

Abstract
The use of catalytic metal carbene methodology with diazoacetates for the construction in high yield of polyether macrocycles having ring sizes greater than 25 has been achieved by preventing access to gamma-C-H positions for intramolecular insertion. Cyclopropanation is the exclusive outcome of reactions performed with dirhodium(II) catalysts, and product yields of greater than 70% are obtained without resorting to high dilution with solvents. With copper(I) catalysts having multiple sites for polyether coordination, intramolecular oxonium ylide formation occurs at the terminal oxygen, followed by [2,3]-sigmatropic rearrangement of the pendant allyl group, in competition with cyclopropanation. Sodium ion coordination with the reactant diazo compound inhibits oxonium ylide formation in copper catalyzed reactions. The composite results are consistent with copper serving as a template for the substrate as well as the site in the ether complex for diazo decomposition and subsequent metal carbene reactions.
Introduction
The formation of macrocycles has been a difficult challenge in organic synthesis.1 As the internal distance between reacting centers increases, the energy difference between intramolecular and intermolecular reactions decreases to zero. Few reactions have been successful in effecting macrocyclization in moderate to high yields2,3 and, despite improvements in ring closing methodologies, the need exists for procedures that provide reliable and selective access to macrocycles4,5 without having to approach infinite dilution in the process.6
Since the initial report of macrocyclization in catalytic metal carbene addition reactions 1995,7 these ring-closing reactions with diazoesters have been demonstrated to be surprisingly effective.8 Intramolecular ring closure by addition to a carbon-carbon double bond,9 carbon-carbon triple bond,10 and aryl group11 or furan12 (aromatic cycloaddition) have been reported to occur in good yields for selected examples up to ring sizes of twenty,9 and high enantiocontrol, matching those for intermolecular reactions of diazoacetates with alkenes using chiral catalysts, often accompanies these reactions.13-17 The major limitation of macrocycle formation by metal carbene addition from diazoacetates has been competitive carbon-hydrogen insertion to form γ-butyrolactones,18 and this competition has prevented further development of this methodology. To control competing C-H insertion we have designed a polyether framework that not only allows cyclopropanation reactions to ring sizes of twenty-eight in high yield, but we have also discovered preferential macrocyclic ylide formation over cyclopropanation with the use of weakly ligated copper(I) catalysts, exemplifying a templating effect around the metal ion to control product formation.
Results and Discussion
In prior studies dirhodium(II) carboxylates and active copper catalysts were found to be suitable for intramolecular cyclopropanation of the triglyme-linked allyl diazoacetate 1,14 but competition from intramolecular carbon-hydrogen insertion forming γ-butyrolactone 4 reduced the macrocyclization pathway to the point of exclusion when dirhodium(II) carboxamidates such as Rh2(MEOX)4 were used, prohibiting the use of these highly enantioselective catalysts for asymmetric macrocyclic cyclopropanation (Scheme 1). Modest amounts of the product from [2,3]-sigmatropic rearrangement (3) were observed with copper catalysts, but not with dirhodium catalysts. Similar outcomes were obtained with homologues of 1 and dirhodium catalysts.14 Since the major limitation to further development of this macrocyclization process for the production of polyethers is intramolecular C-H insertion, we designed a system blocking γ-lactone formation with the goal of enhancing macrocyclic cyclopropanation in dirhodium(II) catalyzed reactions and investigating the alternative ylide pathway for copper carbenes.
Scheme 1.

Since the 1,2-benzenedimethanol linker was shown in earlier studies to inhibit C-H insertion,10,13,15 the placement of this linker at the diazoacetate terminus was expected to prevent γ-butyrolactone formation. We sought a procedure that would allow allyl group placements at various distances from the diazoacetate terminus, thus allowing access to increasingly larger ring sizes. Synthesis of diazoacetate 8 was accomplished linearly, starting from the olefin terminus (Scheme 2) to allow modification of the length of the ethylene glycol linker. The Corey-Myers diazoacetate transfer protocol19 was used in preference to other diazo transfer methods20 because it is a single step process in organic solvent, avoiding exposure of 7 to water and the inherent difficulties in product isolation associated with the removal of water from 8. The syntheses of 9 and 10 were accomplished in a similar manner with the modifications of using allyl bromide in place of 2-allyloxyethyl methanesulfonate (for 9) or penta(ethylene glycol) in place of tetra(ethylene glycol) (for 10) (Scheme 2).
Scheme 2.

Dirhodium(II) catalysts with a known range of reactivity were selected for the diazo decomposition of 8 (eq 1). Chemoselectivity and diastereoselectivity realized from

the employment of each catalyst are summarized in Table 1. Only the 25-membered macrocycles (11) from cyclopropanation were isolated from diazo decomposition of 8 catalyzed by dirhodium(II) tetraacetate, and they were obtained in relatively high yield. Macrocycle 11 was formed as a composite of two HPLC-separable geometrical isomers that were structurally characterized by their 1H NMR chemical shifts, with coupling constants for the cyclopropane hydrogen alpha to the carbonyl group [E (δ 1.69; J = 8.3, 4.6, and 4.3 Hz) and Z (δ 1.89; J = 8.5, 7.7, and 5.9 Hz)] consistent with those seen for E and Z isomers of previously characterized cyclopropanes.13,14 Products were analyzed for geometrical preference on the cyclopropane ring by integration of the characteristic signals observed at 0.89 ppm and 1.10 ppm in the 1H NMR spectrum of the reaction mixture; these isolated signals correspond to the E and Z isomers of 11, respectively. Integration of the UV absorbance from HPLC separation of the isomers of 11 agreed with the E and Z isomer ratios obtained by 1H NMR signal integration.

