

Abstract
Farnesyl diphosphate (FPP) synthase catalyzes the consecutive head-to-tail condensations of isopentenyl diphosphate (IPP, C5) with dimethylallyl diphosphate (DMAPP, C5) and geranyl diphosphate (GPP, C10) to give (E,E)-FPP (C15). The enzyme belongs to a genetically distinct family of chain elongation enzymes that install E-double bonds during each addition of a five-carbon isoprene unit. Analysis of the C10 and C15 products from incubations with avian FPP synthase reveals that small amounts of neryl diphosphate (Z-C10) and (Z,E)-FPP are formed along with the E-isomers during the C5 → C10 and C10 → C15 reactions. Similar results were obtained for FPP synthase from Escherichia coli, Artemisia tridentata (sage brush), Pyrococcus furiosus, and Methanobacter thermautotrophicus and for GPP and FPP synthesized in vivo by E. coli FPP synthase. When (R)-[2-2H]IPP was a substrate for chain elongation, no deuterium was found in the chain elongation products. In contrast, the deuterium in (S)-[2-2H]IPP was incorporated into all of the products. Thus, the pro-R hydrogen at C2 of IPP is lost when the E- and Z-double bond isomers are formed. The synthesis of Z-double bond isomers by FPP synthase during chain elongation is unexpected for a highly evolved enzyme and probably reflects a compromise between optimizing double bond stereoselectivity and the need to exclude DMAPP from the IPP binding site.
Chain elongation is the fundamental building reaction in the isoprenoid pathway.1 During this process the growing hydrocarbon chain in an allylic isoprenoid diphosphate is added to isopentenyl diphosphate (IPP). These reactions are catalyzed by polyprenyl diphosphate synthases, a group of prenyltransferases that can be further subdivided into two families according to the stereochemistry, E or Z, of the double bond in the newly added isoprene unit. Members of the two families utilize the same chemical mechanism for chain elongation but are genetically unrelated.1 Subgroups within each family are selective for the size of the allylic substrate selected for chain elongation and the length of the isoprenoid chain in the final product. Thus, the “trunk” of the isoprenoid biosynthetic pathway is in reality a complex set of trunks that varies from organism to organism rather than a set of linear chain elongation reactions. Members of each family share distinctive highly conserved motifs characteristic of proteins that have evolved from a common ancestor.2,3 Members of the E-family typically synthesize shorter chain isoprenoid diphosphates found early in the pathway, while members of the Z-family synthesize longer chain diphosphates.1
Farnesyl diphosphate (FPP) synthase is the prototypal representative of enzymes in the E-family. FPP synthase catalyzes two reactions – the sequential addition of the hydrocarbon moieties of dimethylallyl diphosphate (C5) and geranyl diphosphate (C10) to IPP (C5) to give FPP (C15). Metabolites derived from FPP are apparently required by all living organisms, and FPP synthase activity appears to be ubiquitous. Because of its central role in isoprenoid metabolism, FPP synthase has served as the platform for studying the structure2,4,5 and mechanism6,7 of E-family enzymes. Structural and genetic studies indicate that the all α-helical “isoprenoid” fold of FPP synthase2 is found in all of the E-family chain elongation enzymes4 and in more distant relatives that catalyze cyclopropanation reactions in the sterol8 and carotenoid9 branches of the pathway and terpenoid cyclization reactions.10 An ancestral FPP synthase or a closely related relative is probably the protein from which the other enzymes evolved.
The stereochemistry of the two reactions catalyzed by FPP synthase was determined by Cornforth and Popjak as part of their classical work on cholesterol biosynthesis.11 They determined the stereochemistry of four distinct events during the chain elongation reaction (see Scheme 1) – (a) the new bond between C1 of the allylic substrate and C4 of IPP is formed with inversion at C1 of the allylic substrate, (b) C1 of the allylic substrate adds to the si-face of the double bond in IPP, (c) a new E-double bond is formed between C2 and C3 in the product, and (d) the pro-R proton is removed from C2 of IPP. Because of the long evolutionary history of chain elongation and its central role in isoprenoid biosynthesis, it has been assumed that FPP synthase catalyzes the reaction with a high degree of stereoselectivity. We now report that a significant amount of Z-double bond isomers are formed during each of the chain elongation steps and present evidence for the conformations of IPP in the active site that lead to the double bond isomers.
Scheme 1.
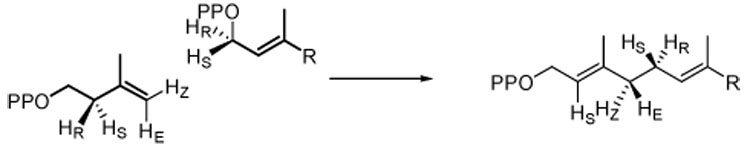
Stereochemistry of chain elongation by FPP synthase
EXPERIMENTAL SECTION
Overproduction and Purification of Recombinant Enzymes
Stock samples of E. coli strains containing the appropriate plasmid (Table 1) were allowed to recover in Luria Bertani (LB) for 90 min and plated on LB containing the appropriate antibiotic. Overnight cultures grown from single colonies (24 h old) were used to inoculate 1 L of LB containing appropriate antibiotics. Cultures were grown at 37 °C and induced with isopropyl β-D-thiogalactoside (IPTG) to a final concentration of 1 mM when the OD600 reached 0.6–0.8. After 4 h, the cells were harvested by centrifugation at 7000 × g and stored at −80 °C until needed. Frozen cells were disrupted by sonication in 50 mM phosphate buffer, pH 8.0, containing 300 mM NaCl and 10 mM imidazole, and the resulting suspension was clarified by centrifugation. Soluble His6-tagged proteins were purified by chromatography on Ni2+-NTA agarose (Qiagen, Chatsworth, CA), according to the manufacturer’s protocol. Fractions containing pure His6-tagged protein were combined, and solid NH4SO4 (47 g/100 mL) was added. Precipitated protein was collected by centrifugation and dissolved in a minimum volume of 20 mM Tris-HCl buffer, pH 8.0, containing 20% glycerol, and 4 mM DTT. The solubilized protein was dialyzed against the same buffer, flash frozen, and stored at −80 °C until needed.
