

Abstract
In the mitochondria of kinetoplastid protozoa, including Trypanosoma brucei, RNA editing inserts and/or deletes uridines from pre-mRNAs to produce mature, translatable mRNAs. RNA editing is carried out by several related multiprotein complexes known as editosomes, which contain all of the enzymatic components required for catalysis of editing. In addition, noneditosome accessory factors necessary for editing of specific RNAs have also been described. Here, we report the in vitro and in vivo characterization of the mitochondrial TbRGG2 protein (originally termed TbRGGm) and demonstrate that it acts as an editing accessory factor. TbRGG2 is an RNA-binding protein with a preference for poly(U). TbRGG2 protein levels are up-regulated 10-fold in procyclic form T. brucei compared with bloodstream forms. Nevertheless, the protein is essential for growth in both life cycle stages. TbRGG2 associates with RNase-sensitive and RNase-insensitive mitochondrial complexes, and a small fraction of the protein co-immunoprecipitates with editosomes. RNA interference-mediated depletion of TbRGG2 in both procyclic and bloodstream form T. brucei leads to a dramatic decrease in pan-edited RNAs and in some cases a corresponding increase in the pre-edited RNA. TbRGG2 down-regulation also results in moderate stabilization of never-edited and minimally edited RNAs. Thus, our data are consistent with a model in which TbRGG2 is multifunctional, strongly facilitating the editing of pan-edited RNAs and modestly destabilizing minimally edited and never-edited RNAs. This is the first example of an RNA editing accessory factor that functions in the mammalian infective T. brucei life cycle stage.
Trypanosoma brucei is a protozoan parasite that causes sleeping sickness in humans and nagana in African wildlife. During their life cycle, T. brucei are transmitted between two different hosts, the tse tse fly insect vector and mammalian host. Due to the resulting drastic changes in environmental growth conditions, the parasite displays differentiation-dependent mechanisms of energy metabolism. In the insect midgut stage (procyclic form (PF)3), energy is generated through cytochrome-mediated oxidative phosphorylation, whereas in the mammalian bloodstream form (BF), energy is generated strictly through glycolysis. Correspondingly, the single mitochondrion of T. brucei undergoes extensive alterations in both morphology and gene expression during differentiation. One aspect of this mitochondrial gene regulation is uridine insertion/deletion RNA editing, a process that is restricted to kinetoplastid protozoa, of which T. brucei is a member. During this process, uridine residues are posttranscriptionally added to and/or deleted from pre-mRNAs to produce translatable mature mRNAs. In T. brucei, the accumulation of many edited RNAs is life cycle stage-dependent, with RNAs encoding the cytochrome components apocytochrome b (CYb) and cytochrome oxidase subunit II (COII) edited only in PF and editing of components of the NADH dehydrogenase complex up-regulated in BF. A further distinction between edited RNAs is the degree to which they are edited. Some RNAs are edited only in discrete domains (CYb, MURF2, and COII), whereas the remainder are edited throughout their length and are thus referred to as pan-edited. Editing relies on largely trans-acting, mitochondrially encoded RNA molecules known as guide RNAs (gRNAs), which contain the base pairing information that specifies uridine insertion and deletion. Editing occurs through the association of the gRNA and pre-mRNA with several related multiprotein complexes known as editosomes, which contain the enzymatic components required for catalysis of RNA editing (1–9). However, due to the complex nature of the RNA-RNA and RNA-protein interactions that must occur during editing, it is likely that a battery of accessory factors that transiently associate with the editosome and/or mitochondrial RNAs are also required for accurate and efficient editing.
The two trypanosome editing accessory factors that have been studied to date are RBP16 and the MRP1/2 complex (10, 11). RBP16 was originally discovered as a oligo(U) binding factor in T. brucei that was associated with ∼30% of the gRNA population (12). Further studies showed that RBP16 is an essential protein in PF T. brucei that mediates the stability and editing of a specific subset of mRNAs (10). RBP16 depletion results in the down-regulation of the never-edited NADH dehydrogenase subunit 4 (ND4) and COI transcripts and the severe inhibition of the editing of CYb RNA (10). A direct role for RBP16 in RNA editing was supported by the demonstration that it stimulates RNA editing in vitro (13). The MRP1/2 complex, like RBP16, is essential for PF growth, and the phenotype of knockdowns is reminiscent of RBP16-depleted cells in that the primary defects are destabilization of ND4 and COI RNAs and a massive decrease in CYb RNA editing (11). Interestingly, the editing of all pan-edited RNAs was unaffected by the depletion of RBP16 or MRP1/2 (10, 11). Thus, additional accessory factors are likely to be involved in the editing of mitochondrial RNAs other than CYb.
One candidate RNA editing accessory factor in T. brucei is a predicted RNA-binding protein (GeneDB number Tb10.406.0050) that is the homologue of the Trypanosoma cruzi protein, TcRGGm (for RGG-containing motifs) (3, 14). TcRGGm was predicted to bind RNA due to the presence of N-terminal RGG boxes as well as a C-terminal RNA recognition motif (RRM) (14). The T. brucei homologue, originally termed TbRGGm, was found associated with immunoaffinity-purified editosomes although not with editosomes isolated through several steps of biochemical purification (3). More recently, TbRGGm was described as a component of a mitochondrial complex of unknown function containing additional RNA-binding proteins (15). These data suggest a role for TbRGGm in RNA editing, probably through transient editosome association and RNA binding. Here, we rename TbRGGm as TbRGG2, to distinguish it from the previously described mitochondrial RNA-binding protein, TbRGG1 (16), and we characterize the role of TbRGG2 in mitochondrial gene regulation using both in vivo and in vitro approaches. In vitro, TbRGG2 binds gRNA and mRNA and displays a preference for poly(U). In vivo, TbRGG2 is 10-fold up-regulated in PF compared with BF T. brucei but is nevertheless essential for growth of both life cycle stages. TbRGG2 forms multiple mitochondrial complexes, some of which are RNA-independent, and a small fraction of the protein co-immunoprecipitates with editosomes. Most importantly, the depletion of TbRGG2 results in the loss of editing of several pan-edited mRNAs in both PF and BF T. brucei, in some cases coupled with an increase in the corresponding pre-edited RNA, indicating that this protein is a bona fide RNA editing accessory factor. These results expand our currently limited knowledge regarding accessory proteins involved in T. brucei editing. They provide the first example of an accessory factor that affects a broad range of RNA targets, as well as the first reported RNA editing accessory factor in the mammalian infective stage of the parasite.