Table 1.
Diazo Decompositiona of 8 with Selected Dirhodium(II) Catalysts
| Catalyst | 11:12b | E:Z (11)a | Yield (11 + 12)c |
|---|---|---|---|
| Rh2(pfb)4 | 94:6 | 52:48 | 41 |
| Rh2(OAc)4 | 100:0 | 36:64 | 71 |
| Rh2(oct)4d | 100:0 | 44:56 | 70 |
| Rh2(4S-DOSP)4 | 100:0 | 40:60 | 62 |
| Rh2(4S-MEOX)4 | 100:0 | 75:25 | 41 |
Reactions were performed by 2 h addition of 8 to refluxing solution of 1 mol % catalyst in 10 mL of dichloromethane. Average of two or more reactions.
Ratios of products obtained by 1H NMR spectroscopy.
Yields were obtained from the mass of the composite isolated products and the integrated 1H NMR signals of 11 and 12.
oct = octanoate.
Cyclopropanation products 11 were also obtained with other dirhodium(II) catalysts (Table 1), showing modest variation in the diastereomer ratio (11E:11Z). The chiral carboxamidate catalyst Rh2(4S-MEOX)4 formed 11 in moderate yield but, despite extensive efforts, the enantioselectivities of the products formed from this reaction could not be obtained due to the lack of enantiomer separation. Dirhodium(II) caprolactamate [Rh2(cap)4] was used for comparison with results from the use of Rh2(4S-MEOX)4, but with Rh2(cap)4 only trace amounts of 11 were formed, and the only additional products that could be identified (20% of total) were those resulting from carbene dimerization (singlets at 6.3 and 6.9 ppm for maleate and fumarate esters, respectively). With use of the highly reactive Rh2(pfb)4 a new product (12), consistent with that from intramolecular oxonium ylide formation and subsequent [2,3]-sigmatropic rearrangement, was observed in minor amounts (eq 1). Overall, the high yields of product obtained in this catalytic procedure that uses regulated addition of the diazo compound, but not high dilution, are quite distinctive.
The results found in Table 1 demonstrate that large rings can be formed exclusively through cyclopropanation in moderate to good yields, and that inhibition of C-H insertion at the gamma position enhances the effectiveness for macrocycle formation of dirhodium(II) catalysts, especially Rh2(4S-DOSP)421 and the chiral carboxamidate Rh2(4S-MEOX)4,20 which are known to effectively promote C-H insertion reactions in competition with cyclopropanation.22,23 The formation of macrocyclic cyclopropane 11 as the exclusive product with both of these catalysts, demonstrates that blocking of the gamma position prevents C-H insertion, providing access to a much larger range of dirhodium(II) catalysts for this important ring-forming methodology. However, with Nishiyama's RuCl2(S,S)iPr-pyBOX catalyst (13),24 [RuCl2(p-cymene)]2 (14), and silver(I) triflate, catalytic diazo decomposition of 8 showed no macrocycle formation.

Copper catalysts also effect good conversion of 8 to macrocyclic products, but oxonium ylide formation, previously found to be a minor process in Cu(CH3CN)4PF6-catalyzed reactions of diazoacetates linked to an allyl group through 1,2-benzenedimethanol9 or naphthalene-1,8-dimethanol,15 is substantially competitive with cyclopropanation. As seen by the results in Table 2, counterions associated with the copper catalyst had minimal influence on reaction chemoselectivity or diastereoselectivity. However, adding the (S,S)-tBu-BOX ligand25 to Cu(CH3CN)4PF6 resulted in no identifiable products even though complete diazo decomposition was observed. Diazo decomposition was also attempted using copper(II) acetylacetonate [Cu(acac)2], but reaction only occurred in refluxing dichloroethane (DCE) without formation of macrocycles 11 or 12. These results suggest that coordination of polyether reactant and/or product with the copper catalyst is occurring; complexation between copper(I) hexafluorophosphate and the acetate of 8, wherein acetate replaces the diazoacetate functionality, was verified by 1H NMR and IR spectroscopy.26
Table 2.
Diazo Decomposition of 8 with Selected Copper Catalystsa
| Catalyst | 11:12b | E:Z (11)b | Yield (11 + 12), %c |
|---|---|---|---|
| [Cu(OTf)]2C6H6 | 62:38 | 57:43 | 76 |
| Cu(CH3CN)4PF6 | 51:49 | 57:43 | 82 |
| Cu(hfacac)2d | 52:48 | 60:40 | 75 |
Reactions were performed by 2 h addition of 8 to refluxing solution of 10 mol% catalyst in 10 mL of dichloromethane. Average of two or more reactions.
Ratios of products obtained by 1H NMR spectroscopy.
