

Abstract
Phosphonic acids encompass a common yet chemically diverse class of natural products that often possess potent biological activities. Here we report that, despite the significant structural differences among many of these compounds, their biosynthetic routes contain an unexpected common intermediate, 2-hydroxyethyl-phosphonate, which is synthesized from phosphonoacetaldehyde by a distinct family of metal-dependent alcohol dehydrogenases (ADHs). Although the sequence identity of the ADH family members is relatively low (34–37%), in vitro biochemical characterization of the homologs involved in biosynthesis of the antibiotics fosfomycin, phosphinothricin tripeptide, and dehydrophos (formerly A53868) unequivocally confirms their enzymatic activities. These unique ADHs have exquisite substrate specificity, unusual metal requirements, and an unprecedented monomeric quaternary structure. Further, sequence analysis shows that these ADHs form a monophyletic group along with additional family members encoded by putative phosphonate biosynthetic gene clusters. Thus, the reduction of phosphonoacetaldehyde to hydroxyethyl-phosphonate may represent a common step in the biosynthesis of many phosphonate natural products, a finding that lends insight into the evolution of phosphonate biosynthetic pathways and the chemical structures of new C–P containing secondary metabolites.
Phosphonic acids are common natural products that are structurally similar to phosphate-esters but contain stable carbon-phosphorus (C–P) bonds in place of the more labile carbon-oxygen-phosphorus bond motif. In many organisms, phosphonates are found as a side group on exopolysaccharides and glycoproteins, whereas in others, they constitute the core of the polar head group of phosphonolipids (1). In the latter case, they may comprise up to 50% of the total phosphorus found in the organisms, suggesting that they fulfill the critical roles in the biology of the organisms in which they are found (2). Many chemically diverse secondary metabolites produced by both fungi and bacteria also contain phosphonate functional groups, including compounds with antibacterial, antiviral, antifungal, and other useful therapeutic traits (3). Notable examples include the antibiotic fosfomycin, which is clinically used in the treatment of cystitis (4, 5), phosphinothricin tripeptide (PTT4 or bialaphos) (6), a potent and widely used herbicide, and the antibiotic dehydrophos, recently shown to contain a highly unusual phosphonate moiety analogous to dehydroalanine (7) (see Fig. 1a).
FIGURE 1.

Biosynthesis of fosfomycin, PTT, and dehydrophos. a, chemical structures of fosfomycin, PTT, and dehydrophos. b, the first three conversion steps catalyzed by PEP mutase, PnPy decarboxylase, and ADH. c, partial gene clusters of fosfomycin, PTT, and dehydrophos. Genes fom1, ppm, and dhpE encode for PEP mutase; fom2, ppd, and dhpF encode for PnPy decarboxylase; fomC, phpC and dhpG encode for ADH; and other encoded enzymes and the complete clusters were described elsewhere (12, 13).
Numerous studies have addressed the biosynthesis of phosphonate natural products, but the complete biosynthetic pathway is known only for aminoethylphosphonate (AEP) (8, 9). Nevertheless, these studies indicate that most if not all phosphonate biosynthetic pathways begin with the same two enzymatic steps, namely the rearrangement of phosphoenolpyruvate (PEP) to phosphonopyruvate (PnPy) catalyzed by PEP phosphomutase (PPM) and the subsequent conversion of PnPy to phosphonoacetaldehyde (PnAA) and CO2 catalyzed by PnPy decarboxylase (PPD) (see Fig. 1b) (9–11). During our recent studies on the biosynthesis of fosfomycin (from Streptomyces wedmorensis) (12) and PTT (from Streptomyces viridochromogenes) (see Fig. 1c) (13, 14), we noted that both pathways appear to require alcohol dehydrogenases (ADHs) (encoded by fomC and phpC, respectively) in addition to PPM and PPD. In the feeding experiments for fosfomycin biosynthesis, AEP fed to a mutant blocked at the FomC step did not complement fosfomycin production, whereas feeding hydroxyethylphosphonate (HEP) to this mutant complemented fosfomycin production (15), indicating that HEP feeds downstream of the FomC blockage. A similar phenomenon was observed with a ΔphpC mutant from the PTT biosynthetic pathway, in which AEP was found to accumulate (14). AEP has been used as a physiological equivalent of PnAA, and the conversion of carbonyl to amino group, as well as the reverse reaction, is thought to be carried out by endogenous aminotransferases (16, 17). These observations indicate that FomC and PhpC likely catalyze the step following PPD in their corresponding pathways.