EXPERIMENTAL PROCEDURES
Cloning and Expression of TbRGG2—The TbRGG2 open reading frame (Tb10.406.0050) was PCR-amplified from oligo(dT)-primed cDNA from procyclic form T. brucei (clone IsTaR1 stock EATRO), using the primers RGG2-5 (5′-GCGAATTCATGAAGCGCACACCTGTTAG-3′) and RGG2-3 (5′-GGAAGCTTTTCCTTCTGACTGGCATC-3′), and the product was cloned into pCR2.1 (TA-TOPO kit; Invitrogen). TbRGG2 was excised from pCR2.1-TbRGG2 and ligated into the EcoRI and HindIII sites of pET42a (Novagen). The resultant pET42-TbRGG2 was then transformed into Rosetta strain Escherichia coli cells (Novagen) for expression. Cells were grown to an OD of 0.6, and protein production was induced with 0.4 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 37 °C. Recombinant protein was purified using a standard GST purification scheme using glutathione-agarose (Invitrogen). GST-MRP2 was produced by PCR amplification from oligo(dT)-primed PF cDNA using oligonucleotides MRP2-5 (5′-GCGAATTCGCCGCTTCTTCCAGTGATG-3′) and MRP2-3 (5′-GGAAGCTTCTCAGATGTGCGAGCGAAGC-3′) to amplify the region of the open reading frame corresponding to amino acids 30–224. The product was digested with EcoRI and HindIII and cloned into pET42a. GST-MRP2 was expressed and purified in a similar fashion as GST-TbRGG2.
Antibodies and Western Blotting—Rabbit polyclonal serum raised against purified recombinant GST-TbRGG2 was produced by Proteintech Group, Inc. (Chicago, IL). Antisera against TbRGG2 recognized a band primarily of 38 kDa in lysates from wild-type T. brucei. Polyclonal antibodies against RBP16 were described previously (12). Anti-Hsp70 antibodies were generously provided by James Bangs (University of Wisconsin). Antibodies against the CTD of RNA polymerase II were a generous gift from Vivian Bellafatto (University of Medicine and Dentistry of New Jersey). TbRGG1 antibodies were generated in rabbits using the C-terminal domain of TbRGG1 linked to maltose-binding protein as an antigen (17). Polyclonal antibodies against MRP2 were raised in rabbits by Proteintech Group, Inc. (Chicago, IL) against recombinant GST-MRP2. Antibodies against Tbmp45 (previously termed REAP-1)4 were generously provided by Steve Hajduk (University of Georgia) (18, 19). Monoclonal antibodies against editosome components KREL1, KREPA1, KREPA2, and KREPA3 were a kind gift from Dr. Kenneth Stuart (Seattle Biomedical Research Institute). For Western analysis, cells were counted using a hemacytometer, and total cells were pelleted, resuspended in SDS-PAGE sample buffer, and boiled prior to SDS-PAGE and immunoblotting.
Cell Cultures and RNAi—PF 29-13 and BF single marker cells (both kindly provided by George Cross, Rockefeller University), each expressing the T7 polymerase under control of a tetracycline-inducible promoter, were grown as indicated previously (10). To construct an RNAi vector for TbRGG2, DNA corresponding to the entire TbRGG2 open reading frame was cloned into the BamHI and HindIII sites of p2T7-177 to yield p2T7-177-TbRGG2. 50 μg of p2T7-177-TbRGG2 was transfected by electroporation into 29-13 or BF single marker cells, and transformants were selected with phleomycin (2.5 μg/ml). Positive transformants were cloned by limiting dilution to produce PF and BF single clonal lines cell expressing inducible RNAi for TbRGG2. Growth effects of TbRGG2 RNAi were monitored for at least 12 days in the absence or presence of 2.5 μg/ml tetracycline in both life cycle stages, and protein down-regulation was verified by immunoblotting.
RNA Binding Analysis—[α-32P]UTP (3000 Ci/mmol) and [α-32P]GTP (800 Ci/mmol) were purchased from PerkinElmer Life Sciences. Homopolymer RNAs were purchased from Sigma. The gRNA gCYb[558] (20) and the mRNA 3′A6U, containing 180 nt at the 3′-end of the RNA (21), were transcribed and body-labeled with [α-32P]UTP and [α-32P]GTP, respectively, using a Maxiscript kit (Ambion). 3′A6E, which contains 440 bases of edited A6 RNA sequence corresponding to the 3′-end of the RNA, was constructed using primers A6E-221-5′ (5′-GCGAATTCGTTGGTGATAGTTTTATGGATG-3′) and A6–3′ exact (5′-GCGGATCCATTTGATCTTATTCTATAACTCC-3′). Edited A6 sequence was amplified from T. brucei cDNA using Pfx polymerase and cloned into the EcoRI-BamHI sites of pBS SKII-(Stratagene). As a negative control for a random RNA, pBS RNA was also transcribed using T7 RNA polymerase, following a SacI digestion of pBS SKII–, to give a 115-nt RNA. All unlabeled RNAs were transcribed using a Megascript kit (Ambion). Cross-linking experiments contained 5 fmol of gCYb[558] or 3′A6U and 1 μm TbRGG2. RNA(s) and protein were incubated in buffer B (25 mm HEPES (pH 7.5), 30 mm KCl, 5 mm MgCl2, 0.1 mm EDTA, 0.1 mm EGTA, 10% glycerol) for 20 min at room temperature. Reactions were then UV-cross-linked using a Stratalinker 2400 (Stratagene) (1 J) for 10 min on ice. Complexes were then subjected to RNase A treatment for 15 min at 37 °C, and reactions were stopped by the addition of SDS-PAGE loading buffer. Labeled protein was resolved on 10% SDS-polyacrylamide gels and visualized by PhosphorImager analysis (Bio-Rad).
