of patients carry overt metastasis at the time of or shortly after primary tumor resection. Others may remain free of clinical evidence of disease for years or even decades after primary tumor removal. However the appearance of tumor relapse after such a prolonged disease free period implies that residual, dormant tumor cells remain in the body and that these cells were resistant to therapy.21 Considering the possibility of single disseminated tumor cells, these cells must have entered a growth arrest and also survive during the entire disease free period. These two characteristics, namely cell growth arrest and cell survival, are the two main hallmarks of the dormant tumor cell. The signals that force cells into dormancy and cause drug resistance are unknown, but it has been proposed that the stress induced by chemotherapy,22 an unfavorable microenvironment23 or even the one imposed by the dissemination may activate stress-signaling pathways and force these solitary cells into a growth arrest mode. Thus, this stage may represent a selective advantage for solitary metastatic cells to evade cell death caused by dissemination or adjuvant therapy and pause until new conditions favor growth. Understanding cancer dormancy is critical to design therapies aimed at inducing dormancy of growing lesions, maintain dormancy of residual disease or inhibiting their survival signaling and eradicating dormant cells without interrupting their growth arrest. The above-mentioned possibilities would be beneficial for patients with disseminated disease.
While clinical evidence for cancer dormancy is abundant, insight into the mechanisms of induction, maintenance and escape from dormancy is scarce.19 More specifically, mechanisms that explain why disseminated tumor cells may enter dormancy are only now being identified.20,24,25 The possibility that tumor cells that had accumulated critical genetic and epigenetic changes required for primary tumor growth suddenly become reprogrammed and enter growth arrest suggests that a tumor cell need not always conform with all the hallmarks of cancer proposed by Hanahan and Weinberg.14 This, in a way, is an exciting option because the possibility that malignancy can be reverted in advanced cancers through the manipulation of specific pathways, suggests that there is still plasticity in malignant cells.
NEGATIVE REGULATION OF GROWTH BY p38SAPK CAN RESULT IN THE INDUCTION OF CARCINOMA DORMANCY
Signaling by p38 has emerged as an important negative regulator of transformation and tumorigenesis.26-29 Although these effects have been reviewed recently9,30 some relevant functions of p38 are summarized here. Activation of p38 occurs mostly in response to exogenous stress that can lead to the upstream activation of MKK3/4/6 and downstream activation of effector kinases such as MAPKAPK2 and several transcription factors (e.g., ATF2, CHOP and MEF2C). Activation of p38 antagonizes Ras-induced transformation in fibroblasts and epithelial cells and in the latter p38 inhibition appears to be a nonclassical target of Ras during transformation.31 Moreover, p38 can exert its negative function on cell transformation by phosphorylating p5332 and frequent overexpression of PPM1D, a p38-phosphatase, is believed to be a mechanism by which tumor cells override p38 induction of growth arrest and apoptosis.33 Further, in rhabdomyosarcoma cells, reversion of malignancy due to terminal differentiation and cell cycle arrest can be achieved by overexpression of MKK6 and activation of p38.26 Finally, recent experiments in MKK3/4/6 null mice fibroblasts have shown that deletion of these three p38-activating MAPKK increased the susceptibility of mouse fibroblasts to MMTV-induced transformation and tumorigenesis in vivo.29 Overall, it appears that p38 is a strong negative regulator of mitogenesis and that acquisition of malignant properties results in a bypass or inhibition of the p38 pathway.29,34
A major obstacle to understanding induction of cancer dormancy and their chemo-resistance has been the lack of proper model systems.10,35 Studies by Ossowski and coworkers and from our laboratory have established a model of tumor dormancy in human squamous carcinoma (HEp3). When these cells are kept in vivo they remain tumorigenic (T-HEp3) and metastatic. However, prolonged in vitro passaging of these cells, results in a nonclonal acquisition of a dormant (D-HEp3) phenotype upon reinoculation in vivo.10,13 These studies have revealed that HEp3 cells maintain a tumorigenic and metastatic phenotype by overexpressing the urokinase receptor (uPAR), which in turn signals through a complex comprising of α 5β1-integrin and EGF receptor. Signaling from the uPAR-integrin-EGFR complex culminates in the activation of ERK mitogenic pathway and the inhibition of p38 stress activated/ growth suppressive pathway. Such cells have high ERK/p38 ratio that favors tumorigenesis.13 Forced or nonclonal spontaneous loss of the uPAR - α5β1- EGFR signaling complex results in p38 activation and in a low ERK/p38 