Table 1.
Enzymes, plasmids, and E. coli host strains.
| Enzyme and Source | Plasmid | Host Strain | Antibiotics (µg/mL) |
Reference |
|---|---|---|---|---|
| Avian FPP synthase | pMJY189 | XA-90 | Amp (100) | 2 |
| Escherichia coli FPP synthase | pET15BEFDS | BL-21 | Amp (100) | 12 |
| Artemisia tridentata FPP synthase | pMPM3BFDS | XA-90 | Amp (100) | 13 |
| Artemisia tridentata CPP Synthase | pMPM3BCDS | XA-90 | Amp (100) | 13 |
| Artemisia tridentata GPP synthase (engineered) | pMPM3BBglCV | XA-90 | Amp (100) | 14 |
| Pyrococcus furiosus FPP/GGPP synthase | pPet-pf1 102 | Rosetta(DE3) pLysS |
Amp (100), Cam(34) |
15 |
| Methanobacter thermautotrophicus FPP/GGPP synthase | pq-mth50 | RosettaBlue | Amp (100), Tet (12.5), Cam(34) |
16 |
Product Studies
Enzyme (200 µg) was added to 35 mM HEPES buffer, pH 7.6, containing 10 mM MgCl2, 5.0 mM β-mercaptoethanol, 200 µM allylic substrate, and 200 µM IPP to a final volume of 400 µL. The mixture was incubated for 2 h at 30 °C (60 °C for the Pyrococcus furiosus and Methanobacterium thermautotrophicus geranylgeranyl diphosphate (GGPP) synthases). After the incubation, 80 µL of 500 mM glycine buffer, pH 10.5, containing 5 mM ZnCl2 and 80 units of calf mucosa alkaline phosphatase (Sigma) were added, followed by incubation at 37 °C for 1 h to hydrolyze the diphosphate esters. Solid NaCl (~0.2 g) was added, and the aqueous phase was extracted three times with 400 µL of tert-butyl methyl ether (TBME). The volume of the extract was reduced to ~50 µl with a stream of dry nitrogen. A 1 µL portion of the extract was analyzed by gas chromatography (GC) on a column 30 m × 0.25 mm (bore size) × 0.25 µm (film thickness) DB-5 capillary column (J & W Scientific) with a temperature gradient from 60 °C to 120 °C at 2 °C per min followed by a temperature gradient of 120 °C to 230 °C at 10 °C per min with a He with a flow rate of 1 mL/min. Samples were also analyzed by GC-MS using the same conditions.
In vivo studies
E. coli BL21(DE3)pLysS and E. coli JM101 were transformed with pET15B-His6-FDS and pKEN-His6-IDI respectively. LB containing ampicillin (100 µg/mL) was inoculated with the transformed cells and incubated on a rotary shaker for 12 h at 37 °C. The overnight culture was diluted 100-fold in the same medium and incubated at 37 °C on a rotary shaker. At OD600 ~ 0.6, cells were induced with 1 mM IPTG and were incubated at 30 °C for 4 h. The cells were collected by centrifugation at 7000 × g, and the pellet was washed three times with double distilled water. The cell pellet was suspended in 35 mM HEPES buffer, pH 7.6 buffer (~3 mL/g of cell paste) and disrupted by sonication. The suspension was centrifuged at 10,000 g for 30 min, and the supernatant was centrifuged at 100,000 g for an hour. To a 5 mL portion of the 100,000 g supernatant was added 1 mL of 0.5 M glycine buffer, pH 10.5, containing 200 units of alkaline phosphatase; 0.8 mL of 100 mM MgCl2 and 1.6 mL of 50 mM ZnCl2. The mixture was incubated at 37 °C for 2 h and then extracted three times with 1 mL of TBME. The combined organic extracts were concentrated to ~50 µL and analyzed by GC and GC-MS.
RESULTS
Analysis of C10 and C15 products
The stereochemistry of the C5 → C10 and C10 → C15 steps of chain elongation was determined for seven E-selective enzymes (see Scheme 2). They include two eukaryotic FPP synthases (avian2 and sagebrush13), a eubacterial FPP synthase (E. coli12), two archaeal bifunctional FPP/GGPP synthases (P. furiosus15 and M. thermautotrophicus16), and two proteins with GPP synthase activity (chrysanthemyl diphosphate (CPP) synthase from Artemisia tridentata speciformis13 and a chimeric enzyme (BglCV) constructed from A. tridentata speciformis FPP synthase and CPP synthase14). The chain elongation enzymes were incubated with equimolar concentrations of IPP and allylic diphosphate followed by treatment with alkaline phosphatase to hydrolyze the diphosphate esters to their corresponding alcohols. The stereochemistries of the newly formed double bonds in the C10 and C15 alcohols were determined by GC and GC-MS analysis with authentic samples for comparison. A typical set of GC traces is shown in Figure 1, and the results are summarized in Table 2. In each instance the expected E-isomer was accompanied by a small, but easily detectable amount of the Z-isomer.