Immunoprecipitation and Glycerol Gradients—Immunoprecipitation of the editosome was performed essentially as described (22), using an equal volume of each editosome monoclonal antibody (directed against KREPA1, KREPA2, KREPA3, and KREL1). For glycerol gradient analysis, mitochondrial lysates from PF strain 29-13 (equivalent to 5 × 109 cells) were purified as indicated previously (23). Mitochondrial vesicles were lysed in lysis buffer (25 mm Tris (pH 8.0), 50 mm KCl, 10 mm MgOAc, 0.2% Nonidet P-40) for 5 min on ice. Following lysis, the supernatant was either treated with 40 units of RNaseOUT or with 15 μg/ml RNase A for 15 min at 37 °C. Lysates were then separated on 10–30% glycerol gradients (25 mm Tris (pH 8.0), 50 mm KCl, 10 mm MgOAc, 100 μm ATP, 10–30% glycerol, one-half tablet of complete protease inhibitor (Roche Applied Science)) at 38,000 × g for 5 h at 4 °C. 500-μl fractions were collected from the top of the gradient, and 10 μl of each fraction was analyzed by Western blot. To establish the sedimentation of 20 S editosome components, Western blotting for KREPA2 and KREL1 was used. To identify the 40 S region of the gradient, monoclonal antibodies were used to detect Tbmp45, which specifically sediments at 40 S (24). These internal size markers were also confirmed using a parallel gradient containing thyroglobulin as a 19 S marker.
Quantitative RT-PCR—Total RNA was extracted from uninduced and induced TbRGG2 RNAi cells using TriZOL reagent (Invitrogen) at 24 and 40 h postinduction for BF and 72 h postinduction for PF. Ten μg of RNA was treated with a DNA-free DNase kit (Ambion) to remove any residual DNA. RNA was reverse transcribed using random hexamer primers and the Taq-Man reverse transcription kit (Applied Biosciences). Resultant cDNA was used in RT-PCRs using primers previously shown to be specific to pre-edited and edited mRNAs from T. brucei (1). Twenty-five-μl real time RT-PCR reactions were set up as established by Carnes et al. (1), and the cDNA was amplified using either a MyiQ single color or an iQ5 real time PCR detection system (Bio-Rad). Results were analyzed using iQ5 software (Bio-Rad) and compared with levels of steady state β-tubulin and/or 18 S ribosomal RNA using the standard curve method. All data shown are normalized to β-tubulin, although most mRNA levels were also confirmed against 18 S rRNA and found to be comparable with those using β-tubulin. The levels of each RNA are represented as the mean and S.E. of 6–15 determinations.
RESULTS
Sequence Analysis of TbRGG2—A protein termed TcRGGm (for RGG-containing motifs) was originally identified in T. cruzi and proposed to be an RNA-binding protein due to the presence of N-terminal RGG boxes and a C-terminal RRM (14). A subsequent study, aimed at characterizing components of the T. brucei editosome, identified a TcRGGm homologue by mass spectrometry of immunopurified editosomes (3), and this protein was coined TbRGGm. The predicted RNA binding capacity and reported editosome association of the T. brucei protein suggested that it might play a role in RNA editing. Here, we present an in vivo and in vitro analysis of the TbRGGm protein, which we have renamed TbRGG2 to differentiate it from the related mitochondrial RNA-binding protein, TbRGG1, which has also been suggested to be involved in editing (16). Initially, we amplified the TbRGG2 open reading frame from T. brucei PF cDNA using primers corresponding to the 5′- and 3′-ends as denoted in the T. brucei genomic data base GeneDB (Sanger Institute) (Tb10.406.0050). The resulting cDNA possessed a 30-nt insert, encoding 10 amino acids, beginning 56 nt from its N terminus compared with the GeneDB entry (Fig. 1B, underline). The corresponding 30-nt insert is also present in the T. brucei gambiense homolog of TbRGG2 identified in GeneDB. The TbRGG2 open reading frame predicts a protein with a molecular mass of 33.1 kDa that contains two putative RNA binding domains: an N-terminal glycine-rich domain with eight RG repeats, four of which are in an RGG context (Fig. 1B, boldface type) (25, 26), and a C-terminal domain with a canonical RRM (Fig. 1). Although the N terminus of the protein is not predicted by computer algorithms to encode a mitochondrial import sequence, visual inspection reveals N-terminal arginine, threonine, and hydrophobic residues that are characteristic of such motifs in kinetoplastids (9). TbRGG2 displays 76% amino acid identity to T. cruzi TcRGGm and 64% amino acid identity to a predicted protein in the Leishmania major data base (LmjF33.0260) (Fig. 1B). Similar genes were also present in all available data bases for kinetoplastid organisms (data not shown). As shown in the ClustalW alignment of the T. brucei, T. cruzi, and L. major proteins in Fig. 1B, the C-terminal RRM is very highly conserved (79.3% identity), and the N-terminal glycine-rich region is somewhat less so. The glycine-rich regions of all three proteins are RG-rich with varying numbers of RGGs (Fig. 1B, boldface type), and all three contain at least eight conserved GWG repeats (12 in the case of (R/G)WG in L. major) (boxed in Fig. 1B). Interestingly, similar glycine-rich sequences containing regularly spaced tryptophans have been described in several proteins of disparate function (27–29), and WG/GW-rich motifs were reported to serve as binding domains for the RNAi-related Argonaute proteins in Arabidopsis (29). The closest human homologue of TbRGG2 is the pre-mRNA splicing factor SFRS1/SF2 (30), although the identity between the two proteins is only 18.8% and is largely restricted to the RRMs. The high level of conservation of the TbRGG2 protein in kinetoplastids suggests an important function in kinetoplastid biology, and the presence of two known RNA binding motifs indicates a potential role in RNA metabolism.
FIGURE 1.