ratio.13 When these cells are inoculated in vivo they enter a state of protracted dormancy characterized by a G0/G1 arrest and no significant apoptosis (Fig. 1B).13 In this stage it appears that the dormant phenotype and p38 signaling are dominant as inhibition of p38 signaling either genetically or pharmacologically was shown to be sufficient to interrupt tumor dormancy and restore the malignant phenotype in vivo. These results implicate a critical role for p38 in the induction of tumor dormancy (Fig. 1B).13 The hypothesis that activation of p38 would restrict metastatic growth would allow predicting that cells devoid of p38 signaling could be more metastatic. In fact MKK4, an activator of p38, is lost in ovarian and prostate cancer and functions as a metastasis, rather than tumor suppressor.36 Mechanistic analysis revealed that restoration of MKK4/MKK6-dependent activation of p38 in ovarian carcinoma36 and MKK4/MKK7-induced activation of JNK in prostate cancer cells,37,38 renders these cells non-metastatic. It remains to be determined whether their activation induced apoptosis or dormancy of metastasis.36 We postulate that stress-dependent activation of p38 during dissemination may eliminate cells through apoptosis, but those that are cells able to cope with stress may resume growth immediately or enter a p38-dependent protracted state of dormancy.10,11
ACTIVATION OF PERK-eIF2α BY p38SAPK AS A MECHANISM TO INDUCE TUMOR CELL DORMANCY
Our work revealed that p38 signaling played a critical role in inducing and or maintaining the dormant phenotype.10,13 However, the mechanisms by which p38 regulated the dormant phenotype remained unknown. To address this problem we performed a proteomic screen for p38-regulated genes in HEp3 cells to identify the mechanisms governing dormancy.12 This analysis revealed the existence of an endoplasmic reticulum (ER) stress response, characterized by the upregulation of BiP/GRP78, PDI/ER60, HSP47 and cyclophilin B and activation of the ER-kinase PERK, phosphorylation of eIF2α and accumulation of spliced XBP-1 protein.12 This finding was intriguing, as the endoplasmic reticulum (ER) stress response is known to allow cells to adapt when the protein folding in the ER is compromised. Perturbations in protein load of the ER lumen triggers a series of pathways that have been extensively reviewed.39 Briefly, under non-stress conditions, BiP is bound to the ER-lumenal domains of, among others, the kinase receptors PERK, IRE1α and a membrane-bound transcription factor ATF6, preventing their activation.39 However, within minutes of client protein overload, BiP dissociates from PERK and IRE1α and translocates to the ER lumen. This results in activation of these receptors.39 Active PERK phosphorylates eukaryotic translation initiator factor 2α (eIF2α) repressing protein synthesis and inducing G0/G1 arrest.39,40 Activation of IRE1α results in XBP-1 mRNA processing which involves the non-canonical splicing of a 26 nt intron present in the mature XBP-1 mRNA.39 XBP-1, in turn, induces unfolded protein response (UPR) genes such as BiP and growth arrest genes (GADD153), which allow the cells to cope with the protein overload and induce growth arrest. To restore translation, the same ISR induces GADD34 (regulatory subunit of PP1c, an eIF2α phosphatase) that relieves translational repression.41 This allows for the UPR-induced mRNAs to be translated and favor proper protein folding. This in turn results in the transcriptional upregulation of proteins that will favor proper protein folding (e.g., BiP, Grp94, HSP47, PDI, cyclophilins).39
It is important to note that activation of PERK signaling during ER stress has previously been shown to induce G0/G1 arrest40 and cell survival,39 the two components of dormancy in our model. Thus, it has been proposed that this response may allow cells to pause and decide their fate depending on the level of damage.39,42 Work from the Diehl laboratory has shown that ER-stress activation of PERK promotes cell cycle arrest at the G1/S boundary and that PERK signaling to eIF2α blocks cyclin D1 translation/stability, providing a mechanistic link between ER-stress signaling and cell cycle arrest.40 Collectively, these findings were intriguing to us because dormant tumor cells enter a G0/G1 arrest in vivo. Work from our laboratory has shown that PERK-mediated phosphorylation of eIF2α is high in dormant cells and that overall protein synthesis is attenuated (Ref. 12 and our unpublished results). Further, our most recent findings suggest that inhibition of eIF2α phosphorylation through expression of a dominant negative PERK, an shRNA to PERK or through overexpression of GADD34 interrupt the dormancy of D-HEp3 cells (Ref. 12 and our unpublished results). These findings led us to conclude that p38 induces changes in the ER-load that result in PERK signaling at a level sufficient to induce growth arrest but not apoptosis (Fig. 2A). Ongoing studies are identifying the source of the ER load and the PERK-dependent mechanism for growth arrest.