Scheme 2.
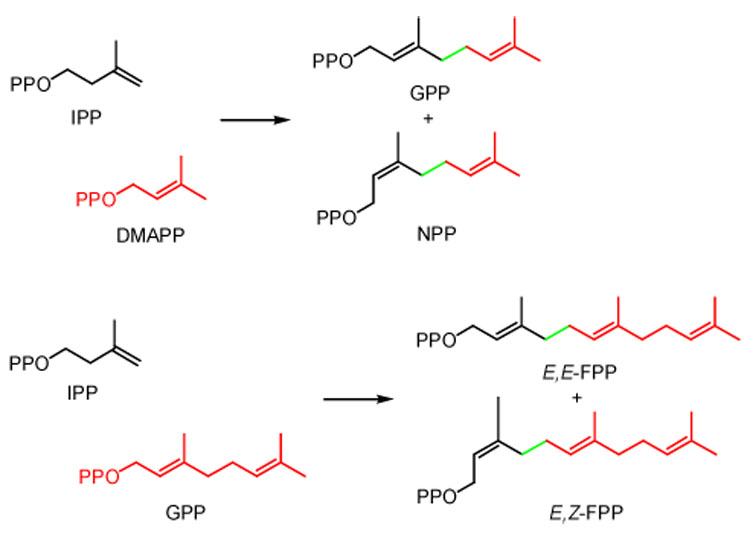
C5 to C10 and C10 to C15 Chain elongation reactions.
Figure 1.
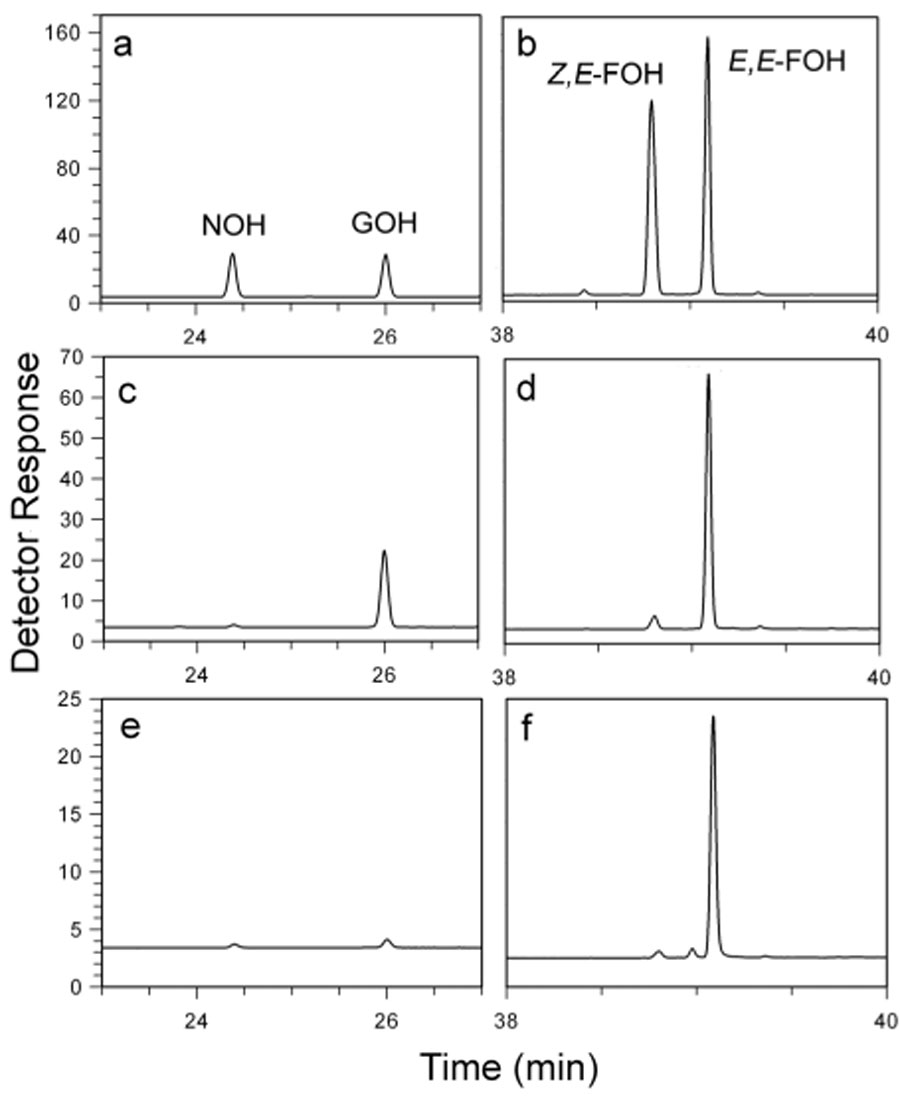
GC analysis of C10 and C15 alcohols after hydrolysis of chain elongation products from avian FPP synthase. Parts a and b: Authentic samples of NOH (Z-C10), GOH (Z-C10), Z,E-FOH and Z,E-FOH. Parts c and d: incubation with IPP and DMAPP. Parts e and f: incubation with IPP and GPP. Minor peaks at ~39.0 and 39.4 min are contaminants seen in control incubations without substrate. Conditions are in the Experimental Section.