Sequence analysis of TbRGG2. A, representation of the domain structure of the TbRGG2 protein. The N-terminal glycine-rich domain and C-terminal RRM are shaded. B, ClustalW alignment of TbRGG2 with homologues from T. cruzi (TcRGGm) and L. major (LmRGGm). The boxed areas indicate the N-terminal (R/G)WG repeats. The RG motifs are indicated in boldface type. The RRM domain of TbRGG2 extends from amino acid 193 to 259. The portion of TbRGG2 present in the sequenced clone that is absent in the corresponding GeneDB entry is underlined. *, identical amino acids; a colon indicates conserved amino acids; a period indicates semiconserved amino acids.
Developmental Regulation and Subcellular Localization of TbRGG2—To begin our analysis of TbRGG2 function, we asked whether expression of the protein is regulated during the T. brucei life cycle. TbRGG2 bearing a GST tag at its N terminus was expressed in E. coli, purified to near homogeneity, and used to produce polyclonal antibodies. Equivalent numbers of cells from PF and BF T. brucei were then subjected to SDS-PAGE and immunoblotted for TbRGG2. A single band of ∼38 kDa was detected in both life cycle stages. This is somewhat larger than the predicted 33 kDa, suggesting that the protein may be posttranslationally modified in vivo. Comparison of TbRGG2 levels in PF and BF revealed that the protein is much more highly expressed in the PF life cycle stage than in BF (Fig. 2). Western blot analysis of 10-fold dilutions indicated that the TbRGG2 protein is up-regulated at least 10-fold in PF. Thus, TbRGG2 is a strongly developmentally regulated protein.
FIGURE 2.

Expression of the TbRGG2 protein in PF and BF life cycle stages of T. brucei. Proteins from either 1 × 106 or 1 × 107 total cells of PF strain 29-13 or BF single marker cells were resolved by 10% SDS-PAGE. The levels of the TbRGG2 protein were determined by Western blot with anti-TbRGG2 antiserum. Levels of Hsp70 were assessed as a loading control.
The N-terminal TbRGG2 sequence suggests that the protein may be mitochondrially localized, and, in agreement with this prediction, the protein was previously reported to be associated with mitochondrial editosomes (3). To definitively ascertain the subcellular localization of TbRGG2, we analyzed subcellular fractions of PF T. brucei by Western blot. Initially, whole cell extract was separated into cytoplasmic and nuclear fractions using a procedure that leaves mitochondria in the cytoplasmic fraction (31).5 We also isolated mitochondria using a standard protocol (32). Five μg of whole cell extract and mitochondrial, cytoplasmic, and nuclear fractions were then analyzed by Western blot with anti-TbRGG2 antisera. TbRGG2 was detected primarily in the mitochondria, with very faint signals present in whole cell extract and cytoplasmic fractions (Fig. 3, top). No TbRGG2 was detected in the nucleus. Analysis of Hsp70 and RNA polymerase II localization confirmed efficient cytoplasmic/nuclear fractionation, and analysis of multiple mitochondrial proteins confirmed the enrichment of mitochondria. The degree to which TbRGG2 is enriched in mitochondria is similar to that of the known mitochondrial proteins RBP16, KREPA2, and KREL1 (Fig. 3, second and third panels). Therefore, we conclude that TbRGG2 is mitochondrially localized, as predicted by its sequence and in keeping with a potential role in mitochondrial gene regulation.
FIGURE 3.

TbRGG2 is a mitochondrially localized protein. Total cell extract from PF 29-13 cells was fractionated into cytoplasmic and nuclear fractions. Mitochondrial extract was produced from the same strain in a separate fractionation procedure. Five μg each of whole cell extract (WCE), mitochondria (Mito), cytoplasmic extract (Cyto), and nuclear extract (Nuclear) was resolved by SDS-PAGE and transferred to nylon membrane. The membrane was then analyzed by Western blot for TbRGG2; mitochondrial markers RBP16, KREL1, and KREPA3; cytoplasmic marker Hsp70; and nuclear marker polymerase (Pol) II CTD.
TbRGG2 Forms Multiple Mitochondrial Complexes—We next investigated whether TbRGG2 is a component of macromolecular complexes and whether it sediments with known editing proteins. Mitochondrial lysates from PF cells were separated on a 10–30% glycerol gradient, followed by immunoblotting for TbRGG2. TbRGG2 displayed a very broad distribution across the gradient fractions (Fig. 4, top, –RNase). A substantial amount of TbRGG2 was detected in fractions of the gradient corresponding to <20 S. However, we also observed a clear peak of TbRGG2 in association with complexes of ∼40 S, as indicated by the sedimentation the 40 S marker, Tbmp45 (previously termed REAP-1 (19)) (Fig. 4, bottom). In addition, smaller amounts of TbRGG2 were present in ∼20 S fractions, co-sedimenting with editosomes (Fig. 4, middle), as well as in fractions higher than 40 S. To determine whether association of TbRGG2 with some or all of these complexes is RNA-mediated, we treated mitochondrial extracts with RNase A prior to glycerol gradient sedimentation and compared its sedimentation with that in untreated samples (Fig. 4, top, +RNase). The 40 S and larger TbRGG2-containing complexes appeared to be partially disrupted by RNase treatment, and increased protein was, correspondingly, present in the upper gradient fractions. This redistribution indicates that a subset of TbRGG2-containing complexes are RNA-dependent. However, significant amounts of TbRGG2 remained in macromolecular complexes following RNase treatment, including a substantial portion of the protein co-sedimenting with editosome components KREPA2 and KREL1 in the 20 S region of the gradient (Fig. 4, middle). Indeed, the subset of TbRGG2 co-sedimenting with editosomes increased upon RNase treatment. These data indicate that although a large percentage of TbRGG2 is present free or in small complexes of <20S, there also exist two larger TbRGG2-containing complexes, an RNA-dependent 40 S complex(es) and an RNA independent 20 S complex(es).
FIGURE 4.

Glycerol gradient sedimentation of TbRGG2. Clarified extract from PF mitochondria (equivalent of 5 × 109 cells) was either treated with RNase A for 15 min (+) or left untreated (–) and subsequently loaded onto a 10–30% glycerol gradient. The indicated size markers were determined by the internal editosome markers, KREPA2 and KREL1 (20 S) and Tbmp45 (40 S). Gradients were centrifuged at 38,000 × g for 5 h and fractionated into 500-μl fractions. Ten μl from each fraction was analyzed by Western blot for the indicated proteins.