Here we show that the dehydrophos biosynthetic cluster from Streptomyces luridus contains another ADH (DhpG). In vitro biochemical characterization of purified FomC, PhpC, and DhpG demonstrates that each enzyme catalyzes the same interconversion between PnAA and HEP. Although the enzymes possess fairly divergent primary sequences (only 34–37% identity), they all belong to the group III iron-dependent ADH family and share common quaternary structure and substrate specificity. Interestingly, the three enzymes characterized here form a monophyletic group with several other group III ADHs. These genes are clustered with genes encoding putative PPM and PPD enzymes. Thus, it seems likely that HEP is a common intermediate in the biosynthesis of numerous C–P-containing natural products, an observation that may prove highly useful in the mining of microbial genomes for novel phosphonate antibiotics and for elucidating the biosynthetic mechanisms of those novel phosphonate antibiotics.
EXPERIMENTAL PROCEDURES
Reagents, bacterial strains, plasmids, cell growth, and cloning of S. wedmorensis fomC and S. luridus dhpG are described in the supplemental data.
Protein Expression and Purification—The purification of S. viridochromogenes PhpC was carried out as described elsewhere (14). For purification of S. wedmorensis FomC and S. luridus DhpG, overnight cultures of BL21 (DE3) cells transformed with the construct pET28-fomC or pET15-DhpG were diluted 1:100 into M medium (9.3 g/liter K2HPO4, 2 g/liter KH2PO4, 6.7 g/liter glucose, 1.3 g/liter (NH4)2SO4, 1.3 g/liter MgSO4, 2.7 g/liter Tris, 5.3 ml glycerol, 13.3 g/liter yeast extract, 20 g/liter LB). 50 μg/ml kanamycin or 100 μg/ml ampicillin was supplemented. Cultures were grown at 37 °C until absorbance at 600 nm reached ∼1.0. Then 0.3 mm isopropyl-β-d-1-thiogalactopyranoside was added to the cultures to induce protein expression. The temperature was reduced to 20 °C, and the cell growth was continued for 14 h.
Cells were harvested by centrifugation at 4 °C for 10 min at 6,000 × g, and the pellets were resuspended in lysis buffer (50 mm HEPES, 500 mm NaCl, and 15% glycerol, pH 7.5). Lysozyme was added at a final concentration of 1 mg/ml before freezing the suspension at -80 °C. The yield was 9–10 g of bacterial wet weight/liter of culture with the M medium. Cell lysate was prepared by thawing the frozen pellets on ice. The suspension was sonicated on ice with a Branson sonicator with a 5- and 10-s interval for 10 min. After centrifugation at 16,000 × g at 4 °C for 30 min, the supernatant was loaded at a flow rate of 2 ml/min onto a column packed with 10 ml of immobilized metal affinity chromatography (IMAC) resin (Talon, Clontech). Protein purification was performed by using a BioLogic LP fast-performance liquid chromatography system (Bio-Rad). The column with the loaded sample was washed with 10 column volumes of lysis buffer followed by 5 column volumes of wash buffer (50 mm HEPES, 500 mm NaCl, 10 mm imidazole, and 15% glycerol, pH 7.5). His6-tagged FomC or His6-tagged DhpG was eluted with elution buffer containing 50 mm HEPES, 500 mm NaCl, 250 mm imidazole, and 15% glycerol, pH 7.5. Due to the relatively low expression level, FomC was only ∼70% pure (DhpG was 90–95% pure and was subjected to dialysis against lysis buffer to remove imidazole). The partially purified FomC was concentrated using an Amicon Ultra-15 centrifugal filter unit (Millipore, Bedford, MA) and loaded onto a Hiload™ 20/26 Superdex™ 200 prep grade column (Amersham Biosciences) previously equilibrated with lysis buffer. The proteins were eluted with the same lysis buffer at a flow rate of 1 ml/min with ∼90% purity.
Protein concentration was determined by the Bradford method (18). The purity of the protein was analyzed by SDS-polyacrylamide gel electrophoresis, and the gel was stained with Coomassie Brilliant Blue.