ROLE OF PERK-eIF2α SIGNALING AND GRP78/BIP EXPRESSION IN STRESS TOLERANCE AND DRUG RESISTANCE OF DORMANT TUMOR CELLS
It is well documented that activation of ER-stress activates sequentially pathways that initially try to restore the unfolding by blocking translation and then more stringent controls are imposed that will decide if the cell should enter apoptosis.39 Thus, initially ER-stress signaling will initiate survival signals to promote restoration of ER homeostasis. PERK signaling can induce survival in response to ER-stress.43,44 The survival mechanism has been linked to the upregulation of genes translated in a phospho-eIF2α -dependent fashion (e.g., ATF4)45 and to direct activation by PERK of the transcription factor Nrf2;46-48 ATF4-/- cells are sensitive to various types of stress.49,50 It appears that the main function of these two transcription factors is to induce genes that promote survival by relieving oxidative stress (e.g., GST).47,48,51 BiP can also serve as a survival factor and its upregulation is linked to resistance to various stresses that affect ER homeostasis.39 Further, elegant studies from the Koumenis laboratory demonstrated that the PERK mediated regulation of eIF2α and ATF4 protects from hypoxia in growing tumors.50 In addition, other reports have shown that overexpression of BiP in HT1080 cells can protect from etoposide and doxorubicin (Dox) treatment,45 while UPR-induced resistance to TOPOII inhibitors has been attributed to a downregulation of the TOPOII enzyme.52 Thus, it is possible that tumor cells might co-opt the ER-stress response to adapt to and survive various types of stress, including conventional chemotherapy.45
A common concept is that residual disease is hard to eradicate because noncycling dormant cells escape the grip of chemotherapeutic agents.12,21 However, it was not known whether dormant cells might in addition tap into other survival pathways to resist treatment.20 It is interesting to note that induction of UPR can desensitize cells to further stress.39 This is evidenced by the persistence of UPR induced genes long after the stress is removed and it is thought that this condition may prepare the cells to resist a second insult. Thus, ER-signaling may be said to cause a transient state in which cells undergoing repair are growth-arrested and survival-competent. Considering that dormant D-HEp3 cells had a basal ER-stress-like response we hypothesized that the p38-dependent upregulation of BiP and activation of PERK-eIF2α signaling might have a survival function for dormant, but not malignant cells.12 If true this could represent an attractive therapeutic target to eradicate residual dormant disease (Figs. 2A-B). Our experiments revealed that dormant D-HEp3, but not malignant T-HEp3 cells, are highly tolerant to ER-stressors such as tunicamycin and thapsigargin and also to low glucose.12 What was more intriguing was that in assays where we measured apoptosis or plating efficiency post drug-treatment, D-HEp3 cells were highly resistant to the DNA damaging agent doxorubicin and etoposide (Ref. 12 and our unpublished data). In contrast, tumorigenic T-HEp3 cells succumbed to a similar chemotherapeutic treatment.12 Because these experiments were done in vitro where both T- and D-HEp3 cells have equal division rate,12 we concluded that it is not the proliferation capacity but rather an inherent survival pathway that distinguished the drug resistance of these cells. To test whether PERK-eIF2α and BiP functions were required for this inherent mechanism, we inhibited PERK-eIF2α signaling by overexpressing a dominant negative PERK or GADD34 and we downregulated BiP with a specific siRNA. All these treatments converted dormant drug resistant D-HEp3 cells into drug-sensitive. Inhibition of BiP in dormant cells and drug-sensitivity was linked to reactivation of the pro-apoptotic protein Bax, which was not activated in cells with high BiP levels.12 We have recently shown that stable shRNA-mediated downregulation of BiP in dormant D-HEp3 cells, renders them susceptible to doxorubicin in vivo, where proliferation is absent (our unpublished results). These results allowed us to conclude that both BiP and PERK propagate survival signals that protect dormant tumor cells from chemotherapy.