Table 2.
C10 and C15 Chain elongation products.
| Allylic Substrate |
Enzyme | C10a | C15b | ||
|---|---|---|---|---|---|
| Zc | Ec | Z,Ec | E,Ec | ||
| DMAPP | Avian FPP Synthase | - | - | 3 | 97 |
| E. coli FPP Synthase | - | - | 4 | 96 | |
| A. tridentata FPP Synthase | - | - | 3 | 97 | |
| P. furiosus FPP/GGPP Synthase | - | - | 12 | 88 | |
| M. thermautotrophicus FPP/GGPP Synthase | - | - | 13 | 87 | |
| A. tridentata CPP Synthase | 8 | 92 | - | - | |
| BglCV GPP Synthase | 4 | 96 | - | - | |
| GPP | Avian FPP Synthase | - | - | 3 | 97 |
| E. coli FPP Synthase | - | - | 3 | 97 | |
| A. tridentata FPP Synthase | - | - | 5 | 95 | |
| P. furiosus FPP/GGPP synthase | - | - | 14 | 86 | |
C10 alcohols were 45–73% and C15 alcohols were 35–50% of the total.
C15 alcohols were 87–94% of the C10 + C15 total.
< ±0.5% for three independent determinations.
For incubations with DMAPP as the starting substrate, the mixture of alcohols, following treatment with phosphatase, included dimethylallyl alcohol from unreacted DMAPP, geraniol (GOH), nerol (NOH), and the E,E- and Z,E-isomers of farnesol (FOH). The stereoselectivity for the C10 → C15 step was determined from the relative amounts of E,E-FOH and Z,E-FOH. E,E-FOH comprised 96–97% of the mixture of C15 alcohols from bacterial and eukaryotic FPP synthases, and 87–88% for the archaeal enzymes. The relative amounts of GOH and NOH for enzymes that synthesized FPP did not accurately reflect the stereoselectivity of the C5 → C10 step because GPP, the normal allylic substrate for the C10 → C15 step, is consumed in preference to neryl diphosphate (NPP). In control experiments where IPP was incubated with GPP and NPP, the rate for conversion of NPP to E,Z-FPP was <5% of that for conversion of GPP to FPP. Thus, analysis of the peak intensities for NOH and GOH overestimates the amount of NPP generated during chain elongation because GPP is consumed preferentially. A peak for E,Z-FOH was not seen in incubations with DMAPP, presumably because of the small amount of NPP formed in the first step of chain elongation and the inefficiency of elongating NPP relative to GPP in the second step. The stereochemistry of the C5 → C10 step was determined for BglCV, an engineered version of sagebrush FPP synthase that did not catalyze the second step of chain elongation, and CPP synthase, an enzyme with C5 → C10 chain elongation activity. The E/Z stereoselectivity measured for BglCV was similar to those seen for FPP synthases. A substantially higher proportion of NPP, the Z-C10 stereoisomer, was seen for the chain elongation catalyzed by CPP synthase.
Stereochemistry at C2 of IPP
The stereochemistry for proton elimination at C2 of IPP during formation of both double bond stereoisomers was determined for the C5 → C10 step for BglCV and the C5 → C10 and C10 → C15 steps for avian, E. coli, and A. tridentata FPP synthases during incubations with (R)-[2-2H]IPP and (S)-[2-2H]IPP. The results of GC-MS analysis of the chain elongation alcohols following hydrolysis of the diphosphate esters are shown in Table 3 (mass spectra are shown in Supporting Information). When unlabelled IPP and DMAPP were used as substrate, the EI MS for GOH and NOH gave peaks at m/z 154 (M+), 139 (M+-CH3), 136 (M+-H2O), 123 (M+-CH2OH), 121 (M+-H2O-CH3). Similarly, the EI MS of Z,E- and E,E-FOH had peaks at m/z 222 (M+), 207 (M+-CH3), 204 (M+-H2O). All of the alcohols gave a m/z 69 base peak for loss of the ω-isoprene unit. For each of the enzymes, deuterium was lost from C2 of (R)-[2-2H]IPP and retained at C2 of (S)-[2-2H]IPP during the C5 → C10 step and the C10 → C15 step, regardless of which double bond isomer was produced. When DMAPP was the allylic substrate, incubation with (R)-[2-2H]IPP gave C10 chain elongation alcohols with m/z at 154, 139, 136, 123, and 121 (BglCV) and C15 alcohols with m/z at 222, 207, 204 (avian, E. coli, A. tridentata). Similar incubations with (S)-[2-2H]IPP gave peaks at m/z 155, 140, 137, 124, and 122 for the C10 alcohols and m/z 224, 209, 206 (addition of two IPP units) for the C15 alcohols. Related behavior was seen for the C10 → C15 step when GPP was the allylic substrate where (R)-[2-2H]IPP gave C15 alcohols whose mass spectra had peaks at m/z 222, 207, 204 (avian, E. coli, A. tridentata) and (S)-[2-2H]IPP gave alcohols with peaks at m/z 223, 208, 205 (addition of one IPP unit).
Table 3.