Determination of Quaternary Structure—To determine the quaternary structure, size exclusion high-performance liquid chromatography (HPLC) was performed using an Agilent 1100 series HPLC system with a Bio-Sil SEC-250 column (300 × 7.8 mm, Bio-Rad) and a mobile phase of 0.1 m Na2HPO4, 0.15 m NaCl, and 0.01 m NaN3, pH 6.8. A protein mass standard (Bio-Rad catalog number 151-1901) was used to standardize the retention time of the column with respect to molecular mass. The flow rate was 1 ml/min with the detector set at 280 nm. Samples of 20 μl of the purified enzyme or the standard were injected manually, and data were collected for 15 min. A standard curve was created by plotting the retention time versus molecular mass (log scale), and the molecular weight of the target protein was obtained by fitting of the standard curve using Origin 5.0 (Microcal Software Inc., Northampton, MA).
Chemical Synthesis of Substrates—PnAA, HEP, and hydroxymethylphosphonate (HMP) were synthesized according to the methods reported elsewhere (14, 19).
31P NMR Method—NMR analyses of samples were carried out in 20% D2O in the NMR laboratory at the University of Illinois at Urbana-Champaign on a Varian Unity U500 spectrometer equipped with a 5-mm Nalorac Quad probe.
Enzyme Kinetics—Initial rates were determined using a Cary 100 Bio UV-visible spectrophotometer (Varian, Palo Alto, CA) at 25 °C. ADH activity was measured by determining NAD(P)H-dependent PnAA reduction to HEP or NAD(P)+-dependent HEP oxidation to PnAA using a molar absorption coefficient of 6.22 × 103 m-1 cm-1 at 340 nm. The substrates were dissolved in reaction buffer (50 mm HEPES, 200 mm NaCl, pH 7.5). Kinetic determinations were performed for each condition with six different concentrations of one substrate at a fixed, saturating concentration of the other substrate. The Km and kcat were determined by non-linear regression using Origin 5.0. Parameters are expressed as the mean for at least three different measurements.
For assays at different pH values, the reactions were performed in universal buffers. Universal buffer I (25 mm acetate/MES/HEPES/borate plus 200 mm NaCl) with pH 5.0–9.5 was used for determining the kinetic parameters for PnAA reduction. Universal buffer II (25 mm HEPES/glycine plus 100 mm NaCl) with pH 7.5–10.5 was used for determining the kinetic parameters for HEP oxidation. To construct the pH profiles, the kinetic parameters kcat and kcat/Km for PnAA and HEP were determined at each pH, and the pH dependence of Y (kcat/Km) was fitted to a bell-shaped curve described by Equation 1, where H is the proton concentration, K1 and K2 are the dissociation constants for the groups that ionize at low and high pH, respectively, and YH is the pH-independent plateau value of Y at intermediate pH. The pH profile for kcat was constructed by connecting data points with smooth lines.
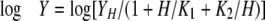 |
(Eq. 1) |
Metal Identification—1,10-Phenanthroline was used to determine the iron content of a protein (20, 21). It forms an orange-colored complex (Fe(phen)3)2+ with Fe2+ and has a maximal absorbance at 510 nm. Each of the ADHs (3–5 μm final concentration) was mixed with 1,10-phenanthroline (1.5–2 mm final concentration) and incubated at 4 °C until the activity was completely abolished (usually 1–2 h). To set up a calibration curve, in parallel, different concentrations (from 0 to 50 μm) of standard Fe2+ solution were prepared and incubated with 1,10-phenanthroline simultaneously. Absorbance was measured at 510 nm, and the iron contents of ADHs were determined by comparison with the calibration curve. Because the iron content of the PhpC sample was determined to be less than 5%, PhpC was considered to contain a different metal. EDTA was used to remove the metal from its active site. Approximately 20–25 μm PhpC was incubated with 20–25 mm EDTA at 4 °C until the activity was completely abolished (usually 1–2 h). Then the mixture was loaded to a PD-10 desalting column obtained from GE Healthcare to remove the metal-EDTA complex and the free EDTA. The inactivated PhpC (10–20 μm) was then incubated with various divalent metals (Ca2+, Co2+, Cu2+, Ni2+, Mn2+, Zn2+, Mg2+, Fe2+) prepared in the reaction buffer (50 mm HEPES, 200 mm NaCl, pH 7.5) at the concentration of 1 mm for 10 min, and the activity of PnAA reduction was measured (Fe2+ solution was prepared in the presence of 100 mm ascorbate). Among all the metals, only Zn2+ could reconstitute the PhpC activity. The methods to determine zinc content were reported elsewhere (22). 4-(2-Pyridylazo)resorcinol (PAR) forms a red-colored complex with zinc, and the complex has a maximal absorbance at 500 nm.6 m guanidine hydrochloride was used to denature PhpC for 10 min, and 10 mm PAR was prepared according to the literature (22). Approximately 20–25 μm denatured PhpC was mixed with 0.5–1 mm PAR, and the absorbance was measured and compared with the calibration curve set up by the reactions with standard Zn2+ solutions. Due to the instability of PhpC, the His6 tag was not removed from the enzyme because the protease digestion step required ∼16–20 h at 4 °C, after which most of the enzyme was precipitated.