Yields were obtained from the mass of the isolated products and the integrated 1H NMR ratios for 11 and 12.
hfacac = hexafluoroacetoacetonate.
The effect of catalyst loading on chemoselectivity and diastereoselectivity (Table 3) was modest, suggesting that the copper species that is the active catalyst does not change significantly either during the course of the reaction or as a result of different molar ratios of 8 to copper. If 8 or one of the reaction products formed a complex with copper that underwent reaction with a “free” diazoacetate (eq 2), one should expect
| (2) |
differences in diastereoselectivity and/or chemoselectivity with changing ratios of reactant to catalyst. That only modest differences are observed, and the absence of macrocyclization from reactions in which copper is coordinated with a ligand that does not undergo exchange with 8, suggests that the major reaction pathway is the one through which the copper-complexed 8 is undergoing internal diazo decomposition and subsequent carbene reactions (eq 3,4). The use of the tetrahedral copper(I) geometry to
| (3) |
| (4) |
hold ligands in an orientation suitable for subsequent macrocyclization is well established;27 the unique role of copper(I) in this study is that the templated copper is the site on which metal carbene reactions occur, a depiction for which is given in eq 5. In the orientation depicted in eq 5 the bound carbene has access to both the carbon-carbon double bond and to the allyl ether oxygen; an alternative orientation that afforded coordination with the allylic ether oxygen would prevent oxonium ylide formation, but not cyclopropanation.

Table 3.
Catalyst Loading and Product Selectivity in Diazo Decomposition of 8a
| Catalyst (mol %) | 11:12b | E:Z (11)b | Yield (11 + 12), %c |
|---|---|---|---|
| 110 | 62:38 | 66:34 | 62 |
| 80 | 63:37 | 63:37 | 54 |
| 10d | 51:49 | 57:43 | 82 |
| 1 | 61:39 | 48:52 | 76 |
Reactions were performed by 2 h addition of 8 to refluxing solution of 10 mol% copper(I) hexafluorophosphate in 10 mL of dichloromethane.
Ratios of products obtained by 1H NMR spectroscopy.
Yields were obtained from the mass of the isolated products and the integrated 1H NMR ratios for 11 and 12.
Reaction repeated three times with same outcome.
Sodium tetraphenylborate effectively coordinates Na+ with 8 as a 1:1 complex (15 and 16 are proposed structures). If reaction selectivity from diazo decomposition is independent of NaBPh4 and its molar ratio with 8, then carbene addition/ylide formation can be attributed to dynamic factors that are independent of copper coordination with 8. If, on the other hand, sodium ion coordination of 8 influences product selectivity, then diazo decomposition and subsequent carbene reactions of the copper-templated reactant can be said to be occurring. Note that intramolecular cyclopropanation and ylide reactions of the metal carbene are inhibited if 16 is the reactive form that undergoes diazo decomposition.
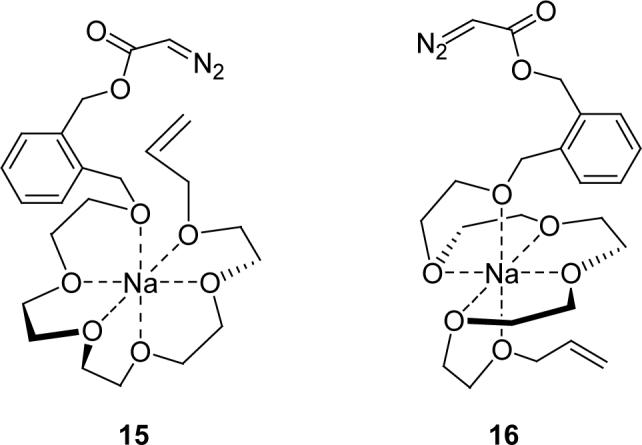
Mixtures of 8 with variable amounts of sodium tetraphenylborate, dissolved in refluxing dichloromethane, were allowed to equilibrate for thirty minutes, and then subjected to diazo decomposition by controlled addition of the 8/NaBPh4 complex to catalyst in dichloromethane, and the results are shown in Table 4. Reactions containing 110 mol % or 80 mol % of sodium tetraphenylborate had 15−30 % lower yields of 11 (and 12) than did those with 10 mol % or no sodium tetraphenylborate, consistent with the involvement of 16. The E :Z ratio of diastereoisomers of 11 did not change with the change in mol % of NaBPh4 for dirhodium(II) tetraacetate catalyzed reactions, but they were substantially different in the copper(I) hexafluorophosphate catalyzed reactions. Formation of oxonium ylide derived product 12 was not observed in the 1H NMR spectrum of the crude reaction mixtures using 110 or 80 mol % of sodium tetraphenylborate, and even 10 mol % of sodium tetraphenylborate impacted chemoselectivity, lowering the amount of 12. Sodium ion coordination with 8 effectively blocks ylide formation by association with the allyl ether oxygen so that one can conclude that the template of copper(I) with 8 is responsible for the production of ylide-derived 12. That there is also a change in diastereoselectivity from addition of sodium tetraphenylborate also suggests that the cyclopropanation pathway occurs with copper coordination (eq 2,3).
Table 4.