The mechanism by which BiP and PERK inhibition restores sensitivity to chemotherapeutic treatment in dormant cells is currently under study. Some possibilities can be expected from published reports (See in this issue Mann and Hendershot; Fu and Lee). PERK may mediate survival through the induction of BiP or through the detoxifying response dependent on Nrf2 and ATF4 transcriptional activation.47,53 Overexpression of BiP may suppress the activation of caspase-7 and caspase-3 dependent apoptosis54,55 or caspase-7-dependent activation of caspase-12, an ER-resident caspase.54 Alternatively the Ca2+-chelating function of BiP combined with its upregulation in the drug resistant cells, could block Ca2+ signaling for apoptosis.56 When Bax localizes to the ER membrane it favors the release of Ca2+. This signal has been shown to induce apoptosis by rapid Ca2+ uptake by the mitochondria after release into the cytosol.56 Thus, it is plausible that mobilization of intracellular Ca2+ from ER stores is inhibited in D-HEp3 cells. Regulation of Ca2+ release is a very likely mechanism as it has been shown that BiP mediates its anti-apoptotic effects by preventing Ca2+ release through its ability to bind up to 25% of the intracellular calcium.57 Thus, even if apoptotic stimuli reach the ER, if Ca2+ is not released it may interrupt apoptotic signaling to the mitochondria. Liang et al recently showed that PERK mediates activation of p38 and JNK kinases after disruption of ER Ca2+ homeostasis58 suggesting that hyper-activation of the pathway may be a mechanism by which BiP downregulation promotes death.
Multiple mechanisms may result in ER-stress induced survival. It is plausible that ER-stress activation of p38 signaling serves also as survival signal45 since p38 was shown to be necessary for the survival to ER-stress both in HeLa human cervical carcinoma cells59 and Saccharomyces cerevisiae.60 An alternative survival mechanism could be attributed to IRE1-mediated activation of TRAF2, which activates the survival transcription factor NF-κB.61,62 The fact that MEFs lacking TRAF2 are more susceptible to ER-stress than their WT counterparts,63 supports this hypothesis. Although p38 signaling may induce apoptosis, these observations suggest that, alternatively, it might also protect and prepare cells to imminent stress. Collectively these findings provide intriguing avenues to explore the mechanisms by which p38, PERK, P-eIF2α and BiP could protect growth arrested dormant cells from chemotherapy-induced damage.
ADAPTATION TO STRESS SIGNALING IN GROWING VS. DORMANT TUMORS
Published evidence supports a role of p38 signaling in inhibiting tumorigenesis and metastasis in various models.9 Further, we have found that PERK signaling is associated with the inhibition rather than the promotion of HEp3 cell growth.12 This is somewhat different from the data from other labs showing that BiP, PERK and IRE-1 signaling are involved in the ability of growing tumors to cope with stress induced by hypoxia (See also in this issue Fels and Koumenis; Fu and Lee;).50,64 The similarity between all these reports is that PERK signaling and BiP upregulation have a pro-survival function. The disparity is that in our model PERK-eIF2α signaling are associated with growth arrest rather than proliferation. A possible explanation to this conundrum may be the context under which these ER-stress signals are generated in growing vs. dormant tumors. Dormant D-HEp3 cells achieve a level of signaling where p38 signaling predominates over ERK and further suppresses ERK activation by establishing a negative feedback loop.10,11,13 Thus, it is not only the PERK signal that may contribute to the arrest but rather a program regulated by p38 which includes ERK inhibition through the transcriptional downregulation of uPAR and its signaling capacity.10,11,13,65 In the Koumenis study for example PERK+/+ and -/- MEFs were transformed by overexpression of K-RasV12 and the growth advantage caused by PERK was attributed to survival signaling and not regulation of cell cycle progression.50 It is possible that in the context of activated Ras overexpression, this strong signal forces the cells into a permanent mitogenic signaling state that may not be reversed by stress signaling through p38 or other pathways. It would be interesting to test whether inhibition of ERK or PI3K and/or activation of p38 in these cells couples PERK to growth arrest. Similar to the Ras-transformed MEFs studies,50 we found that HT1080 cells harboring an N-Ras mutation display, when inoculated in vivo, a transient (~48 hrs) but strong activation of p38 → CHOP signaling that is most likely due to the initial adaptation to a new tissue microenvironment.10,11 Interestingly, these cells are able to suppress p38 → CHOP signaling and resume tumor growth as a primary lesion or as metastasis.11 These findings led us to hypothe-size that cells that have all the genetic and epigenetic alterations to grow at a secondary site will initiate growth after dissemination readily.11 In contrast, tumors that have not fully progressed and acquired all the advantageous traits for metastasis may have the ability to enter a state of dormancy in response to stress caused by dissemination and the target organ microenvironment. This may be an intermediate phenotype with a selective advantage provided by the ability to pause and resume growth later. Collectively, the data suggest that ER-stress signaling will serve as a pro-survival pathway in growing and dormant tumors but that its coupling to growth inhibition may depend on cooperative crosstalk with other pathways in the cells.