Homology Modeling—Using the coordinates for Thermotoga maritima alcohol dehydrogenase (1O2D) and Escherichia coli lactaldehyde-propanediol oxidoreductase (2BL4) from the Protein Data Bank (www.rcsb.org), a structural model was created for FomC, PhpC, and DhpG, respectively, with the automated homology modeling function of Insight II (Accelrys, San Diego, CA). 10 intermediate models were created for each ADH with moderate refinements, and the best intermediate models were chosen for further analysis. Molecular Operating Environment (MOE, Chemical Computing Group Inc., Montreal, Canada) was used to build NAD(P)+ and the iron from 1O2D into the model, and rotamer searches of the important metal ligands were performed. Finally, PnAA or HEP was built into the models and docked manually into the active sites. Hydrogen atoms were added, and the whole structures were subjected to energy minimization using the MMFF94s force field to relieve steric and torsional artifacts from the modeling and docking processes. The final models were inspected for amino acid Ramachandran outliers, van der Waals clashes, and any obvious defects. The substrate binding pockets of FomC, PhpC and DhpG were built by the MOLCAD module in Sybyl (Tripos Inc., St. Louis, MO).
RESULTS
DhpG Is a New Group III Iron-dependent Alcohol Dehydrogenase Required for Synthesis of the Antibiotic Dehydrophos—We recently cloned and sequenced the S. luridus gene cluster responsible for the synthesis of the antibiotic dehydrophos.5 As shown in Fig. 1c, the dehydrophos gene cluster includes genes predicted to encode PEP mutase (encoded by dhpE), PnPy decarboxylase (encoded by dhpF), and a group III iron-dependent alcohol dehydrogenase (encoded by dhpG). The closest homologs of dhpG with known function are the phosphonate biosynthetic genes fomC and phpC. Interestingly, a transposon-induced dhpG mutant was unable to produce dehydrophos and instead accumulated AEP, similar to the phpC mutants described above (Fig. 2). Thus, DhpG, like PhpC and FomC, is likely to be an alcohol dehydrogenase that catalyzes the reduction of PnAA to HEP.
FIGURE 2.

31P-NMR detection of AEP accumulated in the medium by the DhpG disruption mutant. The AEP signal detected in the DhpG disruption mutant overlapped with that of the authentic AEP spiked into the sample.
Biochemical Analysis of FomC, PhpC, and DhpG—To test the hypothesis that FomC, PhpC, and DhpG are PnAA/HEP oxidoreductases, we heterologously expressed each as a fusion protein with a His6 tag. The resulting enzymes were purified and biochemically characterized in vitro. All three enzymes catalyze the reversible conversion of PnAA to HEP, which was verified by 31P NMR (data not shown). As shown in Table 1, FomC has a Km value of 31 μm for PnAA and a kcat value of 1.3 s-1, representing the lowest Km and highest kcat among the three enzymes. Because PhpC and DhpG have a larger Km (185 μm for PhpC and 414 μm for DhpG) and slightly lower kcat (0.41 s-1 for PhpC and 0.46 s-1 for DhpG), their catalytic efficiencies are 19- and 37-fold less than that of FomC. In addition, PhpC is an NADH-dependent enzyme, whereas FomC and DhpG are NADPH-dependent ADHs. No reaction was observed with HMP (one-carbon backbone) and 3-hydroxypropylphosphonate (three-carbon backbone). A possible explanation could be inferred from their structural models as discussed below.
TABLE 1.