Selectivity in Diazo Decomposition of 8 with a Na+ Templatea
| Catalyst | NaBPh4 (mol %) | 11:12b | E:Z (11)b | Yield (11 + 12), %c |
|---|---|---|---|---|
| Rh2(OAc)4 | 110 | 100:0 | 35:65 | 45 |
| (1.0 mol %) | 80 | 100:0 | 38:62 | 47 |
| 10 | 100:0 | 35:65 | 61 | |
| 0 | 100:0 | 36:64 | 71 | |
| Cu(CH3CN)4PF6 | 110 | 100:0 | d | 49 |
| (10 mol %) | 80 | 100:0 | 80:20 | 55 |
| 10 | 63:37 | 72:38 | 73 | |
| 0 | 51:49 | 57:43 | 82 |
Decomposition carried out as 2 h addition of catalyst in dichloromethane to a refluxing dichloromethane solution of 8 and sodium tetraphenylborate.
Ratios of products obtained by 1H NMR. Variability ± 3%.
Yields obtained are based on proton NMR data and assume complete mass recovery after filtration.
Not determined.
Sodium tetraphenylborate also forms a 1:1 complex with macrocycles E-11 and Z-11. These complexes were recovered after reverse phase chromatography of the product mixture resulting from diazo decomposition of the 11/NaBPh4 complex by dirhodium(II) tetraacetate. Evidence for a complex between 8 and either potassium tetraphenylborate or cesium tetraphenylborate was not obtained. Although the solubility of these salts in methylene chloride was lower than that of sodium tetraphenylborate, extensive mixing and greater dilution of the reaction solution did not increase solubility or provide spectral evidence indicative of coordination.
The effect of the length of the linker between the diazoacetate and the allyl group on chemoselectivity and yield was investigated using diazoacetates 8-10 (Scheme 2). These compounds have five (9), six (8), and seven (10) ether oxygen atoms respectively. The difference in available oxygen atoms could produce a difference in chemoselectivity from diazo decomposition, should copper be acting as a template and assuming that coordination is rapid and equilibration between ligated copper species is slow on the reaction time scale. Substrate coordination in a tetrahedral geometry by copper(I) will involve all but one of the ether oxygen atoms of 9, while coordination with 10 will leave three oxygen atoms uncoordinated, allowing more conformational freedom in the substrate. In addition, coordination to the allyl oxygen atom would inhibit ylide formation and, if copper(I) coordination occurs, the difference in selectivity between cyclopropanation and ylide formation with [2,3]-sigmatropic rearrangement should be evident from diazo decomposition of 8-10. Decomposition of these compounds with dirhodium(II) tetraacetate or copper(I) hexafluorophosphate led to cyclopropanes 11, 17, and 18 from carbene addition to the allyl double bond, as well as 12, 19, and 20, resulting from oxonium ylide formation followed by [2,3]-sigmatropic rearrangement (Scheme 3).
Scheme 3.

Diazo decomposition of 10 with Rh2(4S-MEOX)4 did not yield cyclopropanation products in signification quantities in spite of several attempts (Table 5); 1H NMR spectral analysis of the reaction mixture showed only trace amounts of macrocyclic compounds resulting from cyclopropanation. Though Rh2(4S-MEOX)4 catalyzed diazo decomposition of 10 was complete, no additional products could be identified in the 1H NMR spectrum. Copper(II) hexafluoroacetoacetonate [Cu(hfacac)2] was employed because Clark had described the favorable chemoselectivity of this catalyst in oxonium ylide formation followed by [2,3]-sigmatropic rearrangement relative to that from dirhodium(II) tetraacetate and other copper catalysts;28 however, chemoselectivity and diastereoselectivity in the diazo decomposition of 8-10 does not significantly differ from that observed when using copper(I) hexafluorophosphate, strongly suggesting that the hfacac ligand is not bound to copper during the product-forming steps and challenging assumptions made in the uses of this catalyst.29 The cyclopropanes formed from 8-10 using dirhodium(II) tetraacetate exhibit anticipated diastereoselectivity, with E to Z ratios consistent with those seen in intermolecular cyclopropanation using ethyl diazoacetate.14,20 Similar diastereoselectivities are also seen in the decompositions of 8-10 using copper catalysts, but with a discernable trend as a function of ring size. Once again, use of copper hexafluorophosphate with the (S,S)-tBu-BOX ligand25 inhibited diazo decomposition, and only with 9 was a low amount of 15 produced (13 % yield, 50:50 E:Z, no 17). Each reaction was run at 0.25 M in diazoacetate compound; yet the preference for intramolecular reaction over intermolecular reaction was not affected by the length of the linker between the diazoacetate and the allyl group at this concentration.
Table 5.