To determine whether the TbRGG2 does interact with the editosome, as suggested previously (33), we performed co-immunoprecipitation experiments. Editosomes were immunoprecipitated from T. brucei mitochondrial extracts using monoclonal antibodies against four editosome components. As shown in Fig. 5, Western blot analysis of KREPA1 and KREL1 confirmed the presence of editosomes in the bound fraction (Fig. 5, right, editosome IP). We next analyzed unbound and bound fractions of editosome immunoprecipitates for TbRGG2 by Western blot. Although a significant percentage of the total TbRGG2 protein was not bound to the editosome (Fig. 5, left, editosome IP), we did detect a small amount of TbRGG2 associated with editosomes (Fig. 5, right, editosome IP). To confirm the specificity of this interaction, we probed the same fractions for the editing accessory factor MRP2, which was solely in the unbound fraction and not detectable in editosome immunoprecipitates. These data support previous findings that TbRGG2 is a transient component of the editosome but indicate that it is more stably associated than other accessory factors, such as MRP2.
FIGURE 5.

Co-immunoprecipitation of TbRGG2 with components of the editosome. Editosomes were immunoprecipitated from mitochondrial extract derived from PF 29-13 cells using a mix of monoclonal antibodies against KREPA1, KREPA2, KREPA3, and KREL1. For analysis of the unbound or bound immunoprecipitations, either 8% of the unbound fraction or 4% of the bead-bound fraction was analyzed by Western blotting (WB) for editosome components, MRP2, and TbRGG2. As a control, Protein A-Sepharose beads in the absence of antibody were also used for immunoprecipitation (Protein A IP). The asterisk indicates a nonspecific band that reacts with the anti-TbRGG2 polyclonal antibody in the Protein A-alone fractions.
TbRGG2 Binds RNA in Vitro—The RNA-dependent association of
TbRGG2 with some mitochondrial complexes
(Fig. 4), as well as the
presence of putative RNA binding motifs in its sequence
(Fig. 1), strongly suggests
that TbRGG2 has the capacity to bind directly to RNA. To address this, we
performed UV cross-linking analyses with recombinant TbRGG2 and body-labeled
gCYb[558] gRNA (Fig.
6A) (20)
and 3′A6U mRNA (Fig.
6B) (21).
TbRGG2 was capable of binding gCYb[558] gRNA in vitro
(Fig. 6A, lane
2) and also bound other gRNAs and mRNAs in nondenaturing gel shift assays
(data not shown). Once the binding of TbRGG2 to RNA was established, we
performed UV cross-linking competition assays with homoribopolymers to
determine whether the protein displayed any sequence-specific RNA binding
characteristics. As shown in Fig.
6A, the binding of TbRGG2 to gCYb[558] was unaffected by
a 1000-fold excess of either poly(A) or poly(C) homopolymer RNA. However,
poly(G) was able to completely inhibit gRNA binding by TbRGG2 at approximately
a 100-fold excess of cold competitor, whereas poly(U) competed efficiently
with gRNA at about a 10-fold excess (Fig.
6A). Thus, TbRGG2 binds gRNA in vitro and
displays a general RNA binding preference of poly(U) > poly(G) 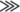 poly(A) or poly(C).
poly(A) or poly(C).
FIGURE 6.

TbRGG2 is an RNA-binding protein. A, TbRGG2 binds to gRNA and displays a preference for poly(U). Five fmol of body-labeled gCYb[558] was incubated with 1 μm purified GST-TbRGG2 for 20 min (lane 2). Lane 1 is 1 μm GST-TbRGG2 in the absence of radiolabeled gRNA. For competitor analyses (lanes 3–6), 5 fmol (lane 3), 50 fmol (lane 4), 500 fmol (lane 5), or 5 pmol (lane 6) of the indicated cold homoribopolymer was added to 1 μm GST-TbRGG2 immediately prior to the addition to 5 fmol gCYb[558]. Reactions were subjected to UV cross-linking and treated with RNase A, and proteins were resolved by 10% SDS-PAGE. Proteins that were cross-linked to radiolabeled gCYb[558] was visualized by PhosphorImager analysis. B, TbRGG2 binds to A6 mRNA in vitro. Five fmol of body-labeled 3′A6U mRNA was incubated with 1 μm purified GST-TbRGG2 as in A. Lane 1, 3′A6U RNA in the absence of TbRGG2 protein. The indicated unlabeled competitor RNAs (lanes 3–5) were added as in A ina1× (5 fmol, lane 3), 10× (50 fmol, lane 4), or 100× molar excess (500 fmol, lane 5) immediately prior to the addition of 5 fmol of radiolabeled 3′A6U.
The RNA binding of TbRGG2 was further characterized to determine whether it associates with mRNAs that undergo editing. For these studies, A6 RNA corresponding to the 3′ 180 nt of the unedited RNA (3′A6U (21)) was body-labeled and subjected to cross-linking analysis as in Fig. 6A. These experiments showed that TbRGG2 binds to 3′A6U RNA in vitro (Fig. 6B, lane 2). For competitor analyses, several unlabeled mRNAs were prepared, including 3′A6U itself, 3′A6E (corresponding to the 3′ 440 nt of the edited A6 RNA), and a 115-nt transcript from the pBS SKII– vector. As seen in Fig. 6B, 3′A6E competed binding ∼5–10-fold better than 3′A6U, whereas pBS showed little ability to compete. The stronger binding of 3′A6E may partially reflect its longer length but is probably also a result of its higher U content (57% compared with 25% in 3′A6U), in agreement with the homopolymer competitions in Fig. 6A.