Steady-state kinetic parameters for FomC, PhpC, and DhpG
| Enzymes | Km, PnAA | kcat | kcat/Km, PnAA | Km, NAD(P)H |
|---|---|---|---|---|
| μm | s–1 | m–1 s–1 | μm | |
| FomC | 31 ± 1 | 1.3 ± 0.1 | 41 | 9.8 ± 1.7a |
| PhpC | 185 ± 16 | 0.41 ± 0.02 | 2.2 | 19.3 ± 3.2b |
| DhpG | 414 ± 21 | 0.46 ± 0.03 | 1.1 | 0.185 ± 0.015a |
Km toward NADPH
Km toward NADH
The pH dependence of enzymatic activity was determined for FomC, PhpC, and DhpG. As illustrated for FomC (supplemental Fig. S1), PnAA reduction is more favorable under neutral conditions (optimal pH at 7.0), whereas HEP oxidation is favored under more basic conditions (optimal pH at 9.0). Due to the thermodynamics of the aldehyde reduction, PnAA reduction is strongly favored at the physiological condition (pH 7.0). The catalytic efficiency of PnAA reduction is 377-fold greater than that of the reverse reaction, and even at pH 9.0, the catalytic efficiency of the forward reaction is still 11-fold greater than that of the reverse reaction.
The group III enzyme family commonly uses iron as a cofactor. Therefore, we measured the iron content for all three ADHs. The stoichiometry between iron and protein was determined to be ∼1 for both FomC and DhpG. However, the iron content of the PhpC sample was less than 5%. To determine its metal content, an enzymatic activity reconstitution experiment was performed. EDTA was used to remove the metal from PhpC, and after removing the excess free EDTA, different divalent metals (Ca2+, Co2+, Cu2+, Ni2+, Mn2+, Zn2+, Mg2+, Fe2+) were mixed with the inactive PhpC. Only Zn2+ could reconstitute the original enzymatic activity (supplemental Fig. S2), indicating that PhpC is a zinc-dependent ADH. The stoichiometry between zinc and PhpC was determined to be 0.82 using the PAR colorimetric assay (22). In fact, PhpC is not the first zinc-dependent ADH from the family. Two family members, Bacillus stearothermophilus glycerol dehydrogenase and Saccharomyces cerevisiae alcohol dehydrogenase IV, were reported to be zinc-dependent (23, 24), and the group III iron-dependent ADH family was suggested to be renamed to the group III metal-dependent ADH family.
The quaternary structures of FomC, PhpC, and DhpG were determined by size exclusion HPLC. Both His6-tagged and non-His6-tagged FomC and DhpG were evaluated, whereas only His6-tagged PhpC was evaluated because it precipitated significantly after overnight protease digestion. Surprisingly, all three enzymes are monomeric (supplemental Fig. S3), which is unprecedented because other enzymes from the same family have oligomeric quaternary structures. For example, B. stearothermophilus glycerol dehydrogenase is an octamer (24), whereas both E. coli lactaldehyde-propanediol oxidoreductase and S. cerevisiae alcohol dehydrogenase IV are dimers (23, 25).
Genome Mining Using PPM, PPD, and ADH as Probes—In addition to S. wedmorensis, S. viridochromogenes, and S. luridus, searching genome and nucleotide databases on the National Center for Biotechnology Information (NCBI) website (www.ncbi.nlm.nih.gov) revealed an additional five organisms containing PPM, PPD, and ADH, including Bacteroides fragilis YCH46, Delta proteobacterium MLMS-1, Amycolatopsis orientalis, Azoarcus sp. BH72, and Frankia alni ACN14a (Fig. 3). Other neighboring genes are labeled by their individual locus tag or gene ID, and the corresponding putative proteins encoded by these open reading frames are listed in supplemental Table S1. Among them, the PPM-PPD-ADH region from A. orientalis is located upstream of the vancomycin group antibiotic biosynthetic cluster (26). The most interesting cluster is from F. alni ACN14a. In addition to PPM, PPD, and ADH, it contains three open reading frames encoding proteins that share a high sequence identity with those from the PTT biosynthetic cluster (13, 14), including FRAAL6372 (47% sequence identity with PhpJ), FRAAL6375 (51% sequence identity with PhpD), and FRAAL6376 (62% sequence identity with PhpE) (supplemental Table S1). Based on the biosynthetic mechanism of the PTT gene cluster, the cluster from F. alni ACN14a would also produce phosphonoformate as an intermediate (14). Nevertheless, the open reading frames that are homologous to many of the remaining PTT biosynthetic genes are absent; thus it seems likely that the operon from F. alni ACN14a encodes a novel phosphonate antibiotic biosynthetic pathway.