Effect of Catalyst and Length of Linker on Product Distributiona
| Catalyst | substrate | Δ:ylide | E:Zb | % yieldc |
|---|---|---|---|---|
| Rh2(OAc)4 | 9 | 100:0 | 40:60 | 69 |
| (1.0 mol %) | 8 | 100:0 | 36:64 | 71 |
| 10 | 100:0 | 35:65 | 68 | |
| Rh2(4S-MEOX)4 | 9 | 100:0 | 44:56 | 39 |
| (1.0 mol %) | 8 | 100:0 | 65:35 | 46 |
| 10 | 100:0 | n.d. | <5 | |
| Cu(CH3CN)4PF6 | 9 | 56:44 | 61:39 | 84 |
| (10 mol %) | 8 | 51:49 | 57:43 | 82 |
| 10 | 55:45 | 47:53 | 76 | |
| Cu(hfacac)2 | 9 | 47:53 | 54:46 | 63 |
| (10 mol %) | 8 | 52:48 | 60:40 | 75 |
| 10 | 51:49 | 54:46 | 66 |
Reactions were performed by 2 h addition of the diazoacetate to a refluxing solution of the catalyst in 10 mL of dichloromethane. Outcome from duplicate or triplicate reactions.
Ratios of products obtained by integration of characteristic 1H NMR absorbances.
Yields were obtained from the mass of the composite isolated products and the integrated 1H NMR signals of cyclopropane and ylide products.
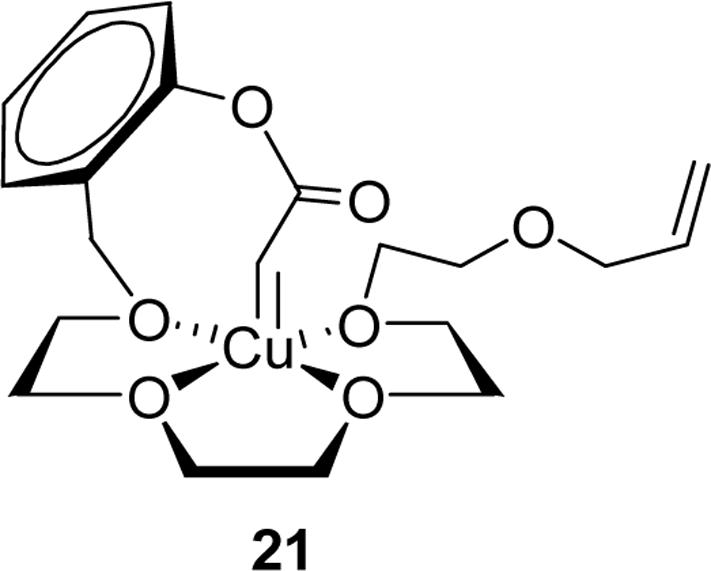
Given the virtually constant ratio products of cyclopropanation to ylide formation/rearrangement in copper ion catalysis for the series of diazoacetates 8-10 where n = 4−6, compared those diazoacetates (Scheme 1) that are incapable of this degree of association, the templating ability of copper(I) in metal carbene reactions is clear. Even the smallest member of the series (9) can form a carbene complex (21) that will facilitate both oxonium ylide formation and cyclopropanation with the same constraints between copper and either the ether oxygen or carbon-carbon double bond as either 8 or 10. The ratio of the two products reflect this.
Substrate design inhibiting γ-lactone formation from C-H insertion opens new possibilities for the use of carbene insertion reactions in macrocycle formation. This inhibition further increases the chemoselectivity of dirhodium(II) catalysts and permits the use of carboxamidate-ligated catalysts not previously viable for macrocycle formation. The good to excellent yields observed and diastereoselectivities consistent with intermolecular reactions demonstrate that a high degree of reaction control is occurring during the process leading even to 28-membered rings, and further development of this chemistry is possible. In the now well established applications of copper catalysts in metal carbene transformations, there has been an implicit assumption that the coordination sphere of copper does not change with copper carbene formation.20,22,23,30 However, the recent structural characterization of N-heterocyclic carbene complexes of copper31 suggests structural flexibility that could account for the unique role of ligand-exchangable copper catalysts in reactions of 8-10.
Experimental Section
(2-(2-(2-(2-(2-(2-Allyloxy-ethoxy)-ethoxy)-ethoxy)-ethoxy)-ethoxymethyl)-phenyl)methyl Diazoacetate (8)
The title compound was synthesized using the Corey-Myers procedure with modifications to the molar ratio of glyoxylic acid chloride p-toluenesulphonylhydrazone and N,N-dimethylaniline to alcohol, reaction time, and purification.32 To a flame-dried round bottom flask was added (2-(2-(2-(2-(2-(2-allyloxy-ethoxy)-ethoxy)-ethoxy)-ethoxy)-ethoxymethyl)-phenyl)-methanol (1.00 g, 2.51 mmol, 1.00 eq) and DCM (5.0 mL). The stirred solution was cooled to 0 °C, and glyoxylic acid chloride p-toluenesulphonylhydrazone (1.18 g, 4.52 mmol, 1.80 eq) was added in one portion followed by addition of N,N-dimethylaniline (0.570 mL, 4.52 mmol, 1.80 eq) as one aliquot, producing a clear, yellow solution. After stirring for 1 h, during which time the solution changed to a deep green color, triethylamine (1.76 mL, 12.6 mmol, 5.00 eq) was added as one aliquot, instantly changing the solution color to deep red, and the reaction mixture was stirred for an additional hour, then concentrated under reduced pressure. Concentration produced a heterogeneous mixture which was dissolved in a minimal amount of methanol and subjected to flash chromatography on silica gel (Et2O:petroleum ether:MeOH 50:50:0 to 48:48:4) to afford diazoacetate 8 as a viscous yellow oil (0.895 g, 1.91 mmol, 76 %): TLC Rf = 0.25 (Et2O: petroleum ether:MeOH, 48:48:4): 1H NMR (400 MHz) δ 7.40−7.29 (comp, 4 H), 5.91 (ddt, J = 17.3, 10.4, 5.7 Hz, 1 H), 5.31 (s, 2 H), 5.26 (ddd, J =17.3, 3.1, 1.3 Hz, 1 H), 5.17 (ddd, J = 10.4, 3.1 1.3 Hz, 1 H), 4.81 (s, broad, 1 H), 4.62 (s, 2 H), 4.02 (dt, J = 1.3, 5.7 Hz, 2 H), 3.67−3.58 (comp, 20 H); 13C NMR (100 MHz) δ 136.5, 134.7, 134.3, 129.2, 129.1, 128.4, 128.0, 117.0, 72.2, 70.9, 70.6, 70.5, 69.6, 69.4, 64.0, 46.2; IR (neat oil): 2111 cm−1 (C=N2), 1692 cm−1 (C=O); HRMS (FAB+) calculated for C23H34O8N2Li: 473.2475; found: 473.2481.