TbRGG2 Is Essential in PF and BF T. brucei—To address the functional importance of TbRGG2 in vivo, we down-regulated its expression in both PF and BF life cycle stages using tetracycline-regulated RNAi (Fig. 7). The entire TbRGG2 open reading frame was cloned between opposing tetracycline-regulated T7 promoters in p2T7-177 (34), the resulting plasmid was linearized and transfected into either PF 29-13 or BF single marker cells, and clonal lines were selected. Down-regulation of TbRGG2 was initiated by the addition of 2.5 μg/ml tetracycline to the growth medium, and depletion of protein levels was confirmed by Western blot (Fig. 7). In both PF and BF life cycle stages, depletion of TbRGG2 had a significant effect on the growth of the parasite. In PF cells, growth slowed by day 4 following RNAi induction, the cells effectively stopped growing by day 6, and cell numbers decreased between days 6 and 12 (Fig. 7A). The decrease in cell growth corresponded with the decrease in TbRGG2 protein levels (78% decrease by day 6 following RNAi induction) (Fig. 7A). In BF cells, the effect of TbRGG2 RNAi was even more pronounced. Growth of BF cells slowed just following the first full day of RNAi induction, and cells ceased growth by day 2. Again, these findings corresponded with the loss of TbRGG2 protein (77% decrease by 48 h following RNAi induction) (Fig. 7B). A decrease in cell numbers continued in subsequent days up to day 7 post-induction, after which cells began to recover from the RNAi, as is often observed in T. brucei upon RNAi analysis of essential genes. From these data, we conclude that TbRGG2 is an essential protein in both PF and BF T. brucei cells.
FIGURE 7.

TbRGG2 is essential in both PF and BF T. brucei life cycle stages. The RNAi vector p2T7-177-TbRGG2 was transfected into both the PF strain 29-13 (A) and the BF single marker strain (B) of T. brucei. RNAi was induced by the addition of 2.5 μg/ml tetracycline to the cell media. Cells from duplicate cultures were counted every 2 days, and 1 × 106 cells were collected and immunoblotted for the presence of TbRGG2 or Hsp70.
Due to the dramatic growth effect observed following TbRGG2 RNAi, we wanted to confirm that other mitochondrial proteins, particularly those known to be involved in editing, were not perturbed secondarily by TbRGG2 depletion. To test this, PF TbRGG2 RNAi cells were either uninduced or induced with tetracycline for 2 or 3 days, and the levels of multiple proteins were analyzed by immunoblotting (Fig. 8A). We examined the levels of four core editosome components, the editing accessory factors RBP16 and MRP2, and TbRGG1, which has been implicated in, but not shown to play a direct role in RNA editing in T. brucei. Hsp70 was included in these analyses as a loading control. Except for the loss of TbRGG2, no other protein analyzed showed any decrease in abundance following down-regulation of TbRGG2. The slight increase in the amount of TbRGG1 on day 3 was not reproducible. These results indicate that the effect of TbRGG2 RNAi was both specific for TbRGG2 (e.g. it did not result in a decrease in the levels of the related TbRGG1), and it did not exert a global effect on other mitochondrial proteins despite a clear growth defect in these cells.
FIGURE 8.

Effect of TbRGG2 down-regulation on steady state levels of other editing related proteins. A, PF TbRGG2 RNAi cells were collected prior to induction and on days 2 and 3 following tetracycline induction and immunoblotted for the indicated proteins. Hsp70 was used as an indicator for equal protein loading. All lanes correspond to 1 × 106 cells, except the editosome components (KREPA1, KREPA2, KREL1, and KREPA3), for which 1 × 107 cells were analyzed. The apparent increase in TbRGG1 on day 6 was not reproducible. B, mitochondrial extracts from TbRGG2 RNAi cells, either uninduced (–) or induced for 3 days (+), were separated on 10–30% glycerol gradients as in Fig. 4. The sedimentation patterns of the 20 S editosome components KREPA1, KREPA2, KREPA3, and KREL1, as well as the 40 S marker, Tbmp45, were detected by Western blot.
Although down-regulation of TbRGG2 did not cause overall effects on the levels of the other proteins analyzed, the possibility remained that the integrity of macromolecular editing complexes might be disrupted by the loss of TbRGG2. To address this, mitochondrial extracts from TbRGG2 RNAi cells either uninduced or induced with tetracycline for 3 days were separated by glycerol gradient sedimentation. Fractions were collected and analyzed for both editosome components and the 40 S marker TbMP45. As shown in Fig. 8B, the sedimentation patterns of both the editosome and Tbmp45 are similar to those of parental 29-13 cells (Fig. 4). Upon RNAi-induced loss of TbRGG2, no discernable change was observed for either the 20 S editosome complex or the Tbmp45 protein (Fig. 8B). Therefore, the loss of the TbRGG2 has no evident effect on the integrity of the core editing complex(es).
TbRGG2 Down-regulation Leads to a Dramatic Decrease in the Levels of Pan-edited mRNAs—Several lines of evidence indicate that TbRGG2 may play a role during RNA editing. TbRGG2 is primarily localized to the mitochondria and binds RNA, and a fraction of the protein co-immunoprecipitates with editosomes. Moreover, the essential nature of TbRGG2 (Fig. 6) suggests that it may play an integral role during the editing process, which is essential in both PF and BF T. brucei (1, 6, 35–37). To investigate the role of TbRGG2 in RNA editing, we extracted total RNA from PF and BF cells either uninduced or induced for TbRGG2 RNAi. RNA was reverse transcribed and analyzed by quantitative real time RT-PCR utilizing primer pairs that have been previously used to analyze mitochondrial RNAs in T. brucei (1, 38) (Fig. 9). Since TbRGG2 is essential, care was taken in determining the time frame of RNA extraction so that it occurred when TbRGG2 protein levels were diminished, but the cells were not experiencing a strong growth effect, thereby limiting secondary effects on RNA metabolism. Based on these criteria, RNA was isolated from PF cells following 3 days of induction (72 h) and from BF cells at two time points (24 and 40 h).
FIGURE 9.

Effect of TbRGG2 down-regulation on mitochondrial RNA levels. A, quantitative RT-PCR analysis of RNAs from PF TbRGG2 RNAi cells at 72 h (3 days) postinduction. B, quantitative RT-PCR of RNAs from BF TbRGG2 RNAi cells at 24 and 40 postinduction. RNA levels represent the mean and S.E. of 6–15 determinations. P, pre-edited; E, edited.