FIGURE 3.

Additional PPM-PPD-ADH containing partial gene clusters identified from mining genome databases. a, A. orientalis (6965 bp). b, F. alni ACN 14a (8080 bp). c, Azoarcus sp. BH72 (13953 bp). d, D. proteobacterium MLMS-1 (8927 bp). e, B. fragilis YCH46 (9112 bp).
FomC, PhpC, and DhpG Define a New Subfamily of Group III Metal-dependent ADHs Involved in Phosphonate Biosynthesis—To decipher the evolutionary origin of FomC, PhpC, and DhpG, phylogenetic analysis was performed on the group III metal-dependent ADHs from bacteria and archaea using the multiple sequence alignment tool CLUSTALW (Biology Workbench). Interestingly, as shown in Fig. 4, DhpG, FomC, PhpC and the ADHs from A. orientalis, Azoarcus sp. BH72, and F. alni ACN14a all cluster together. This indicates that they may have arisen from the same ancestor, from which phosphonate biosynthesis started to diversify.
FIGURE 4.

Phylogenetic analysis of FomC, PhpC, DhpG, and other homologous ADHs from bacteria and archaea.
DISCUSSION
In this work, we biochemically characterized three distinct alcohol dehydrogenases involved in the biosynthesis of an unexpected common intermediate, 2-hydroxyethyl-phosphonate, from three different phosphonate biosynthetic pathways producing fosfomycin, phosphinothricin tripeptide, and dehydrophos. These enzymes are monomeric, require either iron or zinc for activity, and exhibit exquisite substrate specificity.
According to Reid and Fewson (27), the oxidoreductases catalyzing the interconversion of aldehydes, ketones, and alcohols can be classified into three major categories: (i) NAD(P)-dependent alcohol dehydrogenases, (ii) NAD(P)-independent alcohol dehydrogenases that use pyrroloquinoline quinone, heme, or cofactor F420 as a cofactor, and (iii) oxidases that catalyze an essentially irreversible oxidation of alcohols. The first category can, in turn, be divided into three groups. Group I consists of long-chain zinc-dependent dehydrogenases, group II comprises the short-chain zinc-independent dehydrogenases, and group III is made up of iron-dependent alcohol dehydrogenases. Based on protein homology, FomC, PhpC, and DhpG all belong to the group III iron-dependent alcohol dehydrogenase family. A few enzymes from this family have been characterized, such as E. coli lactaldehyde-propanediol oxidoreductase (25), S. cerevisiae alcohol dehydrogenase IV (23), Zymomonas mobilis alcohol dehydrogenase II (28), and B. stearothermophilus glycerol dehydrogenase (24).
The exquisite substrate specificity of FomC, PhpC, and DhpG may be explained by homology modeling. Structural models of these ADHs were built using T. maritima alcohol dehydrogenase and E. coli 1, 2-propanediol oxidoreductase (25), which share 23–34% identity with the three target proteins. As shown in Fig. 5, Asp-189, Gln-193, His-257, and His-271 of FomC, Asp-199, Gln-203, His-268, and His-282 of PhpC, and Asp-189, His-193, His-257, and His-271 of DhpG are predicted to coordinate to their corresponding metals. The orientations of the substrates around the metals would help further polarize the carbonyls (i.e. increase electrophilicity) while placing them close to the nicotinamide rings of NAD(P)Hs, thus catalyzing the hydride transfer. Of note, the NAD(P)H recognition motif Gly-Gly-Gly was found in all three ADHs (supplemental Fig. S4). Gly-35 of FomC, Ser-41 of DhpG, and Gly-40 of PhpC are the corresponding NADPH (FomC and DhpG) or NADH (PhpC) preferring residues in the individual proteins. The reason why PhpC contains a glycine rather than an aspartate (a known NADH preferring residue) (25) but still favors NADH is unknown.
FIGURE 5.

Structure models. a, active site of FomC and its substrate binding pocket. b, active site of PhpC and its substrate binding pocket. c, active site of DhpG and its substrate binding pocket. d, residues of FomC involved in formation of the substrate binding pocket.