Molecular ions (M+) for the chain elongation alcohols from incubations with (R)- and (S)-[2-2H]IPP.
| Enzyme | Allylic Substrate |
GPP | NPP | E,E-FPP | E,Z-FPP | ||||
|---|---|---|---|---|---|---|---|---|---|
| R | S | R | S | R | S | R | S | ||
| Avian | DMAPP | 154 | 155 | 154 | 155 | 222 | 224 | 222 | 224 |
| GPP | - | - | - | - | 222 | 223 | 222 | 223 | |
| E. coli | DMAPP | 154 | 155 | 154 | 155 | 222 | 224 | 222 | 224 |
| GPP | - | - | - | - | 222 | 223 | 222 | 223 | |
| A. tridentata | DMAPP | 154 | 155 | 154 | 155 | 222 | 224 | 222 | 224 |
| GPP | - | - | - | - | 222 | 223 | 222 | 223 | |
| BglCV | DMAPP | 154 | 155 | 154 | 155 | - | - | - | - |
Double bond stereochemistry in vivo
E. coli strain BL21 (DE3)pLysS, transformed with a plasmid that directs expression of E. coli FPP synthase under control of the T7 promoter, was incubated as described in the Experimental Section. The cell free extract was treated with alkaline phosphatase, the resulting alcohols were extracted with MTBE, and extract was analyzed by GC-MS. The GC profile from single ion monitoring at m/z 69, the mass of base peak for E,E- and Z,E-FOH, is shown in Figure 2. The peaks at 38.4 and 38.8 min gave mass spectra that were virtually identical to those of Z,E-FOH and E,E-farnesol, respectively. Other peaks gave mass spectra that were not characteristic of mono- or sesquiterpenoid alcohols. Thus, the minor stereoisomer of FPP is synthesized in vivo by E. coli from plasmid encoded E. coli FPP synthase in a ratio similar to that measured in vitro for the same enzyme.
Figure 2.
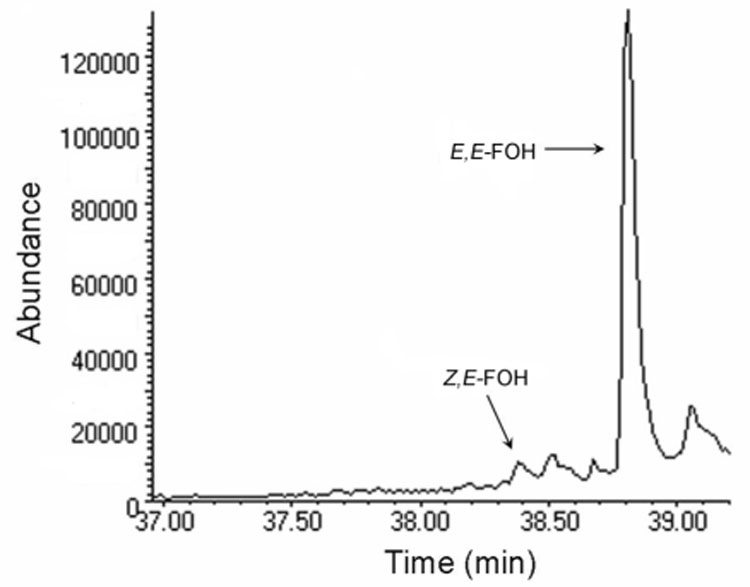
GC trace of MTBE extracts of cell-free homogenate from E. coli strain BL21(DE3)pLysS/pET15BEFDS after treatment with alkaline phosphatase.
DISCUSSION
Isoprenoid chain elongation is generally thought to proceed with a high degree of stereoselectivity characteristic of most enzyme catalyzed reactions. The stereochemistry of the double bonds, particularly the C2–C3 double bond, in the growing hydrocarbon chain of the allylic substrate is important if the intermediate is to serve as a substrate for the next addition of IPP.1 E,E-FPP, which lies at an important multiple branch point in the isoprenoid pathway, is the precursor for biosynthesis of sterols,17 farnesylated proteins,18 and sesquiterpenes.10 FPP is also the allylic primer for the chain elongation enzymes required to synthesize geranylgeranylated proteins,19 dolichols,20 heme a,21 and ubiquinones.22 All of these reactions require the E,E-stereoisomer. IPP and DMAPP are “expensive” substrates for FPP synthase. IPP is synthesized in six steps, and DMAPP in seven steps, from three molecules acetyl-CoA by the mevalonate pathway.23 Both molecules are synthesized in six steps from glyceraldehyde phosphate and pyruvate by the methylerythrose phosphate pathway.24 One might have expected that FPP synthase, with a history that apparently stretches back to the very beginning of cellular life, would have become highly selective for E-chain elongation during the course of its evolution. Our results with eukaryotic, bacterial, and archaeal forms of the enzyme indicate that this is not the case.