FUTURE PERSPECTIVES
A possible interpretation of the acquisition of a dormant phenotype is that in some types of cancer there is still plasticity to reprogram the tumor cells into a nonproliferative phenotype. Results from our and other laboratories allow us to conclude that a candidate pathway to induce dormancy is the p38 pathway. We further propose that concomitant activation of p38 and inhibition of ERK signaling may be a more suitable strategy to enforce a dormant phenotype.10 Progress towards this end is visible as there are several potent MEK inhibitors being tested in clinical trials.66 Such an approach may be useful in tumors highly dependent on MEK signaling such as those with B-Raf mutations,67 although in some melanoma cell lines p38 activation does not inhibit ERK and tumor cell proliferation.10 An additional possibility may be to design and use small molecules aimed at maintaining PERK active or prevent eIF2α dephosphory-lation in tumors. Such an inhibitor of eIF2α dephosphorylation has been reported to work by blocking GADD34-PP1c activity.68 While inducing dormancy of an otherwise untreatable lesion and converting the disease into a chronic asymptomatic condition is a step forward, the undesired effect of inducing dormancy may be its inherent resistance to stress (Fig. 2B). Thus, it is critical to identify and target the specific survival mechanisms in dormant tumor cells to render these cells sensitive to chemotherapy. An interesting link between ER-stress signaling and novel chemotherapeutics derives from the use of proteasome inhibitors for the treatment of various cancers.69 It appears that these inhibitors induce apoptosis by causing strong ER-stress signaling, by virtue of blocking the ERAD pathway.69 Resistance to such inhibitors has been reported70 (See Fribley and Wang, this issue). It will be of interest to determine whether cells with inherent ER-stress signaling are more tolerant to these inhibitors or whether they may induce dormancy and subsequent resistance.
Much work remains to be done to fully elucidate the mechanisms that result in the induction of dormancy and the drug resistance of tumor cells. Promising progress summarized in this review may provide leads about how this phenotype is regulated. It will be interesting to determine whether the adaptive mechanisms that regulate cancer dormancy have any parallel with those regulating the C. elegans dauer stage.4 This organism and its powerful genetics may be useful to identify targets to induce dauer/dormancy or to overcome resistance to stress that can then be mapped onto mammalian models of cancer.
Acknowledgments
This work is supported by a grant from the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program and the NIH/ National Cancer Institute grant CA109182 (to J.A. Aguirre-Ghiso), and Ruth L. Kirschstein National Research Service Award (NIH/National Cancer Institute) Fellowship (to A.C. Ranganathan). We thank Dr. Liliana Ossowski (Mount Sinai School of Medicine, NY) for helpful comments on the manuscript.