More importantly, the size of the substrate binding pocket in FomC, PhpC, or DhpG appears to be optimal for coordinating HEP or PnAA only. 3-Hydroxypropylphosphonate, a substrate with a longer backbone, will not fit into the substrate binding pocket, whereas HMP, a substrate with a shorter backbone, may not be held tightly by the substrate binding pocket. This may be because either the residues involved in forming hydrogen bonds with the substrate are too far to interact with HMP or possibly the distance from the leaving hydrogen on HMP to C4 of NAD(P)+ is too long. The latter is more likely because it was observed that the reaction rate of HEP oxidation catalyzed by FomC was reduced in the presence of an equal mole of HMP (data not shown). The residues that are involved in forming the substrate binding pocket for FomC are shown in Fig. 5d. Among them, Ser-157 and Tyr-261 form hydrogen bonds with the phosphonate group of the substrate.
Phylogenetic analysis showed that DhpG, FomC, PhpC, and the ADHs from A. orientalis, Azoarcus sp. BH72, and F. alni ACN14a all cluster together, indicating that they might share the same ancestor. More interestingly, the Ser-157 and Tyr-261 of FomC that are believed to form hydrogen bonds with the phosphonate group of the substrate are conserved in all the clustered ADHs but not in the non-clustered ADHs, indicating that the Ser-157 and Tyr-261 may be responsible for the phosphonate substrate selectivity (Fig. 6). This unique sequence motif might therefore be useful as a bioinformatic tool to identify additional phosphonate biosynthetic clusters that utilize HEP as an intermediate.
FIGURE 6.

Multiple sequence alignment to show the conservation of Ser-157 and Tyr-261 (FomC numbering) in all the clustered ADHs identified from phylogenetic analysis. The conserved serine and tyrosine are indicated by boxes. Pelobacter propionicus ADH, Staphylothermus marinus ADH, and Paracoccus denitrificans ADH belong to the group III metal-dependent ADH family, but are not clustered along with FomC, PhpC, and DhpG.
In conclusion, we identified three distinct alcohol dehydrogenases that are involved in the biosynthesis of an unexpected intermediate, 2-hydroxyethyl-phosphonate, common to multiple phosphonate biosynthetic pathways. These enzymes exhibit exquisite substrate specificity that is consistent with homology modeling analysis. Further phylogenetic analysis suggests that they are evolutionarily related, forming a monophyletic group with several other group III ADHs, and all these genes are clustered with genes encoding putative PPM and PPD enzymes. Thus, the reduction of phosphonoacetaldehyde to hydroxyethylphosphonate may represent a common step in the biosynthesis of many phosphonate natural products, an observation that may lend insight into the evolution of phosphonate biosynthetic pathways and may prove highly useful in the mining of microbial genomes for novel phosphonate antibiotics and for suggesting the potential chemical structures of the products of these gene clusters.
Supplementary Material
Acknowledgments
We thank Ryan P. Sullivan, Yoo Seong Choi, and Nikhil U. Nair for help with the structure modeling.
This work was supported, in whole or in part, by National Institutes of Health Grants GM059334, GM067725, and GM077596 through the NIGMS. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental information, four supplemental figures, and one supplemental table.
Footnotes
The abbreviations used are: PTT, phosphinothricin tripeptide; AEP, aminoethylphosphonate; HEP, hydroxyethylphosphonate; HMP, hydroxymethylphosphonate; PEP, phosphoenolpyruvate; PPM, PEP phosphomutase; PnPy, phosphonopyruvate; PPD, PnPy decarboxylase; PnAA, phosphonoacetaldehyde; ADH, alcohol dehydrogenase; HPLC, high pressure liquid chromatography; PAR, 4-(2-pyridylazo)resorcinol; MES, 4-morpholineethanesulfonic acid.