A biosynthetic pathway that uses IPP and DMAPP in the first step of chain elongation presents special challenges to FPP synthase. For maximal efficiency, the regions of the active site that bind IPP and DMAPP must distinguish between the two C5 isomers. Although IPP and DMAPP can both bind to the allylic site,25,26 this mode of IPP binding does not appear to present a problem. At physiological concentrations of IPP and DMAPP, substrate inhibition due to IPP binding in the allylic site does not appear be important.25 In addition, the homoallylic moiety in IPP cannot participate in chain elongation by the well-established step-wise electrophilic alkylation mechanism shown in Scheme 3.6,7 In contrast, the double bond in DMAPP is susceptible to electrophilic alkylation. FPP synthase apparently has the ability to exclude that molecule from the IPP binding region to prevent a reaction with itself.14, 26 The X-ray structure of the E. coli enzyme complexed with IPP and an unreactive thio analogue of DMAPP,5 provides an explanation for the discrimination against DMAPP at the IPP site based on differences in the conformational flexibility of the two substrates. The orientation of IPP and the thioDMAPP analogue in the E. coli FPP synthase·IPP·thioDMAPP complex (see Figure 3) would produce the E-stereochemistry during chain elongation by FPP synthase. The hydrocarbon unit of DMAPP is located on the si-face of the C3–C4 double bond in IPP with C1 positioned for an inversion of configuration upon alkylation. The C1–C2/C3–C4 dihedral angle in IPP,Θ is 118°. In this orientation, a “least motion” elimination of a proton from C2 would give an E-double bond in the allylic product. In addition, the HR proton at C2 in IPP is suitably positioned for elimination, presumably assisted by a non-bridging oxygen atom in the nearby molecule of inorganic pyrophosphate.1 DMAPP has a C2–C3 double bond, and as a consequence the C1–C2/C3–C4 dihedral angle Θ s restricted to 180°. The inability of FPP synthase to bind DMAPP at the IPP site can be explained conformational differences between the two substrates; whereas, IPP cannot be readily excluded from the DMAPP site because IPP is a more flexible molecule that can adopt conformations whose topologies are very similar to any of those accessible to DMAPP. This is illustrated in Figure 4 using space-filling representations of IPP and DMAPP. The shape of the conformer of IPP where the five carbon atoms are in the same plane (a and c) is very similar to that of DMAPP (b and d). In contrast, rotation about the C2–C3 bond in IPP, which in not possible for DMAPP, gives a substantially different shape (d and e).
Scheme 3.
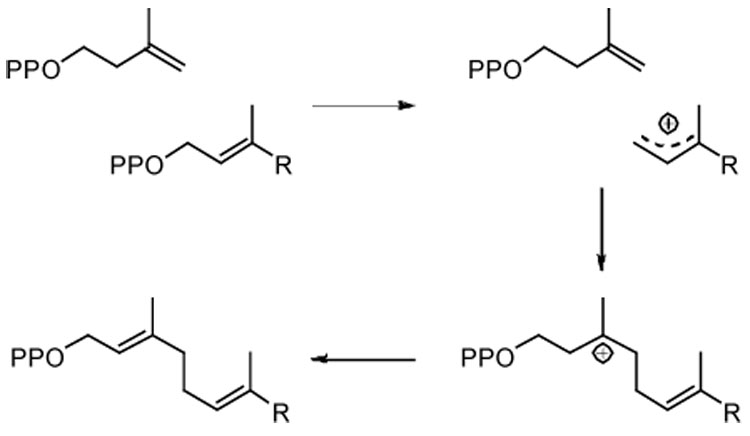
Mechanism for chain elongation
Figure 3.
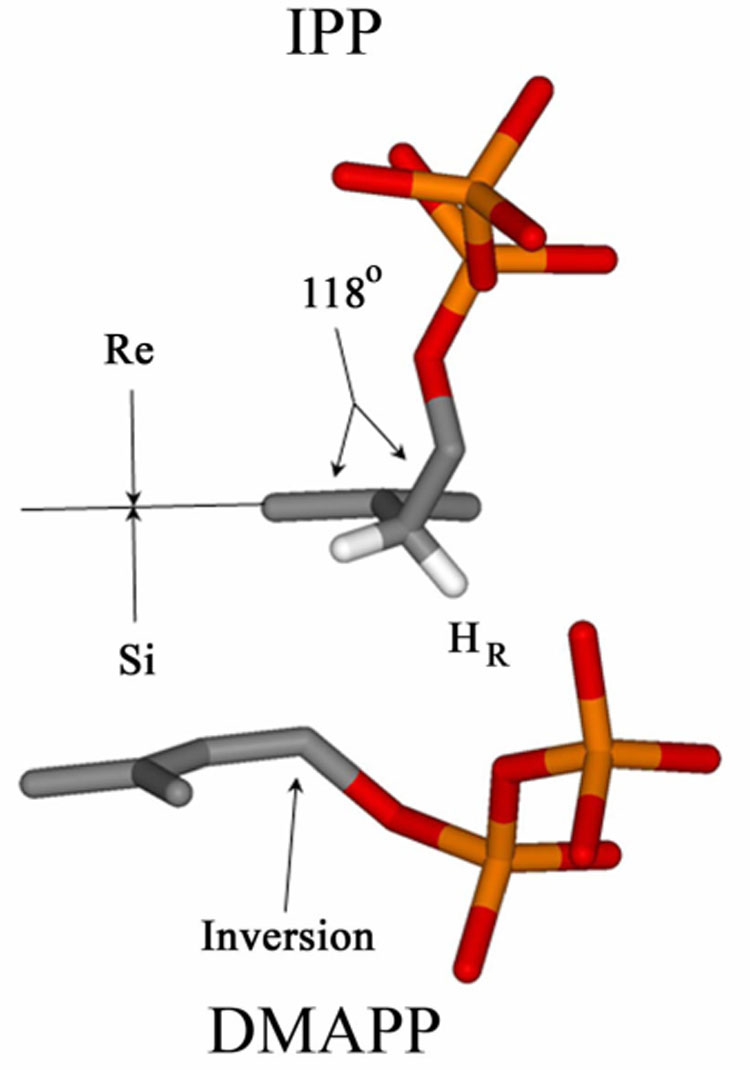
Orientation of IPP and DMAPP in the active site of E. coli FPP synthase highlighting stereochemical features important for E-selective chain elongation.