Example Procedure for Diazo Decomposition of 8, 9, and 10 with Dirhodium(II) Catalysts
An oven dried flask was charged with Rh2(OAc)4 (1.7 mg, 2.1 μmol, 0.010 eq) and DCM (5 mL) and brought to reflux. To the refluxing solution was added a solution of 8 (100 mg, 0.21 mmol, 1.0 eq) dissolved in anhydrous DCM (5 mL) over 2 h using a Kazel syringe pump. The resultant yellow solution was allowed to reflux for an additional 2 h, cooled to room temperature, and concentrated under reduced pressure. The crude reaction mixture was filtered through a glass pipet loaded with 2 inches of silica gel with a solution of EtOAc:Et2O (3:1, 15 mL) to remove the catalyst. The filtrate was concentrated under reduced pressure, to yield a clear oil (82 mg, 91 %), and a 1H NMR spectrum was obtained immediately. 1H NMR spectra of the crude decomposition mixtures do not reveal any signals between 2.5 and 3.3 ppm that would correlate with an intramolecular C-H insertion product, and no other monomeric product from 8, including those from aromatic cycloaddition or formal aromatic substitution, could be identified. Other materials were observed, but their origins were not determined.
Example Procedure for Diazo Decomposition of 8, 9, and 10 with Copper(I) Catalysts
An oven dried flask was charged with copper (I) hexafluorophosphate (7.8 mg, 22 μmol, 0.10 eq) and DCM (5 mL) and brought to reflux. To the refluxing solution was added a solution of 10 (120 mg, 0.22 mmol, 1.0 eq) dissolved in anhydrous DCM (5 mL) over 2 h using a Kazel syringe pump. The resultant yellow solution was allowed to reflux for an additional 2 h, and products were isolated as previously described to yield a clear oil (88 mg, 84 %), and a 1H NMR spectrum was obtained immediately.
Z-3,9,12,15,18,21,24-Heptaoxa-tricyclo[24.4.0.05,7]triaconta-1(26),27,29-trien-4-one (Z-11)
1H NMR (400 MHz) δ 7.46 (dd, J = 9.0, 3.7 Hz, 1 H), 7.42 (dd, J = 9.0, 3.7 Hz, 1 H), 7.33 (dd, J = 9.0, 3.7 Hz, 2 H), 5.32 (d, J = 12.7 Hz, 1 H), 5.25 (d, J = 12.7 Hz, 1 H), 4.72 (d, J = 12.0, 1 H), 4.68 (d, J = 12.0 Hz, 1 H), 3.87 (dd, J = 10.6, 5.1 Hz, 1 H), 3.75−3.48 (comp, 21 H), 1.89 (ddd, J = 8.5, 7.7, 5.9 Hz, 1 H), 1.65 (dddt, J = 8.5, 8.4, 7.0, 5.1, 1 H), 1.14−1.08 (comp, 2 H); 13C NMR (100 MHz) δ 172.3, 136.7, 134.8, 129.6, 129.3, 128.2, 128.0, 71.1, 70.7, 70.5, 70.0, 69.8, 68.6, 63.9, 21.2, 17.6, 11.5; HRMS (FAB+) calculated for C23H34O8: 439.2332; found: 439.2338.