A. C. Eliot and W. W. Metcalf, unpublished data.
References
- 1.Hilderbrand, R. L. (1983) The Role of Phosphonates in Living Systems, CRC press, Boca Raton, FL
- 2.Quin, L. D. (1965) Biochemistry 4 324-330 [Google Scholar]
- 3.Fields, S. C. (1999) Tetrahedron 55 12237-12272 [Google Scholar]
- 4.Reeves, D. S. (1992) Infection 20 S313-S316 [DOI] [PubMed] [Google Scholar]
- 5.Stein, G. E. (1999) Int. J. Fertil. Womens Med. 44 104-109 [PubMed] [Google Scholar]
- 6.Thompson, C. J., and Seto, H. (1995) Biotechnology 28 197-222 [DOI] [PubMed] [Google Scholar]
- 7.Whitteck, J. T., Ni, W., Griffin, B. M., Eliot, A. C., Thomas, P. M., Kelleher, N. L., Metcalf, W. W., and van der Donk, W. A. (2007) Angew. Chem. Int. Ed Engl. 46 9089-9092 [DOI] [PubMed] [Google Scholar]
- 8.Barry, R. J., Bowman, E., Mcqueney, M., and Dunawaymariano, D. (1988) Biochem. Biophys. Res. Commun. 153 177-182 [DOI] [PubMed] [Google Scholar]
- 9.Seto, H., and Kuzuyama, T. (1999) Nat. Prod. Rep. 16 589-596 [DOI] [PubMed] [Google Scholar]
- 10.Seidel, H. M., Freeman, S., Seto, H., and Knowles, J. R. (1988) Nature 335 457-458 [DOI] [PubMed] [Google Scholar]
- 11.Nakashita, H., Watanabe, K., Hara, O., Hidaka, T., and Seto, H. (1997) J. Antibiot. (Tokyo) 50 212-219 [PubMed] [Google Scholar]
- 12.Woodyer, R. D., Shao, Z., Thomas, P. M., Kelleher, N. L., Blodgett, J. A., Metcalf, W. W., van der Donk, W. A., and Zhao, H. M. (2006) Chem. Biol. 13 1171-1182 [DOI] [PubMed] [Google Scholar]
- 13.Blodgett, J. A. V., Zhang, J. K., and Metcalf, W. W. (2005) Antimicrob. Agents Chemother. 49 230-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blodgett, J. A., Thomas, P. M., Li, G., Velasquez, J. E., van der Donk, W. A., Kelleher, N. L., and Metcalf, W. W. (2007) Nat. Chem. Biol. 3 480-485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodyer, R. D., Li, G. Y., Zhao, H. M., and van der Donk, W. A. (2007) Chem. Commun. 359-361 [DOI] [PubMed]
- 16.Imai, S., Seto, H., Ogawa, H., Satoh, A., and Otake, N. (1985) Agric. Biol. Chem. 49 873-874 [Google Scholar]
- 17.Alijah, R., Dorendorf, J., Talay, S., Puhler, A., and Wohlleben, W. (1991) Appl. Microbiol. Biotechnol. 34 749-755 [DOI] [PubMed] [Google Scholar]
- 18.Bradford, M. M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 19.Isbell, A. F., Englert, L. F., and Rosenberg, H. (1969) J. Org. Chem. 34 755 [Google Scholar]
- 20.Agudelo, M. I., Kustin, K., Mcleod, G. C., Robinson, W. E., and Wang, R. T. (1983) Biol. Bull. (Woods Hole) 165 100-109 [Google Scholar]
- 21.Farh, L., Hwang, S. Y., Steinrauf, L., Chiang, H. J., and Shiuan, D. (2001) J. Biochem. (Tokyo) 130 627-635 [DOI] [PubMed] [Google Scholar]
- 22.Crow, J. P., Sampson, J. B., Zhuang, Y., Thompson, J. A., and Beckman, J. S. (1997) J. Neurochem. 69 1936-1944 [DOI] [PubMed] [Google Scholar]
- 23.Drewke, C., and Ciriacy, M. (1988) Biochim. Biophys. Acta 950 54-60 [DOI] [PubMed] [Google Scholar]
- 24.Ruzheinikov, S. N., Burke, J., Sedelnikova, S., Baker, P. J., Taylor, R., Bullough, P. A., Muir, N. M., Gore, M. G., and Rice, D. W. (2001) Structure (Camb.) 9 789-802 [DOI] [PubMed] [Google Scholar]
- 25.Montella, C., Bellsolell, L., Perez-Luque, R., Badia, J., Baldoma, L., Coll, M., and Aguilar, J. (2005) J. Bacteriol. 187 4957-4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Wageningen, A. M. A., Kirkpatrick, P. N., Williams, D. H., Harris, B. R., Kershaw, J. K., Lennard, N. J., Jones, M., Jones, S. J. M., and Solenberg, P. J. (1998) Chem. Biol. 5 155-162 [DOI] [PubMed] [Google Scholar]
- 27.Reid, M. F., and Fewson, C. A. (1994) Crit. Rev. Microbiol. 20 13-56 [DOI] [PubMed] [Google Scholar]
- 28.Conway, T., and Ingram, L. O. (1989) J. Bacteriol. 171 3754-3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.