References
- 1.Cowan KJ, Storey KB. Mitogen-activated protein kinases: New signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. 2003;206:1107–15. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- 2.Lee M, Choi I, Park K. Activation of stress signaling molecules in bat brain during arousal from hibernation. J Neurochem. 2002;82:867–73. doi: 10.1046/j.1471-4159.2002.01022.x. [DOI] [PubMed] [Google Scholar]
- 3.Koornneef M, Bentsink L, Hilhorst H. Seed dormancy and germination. Curr Opin Plant Biol. 2002;5:33–6. doi: 10.1016/s1369-5266(01)00219-9. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Kim SK. Global analysis of dauer gene expression in Caenorhabditis elegans. Development. 2003;130:1621–34. doi: 10.1242/dev.00363. [DOI] [PubMed] [Google Scholar]
- 5.Solomon A, Bandhakavi S, Jabbar S, Shah R, Beitel GJ, Morimoto RI. Caenorhabditis elegans OSR-1 regulates behavioral and physiological responses to hyperosmotic environments. Genetics. 2004;167:161–70. doi: 10.1534/genetics.167.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondo M, Yanase S, Ishii T, Hartman PS, Matsumoto K, Ishii N. The p38 signal transduction pathway participates in the oxidative stress-mediated translocation of DAF-16 to Caenorhabditis elegans nuclei. Mech Ageing Dev. 2005;126:642–7. doi: 10.1016/j.mad.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Yee D. Targeting insulin-like growth factor pathways. Br J Cancer. 2006;94:465–8. doi: 10.1038/sj.bjc.6602963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibrahim YH, Yee D. Insulin-like growth factor-I and breast cancer therapy. Clin Cancer Res. 2005;11:944s–50s. [PubMed] [Google Scholar]
- 9.Bulavin DV, Fornace AJ., Jr p38 MAP kinase’s emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. doi: 10.1016/S0065-230X(04)92005-2. [DOI] [PubMed] [Google Scholar]
- 10.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–95. [PubMed] [Google Scholar]
- 11.Aguirre-Ghiso JA, Ossowski L, Rosenbaum SK. Green fluorescent protein tagging of extra-cellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res. 2004;64:7336–45. doi: 10.1158/0008-5472.CAN-04-0113. [DOI] [PubMed] [Google Scholar]
- 12.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced Upregulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–11. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–79. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 15.Folkman J, Hanahan D. Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symp. 1991;22:339–47. [PubMed] [Google Scholar]
- 16.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 17.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, Herlyn D, Terstappen LW, Fehm T, Tucker TF, Lane N, Wang J, Uhr JW. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 18.Naumov GN, MacDonald IC, Chambers AF, Groom AC. Solitary cancer cells as a possible source of tumour dormancy? Semin Cancer Biol. 2001;11:271–6. doi: 10.1006/scbi.2001.0382. [DOI] [PubMed] [Google Scholar]
- 19.Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, Chambers AF. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 2002;62:2162–8. [PubMed] [Google Scholar]
- 20.Chambers AF, Naumov GN, Varghese HJ, Nadkarni KV, MacDonald IC, Groom AC. Critical steps in hematogenous metastasis: An overview. Surg Oncol Clin N Am. 2001;10:243–55. vii. [PubMed] [Google Scholar]
- 21.Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 22.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–15. [PubMed] [Google Scholar]
- 23.Kauffman EC, Robinson VL, Stadler WM, Sokoloff MH, Rinker-Schaeffer CW. Metastasis suppression: The evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site. J Urol. 2003;169:1122–33. doi: 10.1097/01.ju.0000051580.89109.4b. [DOI] [PubMed] [Google Scholar]
- 24.White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–70. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 25.Shachaf CM, Felsher DW. Tumor dormancy and MYC inactivation: Pushing cancer to the brink of normalcy. Cancer Res. 2005;65:4471–4. doi: 10.1158/0008-5472.CAN-05-1172. [DOI] [PubMed] [Google Scholar]
- 26.Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, Feramisco JR, Karin M, Wang JY. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000;14:574–84. [PMC free article] [PubMed] [Google Scholar]
- 27.Haq R, Brenton JD, Takahashi M, Finan D, Finkielsztein A, Damaraju S, Rottapel R, Zanke B. Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 2002;62:5076–82. [PubMed] [Google Scholar]
- 28.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback for Ras proliferative signaling. J Biol Chem. 2000;275:38973–80. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 29.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–78. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engelberg D. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin Cancer Biol. 2004;14:271–82. doi: 10.1016/j.semcancer.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J Biol Chem. 2002;277:31808–17. doi: 10.1074/jbc.M203964200. [DOI] [PubMed] [Google Scholar]