E-3,9,12,15,18,21,24-Heptaoxa-tricyclo[24.4.0.05,7]triaconta-1(26),27,29-trien-4-one (E-11)
1H NMR (400 MHz) δ 7.43−7.36 (comp, 2 H), 7.33 (dt, J = 6.0, 2.7 Hz, 2 H), 5.29 (d, J = 12.5 Hz, 1 H), 5.25 (d, J = 12.5 Hz, 1 H), 4.71 (d, J = 12.5 Hz, 1 H), 4.68 (d, J = 12.5 Hz, 1 H), 3.74−3.61 (comp, 21 H), 3.23 (dd, J = 10.6, 7.5 Hz, 1 H), 1.76 (ddddd, J = 8.5, 7.5, 6.9, 4.6, 4.3 Hz, 1 H), 1.69 (dt, J = 8.3, 4.3 Hz, 1 H), 1.25 (ddd, J = 9.1, 4.6, 4.6 Hz, 1 H), 0.89 (ddd, J = 8.3, 6.3, 4.3 Hz, 1 H); 13C NMR (100 MHz) δ 173.6, 137.0, 134.6, 129.7, 129.2, 128.4, 128.0, 72.5, 71.2, 70.93, 70.85, 70.81, 70.78, 70.75, 70.72, 70.67, 70.1, 69.8, 64.3, 22.0, 18.9, 12.6; HRMS (FAB+) calculated for C23H34O8: 439.2332; found: 439.2338.
Purification of the crude reaction mixture containing Z-11 and E-11 was achieved using semi-preparative reverse-phase chromatography at a flow rate of 3.0 mL/min with water:acetonitrile (60:40) for 18 min, ramped at 2.4 %/min to water:acetonitrile (0:100) and maintained 15 min. Z-11 was eluted at 23.1 min and E-11 eluted at 24.7 min. The collected fractions were concentrated under reduced pressure to remove acetonitrile, frozen, and residual water was sublimed under reduced pressure.
Example Procedure for Diazo Decomposition of 8, 9, and 10 with NaBPh4
To a 1.5 dram vial with a Teflon cap liner was added 8 (0.101 g, 0.214 mmol, 1.00 eq), then DCM (1.0 mL), and finally NaBPh4 (81.8 mg, 0.235 mmol, 1.10 eq). The solution was shaken for 1 min. and allowed to stand for 30 min. Next, an oven dried flask was charged with the appropriate catalyst and DCM (5 mL) and brought to reflux. To the refluxing solution was added the solution of 8 and NaBPh4 diluted with DCM to a total volume of 5 mL over 2 h using a Kazel syringe pump. The resultant yellow/brown solution was allowed to reflux for an additional 2 h and products were isolated as previously described to yield a cloudy hetereogeneous mixture; integration of 1H NMR spectra of the crude reaction mixture was used to determine relative yields. Purification of the crude reaction mixture was achieved using semi-preparative reverse phase HPLC at a flow rate of 3.0 mL/min with water:acetonitrile (60:40) for 28 min, ramped at 2.7%/min to water:acetonitrile (0:100) and maintained 15 min. Z-11 and E-11 were eluted as a mixture at 20.5−22.8 min along as their NaBPh4 salts. The collected fractions were concentrated under reduced pressure to remove acetonitrile, frozen, and residual water was sublimed under reduced pressure.
Supplementary Material
Acknowledgment
The financial support of the National Institutes of Health (GM 46503) and the National Science Foundation is gratefully acknowledged.
Footnotes
Supporting Information Available. Additional experimental details, 1H and 13C NMR spectra, and other spectral details for reactants and products. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.a Meng Q, Hesse M. In: Topics in Current Chemistry: Macrocycles. Dewar MJS, Dunitz JD, Hafner K, Ito S, Lehn J-M, Niedenzu K, Raymond KN, Rees CW, Vögtle F, editors. Vol. 161. Springer-Verlag; New York: 1992. pp. 107–176. [Google Scholar]; b Parker D, editor. Macrocycle Synthesis: A Practical Approach. Oxford University Press; Oxford, New York: 1996. [Google Scholar]
- 2.For reviews, see: Roxburgh CJ. Tetrahedron. 1995;36:9767. Griesbeck AG, Henz A, Hirt J. Synthesis. 1996:1261.
- 3.For recent examples: Molander GA, Dehmel F. J. Am. Chem. Soc. 2004;126:10313. doi: 10.1021/ja047190o. Lee CW, Choi TL, Grubbs RH. J. Am. Chem. Soc. 2002;124:3224. doi: 10.1021/ja017790c. Lee XH, Upton TG, Gibb CLD, Gibb BC. J. Am. Chem. Soc. 2003;125:650. doi: 10.1021/ja029116g. Hodgson DM, Angrish D. Chem. Commun. 2005:4902. doi: 10.1039/b510829b. Li G-Y, Che C-M. Org. Lett. 2004;6:1621. doi: 10.1021/ol049626a.