We analyzed multiple classes of RNA in PF TbRGG2 RNAi cells, including minimally edited (CYb, COII, and MURF2), pan-edited (A6, COIII, ND7, and RPS12), and never-edited (ND4 and COI) RNAs. These analyses revealed an intriguing pattern. Upon TbRGG2 depletion, the never-edited RNAs encoding ND4 and COI were up-regulated ∼2-fold, suggesting that TbRGG2 contributes to destabilization of these RNAs. We also found that all three of the minimally edited RNAs examined were up-regulated 1.5–2.5-fold in TbRGG2-depleted cells. This modest stabilization was evident for both the pre-edited and edited versions of CYb, COII, and MURF2 RNAs, again pointing toward a role for TbRGG2 in RNA destabilization. For these edited RNAs, we cannot distinguish whether both preedited and edited RNAs are stabilized directly or whether a stabilization of the pre-edited RNA leads indirectly to an increase in the corresponding edited RNA. Interestingly, when we analyzed pan-edited RNAs, we observed a completely different pattern. All four pan-edited RNAs tested exhibited a very significant decrease in edited RNA and, in some cases, an increase in the corresponding pre-edited RNA (Fig. 9A). The RNA that was most dramatically affected by TbRGG2 depletion was A6, for which levels of edited RNA were decreased 10–20-fold. Edited RPS12 RNA also decreased almost 10-fold, and edited COIII and ND7 RNAs were decreased 7–8-fold. Pre-edited A6 and ND7 RNAs were increased almost 2-fold, as expected if their editing was decreased by the loss of TbRGG2. Levels of pre-edited COIII and RPS12 RNAs were nearly unaffected; however, it is common that pre-edited RNAs fail to accumulate, even when editing is inhibited by depletion of core editing components (1, 2, 5, 35). Together, these data are consistent with a model in which TbRGG2 is multifunctional, strongly facilitating the editing of pan-edited RNAs and modestly destabilizing minimally edited and never-edited RNAs in PF T. brucei.
We next analyzed mitochondrial RNAs from BF stage cells depleted for TbRGG2. Similar to what was observed in PF cells, never-edited ND4 RNA and minimally edited MURF2 RNAs were somewhat stabilized by TbRGG2 down-regulation (Fig. 9B). Pre-edited and edited MURF2 RNAs displayed increased levels at both 24 and 40 h following RNAi induction, although the increases were more dramatic at the 40 h time point than at the 24 h time point, implying an increase in stabilization of these RNAs as TbRGG2 levels decrease. We next analyzed several pan-edited RNAs in BF, and similar to what was observed in PF, the edited forms of all pan-edited RNAs were decreased, in most cases very dramatically. Levels of edited A6 and RPS12 RNAs were decreased 9–10-fold, and edited ND8 RNA was decreased 5-fold, whereas edited COIII RNA levels were least affected. In contrast to PF cells, however, the pre-edited forms of some of these RNAs accumulated significantly in TbRGG2-depleted cells. In particular, pre-edited ND8 and RPS12 RNA levels in induced BF cells reached 4–5-fold those in uninduced cells. Thus, our data demonstrate that TbRGG2 acts as an RNA editing accessory factor with a significant role in both PF and BF T. brucei and that it affects specifically pan-edited RNAs. This is the first description of an RNA editing accessory factor that functions in the BF life cycle stage of the organism.
DISCUSSION
In this report, we show that TbRGG2 is a mitochondrial RNA-binding protein that functions as an RNA editing accessory factor for pan-edited RNAs in both the PF and BF life cycle stages of T. brucei. TbRGG2 is strongly developmentally regulated, with protein levels in PF reaching 10 times those in BF. Nevertheless, depletion of TbRGG2 resulted in decreases in all four pan-edited RNAs tested in BF as well as in a slightly different set of four pan-edited RNAs in PF. In the majority of cases, decreases in edited RNA levels were very dramatic, often nearly 10-fold or more. TbRGG2 is essential for growth in both life cycle stages, presumably due to its very significant role in RNA editing. In contrast to the effect on pan-edited RNAs, TbRGG2 down-regulation led to a modest stabilization of never-edited and minimally edited RNAs, suggesting that the protein may also have a general function in RNA destabilization. TbRGG2 is the first reported RNA editing accessory factor that functions in the BF stage of the T. brucei life cycle. Previously studied accessory factors include RBP16 and the MRP1/2 complex, both of which are essential for editing of CYb RNA in PF T. brucei (10, 11). The role of RBP16 has not been studied in BF cells. BF cells in which both alleles of MRP1 were knocked out grew normally and displayed no editing defects, although some effects on RNA stabilization were observed (39). To date, the reported effects of RBP16 and MRP1/2 on RNA editing are primarily restricted to CYb RNA, which is only edited in PF. Thus, in addition to its unique role in BF, the impact of TbRGG2 on RNA editing is much broader than that of the previously described accessory factors, since it strongly facilitates editing of many RNAs in both life cycle stages.
TbRGG2 is a component of a 40 S RNA-dependent complex and a 20 S RNA-independent complex as well as being present as either a free protein or as a component of smaller complexes. These complexes probably mediate the multifaceted roles of TbRGG2 in mitochondrial gene expression. The association of TbRGG2 with 20 S complexes may represent a transient association with editosomes. Indeed, TbRGG2 (then termed TbRGGm) was first reported in T. brucei as a protein identified by mass spectrometry in editosomes isolated by immunoprecipitation (3), and we show here that a small fraction of the protein can be detected by Western blot in immunoprecipitated editosomes. TbRGG2 is clearly not a stable or core editosome component, however, since it is not present in editing complexes isolated biochemically or through TAP tagging (40). TbRGG2 was also recently reported to be a component of a mitochondrial 30–40 S complex of unknown function (15). This complex was termed the putative mitochondrial RNA-binding complex 1 (put-MRB complex 1) based on the presence of five putative RNA-binding proteins, including TbRGG2. The RNase sensitivity of put-MRB complex 1 was not investigated. It is possible that the RNase-sensitive TbRGG2-containing complex that we observe at ∼40 S by glycerol gradient sedimentation may be the put-MRB complex 1. The RNase sensitivity of the TbRGG2-containing ∼40 S complex is also reminiscent of the behavior of MRP1/2 in Leishmania tarentolae, which associate with high molecular weight complexes in an RNase-sensitive manner (41). Further studies are required to determine the roles of the various TbRGG2-containing multiprotein and ribonucleoprotein complexes in trypanosome mitochondrial gene expression.