Figure 4.
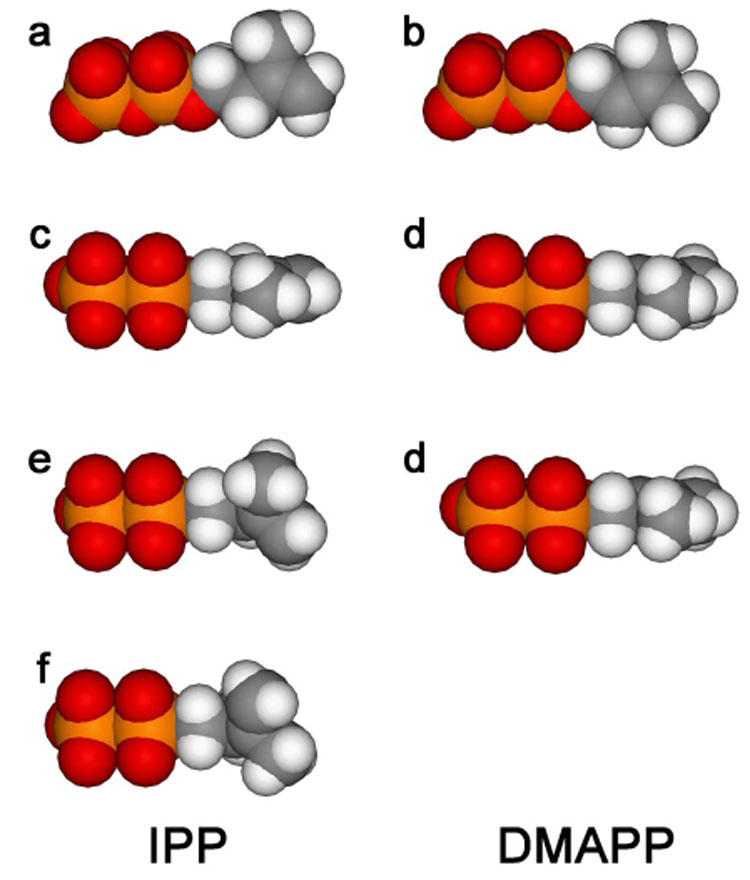
Space-filling representations of IPP and DMAPP. Parts a and b; both molecules in conformations that allow maximum spatial overlap, viewed from top face of isoprenoid units. Parts c and d; edge-on view of the conformers shown in parts a and b. Part e; same orientation as part c except the C1–C2/C3–C4 dihedral angle is 118°. Part f; same orientation as part c except the C1–C2/C3–C4 dihedral angle is 62°.
Why is the degree of stereocontrol exerted by FPP synthase during chain elongation not higher? The observation that the HR in IPP is removed when either double bond isomer is formed provides a clue. Presumably the electrophilic alkylation of IPP proceeds with the initial formation of a π-complex between C1 of the allylic cation and the double bond, followed by collapse to the C3 tertiary cation.27,28,29 The relative positions of C1 in the analog and the C3–C4 IPP double bond in the FPP synthase·IPP·thioDMAPP complex are consistent with this scenario. A 180° rotation about the C2–C3 bond in IPP gives a conformer, Θ=62° that is topologically similar to the Θ=118° conformer (see Figure 4, parts e and f) but which would produce a Z-double bond in the chain elongation product. It might be difficult to distinguish between the Θ=62° and Θ=118° conformers of IPP during binding because of their similar topologies. Thus, a relatively small movement of the dimethylallyl cation during alkylation would suffice to alkylate either conformer of IPP with subsequent elimination of HR, in agreement with our observations.
Bacterial and eukaryotic FPP synthases “waste” approximately 4% of the available IPP and 2% of the DMAPP during synthesis of FPP. These numbers increase substantially to 30% and 15%, respectively, for the thermophilic archaeal enzymes. With the possible exception of plants, the Z-C10 and Z,E-C15 double bond isomers do not appear to be utilized in subsequent biosynthetic reactions and are presumably degraded. While this amount of waste is burdensome for synthesis of FPP, it is unsustainable for the longer chain isoprenoid diphosphates required for biosynthesis of ubiquinones or dolicohols. Given the ancient origins of FPP synthase, it seems doubtful that the enzyme can achieve a greater degree of discrimination for different conformers of IPP through continued evolution. In this regard it is interesting to note that the long chain polyprenyl diphosphate synthases use FPP or GGPP instead of DMAPP as the allylic substrate to initiate elongation. Perhaps greater stereocontrol is possible in the long chain enzymes through better immobilization of the carbocationic intermediate as a result of binding interactions between the enzyme and the longer isoprenoid chain.
Supplementary Material
Supporting Information Available: Supporting information is available for general Materials and Methods; synthesis of (E,E)- and (Z,E)-farnesol; and mass spectra of (E,E)- and (Z,E)-farnesol (unlabeled, monodeuteriated, and dideuteriated), geraniol (unlabeled and monodeuteriated), and nerol (unlabeled and monodeuteriated). This material is available free of charge via the Internet at http://pubs.acs.org.
ACKOWLEDGEMENTS
We thank Professor John Vederas for generous samples of (R)- and (S)-[2-2H]IPP and Ms. Mo Chen for providing the plasmid for E coli FDS. This work was supported by National Institutes of Health grant GM 21328.