- 4.Raymo FM, Stoddart JF, Fyfe MCT, Stoddart JF. Chem. Rev. Acc. Chem. Res. 1999;1997;9930:1643, 393. doi: 10.1021/cr970081q. [DOI] [PubMed] [Google Scholar]
- 5.Adams RD. Acc. Chem. Res. 2000;33:171. doi: 10.1021/ar980092l. [DOI] [PubMed] [Google Scholar]
- 6.Namba K, Kishi Y. J. Am. Chem. Soc. 2005;127:15382. doi: 10.1021/ja055966v. [DOI] [PubMed] [Google Scholar]
- 7.Doyle MP, Hu W. Synlett. 2001:1364. [Google Scholar]
- 8.Doyle MP, Protopopova MN, Poulter CD, Rogers DH. J. Am. Chem. Soc. 1995;117:7281. [Google Scholar]
- 9.Doyle MP, Peterson CS, Protopopova MN, Marnett AB, Parker DL, Jr., Ene DG, Lynch V. J. Am. Chem. Soc. 1997;119:8826. [Google Scholar]
- 10.Doyle MP, Peterson CS, Parker DL., Jr. Angew. Chem. Int. Ed. Engl. 1996;35:1334. [Google Scholar]
- 11.Doyle MP, Protopopova MN, Peterson CS, Vitale JP, McKervey MA, Garcia CF. J. Am. Chem. Soc. 1996;118:7865. [Google Scholar]
- 12.Doyle MP, Chapman BJ, Hu W, Peterson CS, McKervey MA, Garcia CF. Org. Lett. 1999;1:1327. [Google Scholar]
- 13.Doyle MP, Hu W, Chapman B, Marnett AB, Peterson CS, Vitale JP, Stanley SA. J. Am. Chem. Soc. 2000;122:5718. [Google Scholar]
- 14.Doyle MP, Hu W. J. Org, Chem. 2000;65:8839. doi: 10.1021/jo005589z. [DOI] [PubMed] [Google Scholar]
- 15.Doyle MP, Ene DG, Forbes DC, Pillow TH. Chem. Commun. 1999:1691. [Google Scholar]
- 16.Doyle MP, Davies SB, Hu W. Chem. Commun. 2000:867. [Google Scholar]
- 17.Doyle MP, Hu W. Tetrahedron Lett. 2000;41:6265. [Google Scholar]
- 18.Doyle MP, Phillips IM. Tetrahedron Lett. 2001;42:3155. [Google Scholar]
- 19.Corey EJ, Myers AG. Tetrahedron. Lett. 1984;25:3359. [Google Scholar]
- 20.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: from Cyclopropanes to Ylides. Wiley-Interscience; New York: 1998. [Google Scholar]
- 21.Davies HML, Bruzinski PR, Hutcheson DK, Kong N, Fall MJ. J. Am. Chem. Soc. 1996;118:6897. [Google Scholar]
- 22.Davies HML, Beckwith RE. J. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 23.Timmons D, Doyle MP. In: Multiple Bonds Between Metal Atoms. Third Edition. Cotton FA, Murillo CA, Walton RA, editors. Springer; New York: 2005. Chapter 13. [Google Scholar]
- 24.Nishiyama H, Itoh Y, Matsumoto H, Park S-B, Itoh K. J. Am. Chem. Soc. 1994;116:2223. [Google Scholar]
- 25.Evans DA, Woerpel KA, Hinman MM, Faul MM. J. Am. Chem. Soc. 1991;113:726. [Google Scholar]
- 26.Although there is evident broadening of lines with the use of an equivalent amount of copper(I) hexafluorophosphate, the chemical shift of allylic, phenyl, acetyl, and benzylic hydrogens change less than 0.05 ppm. However, the ether ethylene hydrogens, originally compacted in the region 3.60−3.72 ppm, are spread from 3.60−4.20 ppm. In contrast, the complex with sodium tetraphenylborate produces a decrease in the chemical shift for ether hydrogens to 3.18−3.70; the allylic methylene group shifted from 4.05 to 3.95 ppm, and phenyl, acetyl, and benzylic hydrogens were only modestly affected.
- 27.For recent examples: Perret-Aebi L-E, von Zelewsky A, Dietrich-Buchecker K, Sauvage J-P. Angew. Chem. Int. Ed. 2004;43:4482. doi: 10.1002/anie.200460250. Jimênez MC, Dietrich-Buchecker K, Sauvage J-P. Angew. Chem. Int. Ed. 2000;39:3284. doi: 10.1002/1521-3773(20000915)39:18<3284::aid-anie3284>3.0.co;2-7. Kwan HP, Swager TM. J. Am. Chem. Soc. 2005;127:5902. doi: 10.1021/ja042535o. Weber N, Hamann C, Kern J-M, Sauvage J-P. Inorg. Chem. 2003;42:6780. doi: 10.1021/ic0347034.
- 28.Clark JS, Krowiak SA, Street LJ. Tetrahedron Lett. 1993;34:4385. [Google Scholar]
- 29.a Roskamp EJ, Johnson CR. J. Am. Chem. Soc. 1986;108:6062. doi: 10.1021/ja00279a077. [DOI] [PubMed] [Google Scholar]; b Brogan JB, Bauer CB, Rogers RD, Zercher CK. Tetrahedron Lett. 1996;37:5053. [Google Scholar]; c Wright DL, Weekly RM, Groff R, McMills MC. Tetrahedron Lett. 1996;37:2165. [Google Scholar]
- 30.Fraile JM, García JI, Martínez-Merino V, Mayoral JA, Salvatella L. J. Am. Chem. Soc. 2001;123:7616. doi: 10.1021/ja003695c. [DOI] [PubMed] [Google Scholar]
- 31.Hu X, Castro-Rodriguez I, Meyer K. J. Am. Chem. Soc. 2003;125:12237. doi: 10.1021/ja036880+. [DOI] [PubMed] [Google Scholar]
- 32.Corey EJ, Myers AG. Tetrahedron Lett. 1984;25:3359. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.