- 32.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace AJ, Jr, Appella E. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 33.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, Appella E, Fornac AJ., Jr Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 34.Teng DH, Perry WL, IIIrd, Hogan JK, Baumgard M, Bell R, Berry S, Davis T, Frank D, Frye C, Hattier T, Hu R, Jammulapati S, Janecki T, Leavitt A, Mitchell JT, Pero R, Sexton D, Schroeder M, Su PH, Swedlund B, Kyriakis JM, Avruch J, Bartel P, Wong AK, Tavtigian SV, et al. Human mitogen-activated protein kinase kinase 4 as a candidate tumor suppressor. Cancer Res. 1997;57:4177–82. [PubMed] [Google Scholar]
- 35.Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, Riethmuller G, Eils R, Klein CA. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA. 2003;100:7737–42. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006;66:2264–70. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida BA, Dubauskas Z, Chekmareva MA, Christiano TR, Stadler WM, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 17. Cancer Res. 1999;59:5483–7. [PubMed] [Google Scholar]
- 38.Vander Griend DJ, Kocherginsky M, Hickson JA, Stadler WM, Lin A, Rinker-Schaeffer CW. Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005;65:10984–91. doi: 10.1158/0008-5472.CAN-05-2382. [DOI] [PubMed] [Google Scholar]
- 39.Rutkowski DT, Kaufman RJ. A trip to the ER: Coping with stress. Trends Cell Biol. 2004;14:20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci USA. 2000;97:12625–30. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niwa M, Walter P. Pausing to decide. Proc Natl Acad Sci USA. 2000;97:12396–7. doi: 10.1073/pnas.250476097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 44.Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–63. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 45.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: Friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 46.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38:317–32. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 47.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–17. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 48.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. Embo J. 2005;24:3470–81. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–13. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray MD, Mann M, Nitiss JL, Hendershot LM. Activation of the unfolded protein response is necessary and sufficient for reducing topoisomerase IIalpha protein levels and decreasing sensitivity to topoisomerase-targeted drugs. Mol Pharmacol. 2005;68:1699–707. doi: 10.1124/mol.105.014753. [DOI] [PubMed] [Google Scholar]
- 53.He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2- interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276:20858–65. doi: 10.1074/jbc.M101198200. [DOI] [PubMed] [Google Scholar]
- 54.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 55.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–42. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 56.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lievremont JP, Rizzuto R, Hendershot L, Meldolesi J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+ J Biol Chem. 1997;272:30873–9. doi: 10.1074/jbc.272.49.30873. [DOI] [PubMed] [Google Scholar]
- 58.Liang SH, Zhang W, McGrath BC, Zhang P, Cavener DR. PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem J. 2006;393:201–9. doi: 10.1042/BJ20050374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamamoto K, Hamada H, Shinkai H, Kohno Y, Koseki H, Aoe T. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J Biol Chem. 2003;278:34525–32. doi: 10.1074/jbc.M304188200. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Feldman DE, Deng C, Brown JA, De Giacomo AF, Gaw AF, Shi G, Le QT, Brown JM, Koong AC. Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol Cancer Res. 2005;3:669–77. doi: 10.1158/1541-7786.MCR-05-0181. [DOI] [PubMed] [Google Scholar]
- 61.Leonardi A, Vito P, Mauro C, Pacifico F, Ulianich L, Consiglio E, Formisano S, Di Jeso B. Endoplasmic reticulum stress causes thyroglobulin retention in this organelle and triggers activation of nuclear factor-kappa B via tumor necrosis factor receptor-associated factor 2. Endocrinology. 2002;143:2169–77. doi: 10.1210/endo.143.6.8825. [DOI] [PubMed] [Google Scholar]
- 62.Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol Pharm Bull. 2003;26:931–5. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- 63.Mauro C, Crescenzi E, De Mattia R, Pacifico F, Mellone S, Salzano S, de Luca C, D’Adamio L, Palumbo G, Formisano S, Vito P, Leonardi A. Central role of the scaffold protein tumor necrosis factor receptor-associated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:2631–8. doi: 10.1074/jbc.M502181200. [DOI] [PubMed] [Google Scholar]
- 64.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–7. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 65.Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell. 2002;1:445–57. doi: 10.1016/s1535-6108(02)00072-7. [DOI] [PubMed] [Google Scholar]
- 66.Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–6. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 67.Gill M, Celebi JT. B-RAF and melanocytic neoplasia. J Am Acad Dermatol. 2005;53:108–14. doi: 10.1016/j.jaad.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 68.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 69.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006 doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA. 2003;100:9946–51. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]