Abbreviations
- DMAPP
dimethylallyl diphosphate
- FOH
farnesol
- FPP
farnesyl diphosphate
- GOH
geraniol
- GPP
geranyl diphosphate
- GGPP
geranylgeranyl diphosphate
- IPP
isopentenyl diphosphate
- IPTG
isopropyl β-D-thiogalactoside
- LB
Luria Bertani
- NOH
nerol
- NPP
neryl diphosphate
- TBME
tert-butyl methyl ether
REFERENCES
- 1.Poulter CD. Phytochem. Reviews. 2006;5:17–26. [Google Scholar]
- 2.Tarshis LC, Yan M, Poulter CD, Sacchettini JC. Biochemistry. 1994;33:10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 3.Fujuhashi M, Zhang Y-W, Higuchi Y, Li X-Y, Koyama T, Miki K. Proc Natl. Acad. Sci, USA. 2001;98:4337–4342. doi: 10.1073/pnas.071514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Proc. Natl. Acad. Sci, USA. 1996;93:15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. J. Biol. Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 6.Poulter CD, Rilling HC. Acc. Chem. Res. 1978;11:307–313. [Google Scholar]
- 7.Poulter CD, Wiggins PL, Le AT. J. Am. Chem. Soc. 1981;103:3926–3927. [Google Scholar]
- 8.Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ. J. Biol Chem. 2000;275:30610–30617. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 9.Blagg BSJ, Jarstfer MB, Rogers DH, Poulter CD. J. Am. Chem. Soc. 2002;124:8846–8853. doi: 10.1021/ja020411a. [DOI] [PubMed] [Google Scholar]
- 10.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 11.Popjak G, Cornforth JW. Biochem. J. 1966;101:553–568. [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, Poulter CD. unpublished results. [Google Scholar]
- 13.Hemmerlin A, Rivera SB, Erickson H, Poulter CD. J. Biol. Chem. 2003;34:32132–32140. doi: 10.1074/jbc.M213045200. [DOI] [PubMed] [Google Scholar]
- 14.Erickson H, Poulter CD. J. Am. Chem. Soc. 2003;23:6885–6888. doi: 10.1021/ja034520g. [DOI] [PubMed] [Google Scholar]
- 15.Kidd MT, Poulter CD. unpublished results. [Google Scholar]
- 16.Chen A, Poulter CD. Arch. Biochem. Biophys. 1994;314:399–404. doi: 10.1006/abbi.1994.1459. [DOI] [PubMed] [Google Scholar]
- 17.Poulter CD, Rilling HC. Conversion of farnesyl pyrophosphate to squalene. In: Porter JW, editor. Biosynthesis of Isoprenoid Compounds. Chapter 8. Volume 1. New York: John Wiley & Sons; 1981. pp. 414–441. [Google Scholar]
- 18.Mayer MP, Prestwich GD, Dolence JM, Bond PD, Wu H-y, Poulter CD. Gene. 1993;132:41–47. doi: 10.1016/0378-1119(93)90512-2. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Proteau P, Poulter CD, Ferro-Novick S. J. Biol. Chem. 1995;270:21793–21799. doi: 10.1074/jbc.270.37.21793. [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka S, Sagami H, Kurisaki A, Ogura K. J. Biol Chem. 1991;266:3464–3468. [PubMed] [Google Scholar]
- 21.Weinstein JD, Branchaud R, Beele SI, Bemet WJ, Sinclair PR. Arch. Biochem. Biophys. 1986;245:44–50. doi: 10.1016/0003-9861(86)90188-8. [DOI] [PubMed] [Google Scholar]
- 22.Ashby MN, Edwards PA. J. Biol. Chem. 1990;265:13157–13164. [PubMed] [Google Scholar]
- 23.Qureshi N, Porter JW. Conversion of acetyl-coenzyme A to isopentenyl pyrophosphate. In: Porter JW, Spurgeon SL, editors. Biosynthesis of Isoprenoid Compounds. Volume 1. New York: John Wiley and Sons; 1981. pp. 47–94. [Google Scholar]
- 24.Rohdich F, Kis K, Bacher A, Eisenreich W. Curr. Opinion Chem. Biol. 2001;5:535–540. doi: 10.1016/s1367-5931(00)00240-4. [DOI] [PubMed] [Google Scholar]
- 25.Laskovics FM, Poulter CD. Biochemistry. 1981;20:1893–1901. doi: 10.1021/bi00510a027. [DOI] [PubMed] [Google Scholar]
- 26.King HL, Rilling HC. Biochemistry. 1977;16:3815–3819. doi: 10.1021/bi00636a015. [DOI] [PubMed] [Google Scholar]
- 27.Dewar MJS, Reynolds CH. J. Am. Chem. Soc. 1984;106:1744–1750. [Google Scholar]
- 28.Farcasiu D, Norton SH. J. Org. Chem. 1997;62:5374–5379. [Google Scholar]
- 29.Farcasiu D, Norton SH, Hancu D. J. Am. Chem. Soc. 2000;122:668–676. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Supporting information is available for general Materials and Methods; synthesis of (E,E)- and (Z,E)-farnesol; and mass spectra of (E,E)- and (Z,E)-farnesol (unlabeled, monodeuteriated, and dideuteriated), geraniol (unlabeled and monodeuteriated), and nerol (unlabeled and monodeuteriated). This material is available free of charge via the Internet at http://pubs.acs.org.