The mechanism by which TbRGG2 impact is restricted to pan-edited RNAs during RNA editing is unknown. We envision two possible models. First, TbRGG2 may be specific for certain types of mRNAs, in this case pan-edited mRNAs. Specificity could be conferred by RNA binding determinants in TbRGG2 itself or through an associated RNA-binding protein or protein complex with affinity for distinct RNAs. Detailed RNA binding studies will reveal the affinity of the protein for specific mitochondrial RNAs. A second model, which we favor, is that TbRGG2 is a transiently associated editing factor required for the processivity of RNA editing. This model would explain the observed changes following TbRGG2 RNAi for all pan-edited mRNAs. If down-regulation of TbRGG2 results in its becoming a limiting factor for RNA editing, it is feasible that the editing of pan-edited mRNAs would be affected first, resulting in their decrease. Since RNAi does not result in the complete loss of the intended target mRNA, sufficient amounts of TbRGG2 protein most likely remain in the cell to permit editing of minimally edited mRNAs before the loss of TbRGG2 results in cell death. An effect of TbRGG2 on the processivity of the editing process could be manifested at several levels. This includes enhancing progression of the editosome from one editing site to the next, facilitating usage of sequential gRNAs, and/or coordinating access of the RNA substrates to different editosome subtypes, perhaps upon switching from insertion to deletion sites. It may be enlightening in this respect to determine precisely how far 5′-editing can proceed along a given RNA under TbRGG2 limiting conditions. The biochemical mechanism of TbRGG2 action is also not known. The presence of a basic RG-rich region suggests that the protein may possess RNA annealing activity (42–45), similar to that recently described for RBP16 (46). Given the complex RNA-RNA and RNA-protein interactions that must take place during editing, it would not be unexpected that there is a requirement for multiple proteins that can facilitate changes in RNA structure and interactions during the editing process.
Overall, the data presented here identify TbRGG2 as a new member of the class of kinetoplastid RNA editing accessory factors, those proteins that are not core editosome components but dramatically affect the extent of editing of specific RNAs or classes of RNAs. TbRGG2 is unique compared with known editing accessory factors in two ways. First, it is the only accessory factor shown to affect a broad range of RNAs, and second, it is the first to be described in the mammalian infective stage of the parasite. Future studies addressing the molecular mechanisms of TbRGG2 action and the basis for its preferential effects on pan-edited RNAs will provide insight into the complex process of editing and potentially into the mechanisms by which this process can be regulated.
Acknowledgments
We thank Jay Bangs, Vivian Bellofatto, Steve Hajduk, and Ken Stuart for providing antibodies. We are also grateful to Jason Carnes and Dave Feliciano for advice on quantitative RT-PCR.
This work was supported, in whole or in part, by National Institutes of Health Grant AI061580 (to L. K. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PF, procyclic form; BF, bloodstream form; CYb, apocytochrome b; COI, COII, and COIII, cytochrome oxidase subunit I, II, and III, respectively; ND4, ND7, and ND8, NADH dehydrogenase subunit 4, 7, and 8, respectively; A6, ATPase subunit 6; RPS12, ribosomal protein S12; MURF2, mitochondrial unknown reading frame 2; gRNA, guide RNA; RRM, RNA recognition motif; GST, glutathione S-transferase; RNAi, RNA interference; nt, nucleotide(s); RT, reverse transcription.
Personal communication, S. Madison-Antennuci.
J. C. Fisk, M. Pelletier, and L. K. Read, unpublished results.
References
- 1.Carnes, J., Trotter, J. R., Ernst, N. L., Steinberg, A., and Stuart, K. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trotter, J. R., Ernst, N. L., Carnes, J., Panicucci, B., and Stuart, K. (2005) Mol. Cell 20 403–412 [DOI] [PubMed] [Google Scholar]
- 3.Panigrahi, A. K., Allen, T. E., Stuart, K., Haynes, P. A., and Gygi, S. P. (2003) J. Am. Soc. Mass Spectrom. 14 728–735 [DOI] [PubMed] [Google Scholar]
- 4.Ernst, N. L., Panicucci, B., Igo, R. P., Jr., Panigrahi, A. K., Salavati, R., and Stuart, K. (2003) Mol. Cell 11 1525–1536 [DOI] [PubMed] [Google Scholar]
- 5.Wang, B., Ernst, N. L., Palazzo, S. S., Panigrahi, A. K., Salavati, R., and Stuart, K. (2003) Eukaryot. Cell 2 578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnaufer, A., Panigrahi, A. K., Panicucci, B., Igo, R. P., Jr., Wirtz, E., Salavati, R., and Stuart, K. (2001) Science 291 2159–2162 [DOI] [PubMed] [Google Scholar]
- 7.Huang, C. E., Cruz-Reyes, J., Zhelonkina, A. G., O'Hearn, S., Wirtz, E., and Sollner-Webb, B. (2001) EMBO J. 20 4694–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang, X., Rogers, K., Gao, G., Falick, A. M., Zhou, S., and Simpson, L. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 1017–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McManus, M. T., Shimamura, M., Grams, J., and Hajduk, S. L. (2001) RNA (N. Y.) 7 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelletier, M., and Read, L. K. (2003) RNA (N. Y.) 9 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vondruskova, E., van den Burg, J., Zikova, A., Ernst, N. L., Stuart, K., Benne, R., and Lukes, J. (2005) J. Biol. Chem. 280 2429–2438 [DOI] [PubMed] [Google Scholar]
- 12.Hayman, M. L., and Read, L. K. (1999) J. Biol. Chem. 274 12067–12074 [DOI] [PubMed] [Google Scholar]
- 13.Miller, M. M., Halbig, K., Cruz-Reyes, J., and Read, L. K. (2006) RNA (N. Y.) 12 1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouaissi, A., Vergnes, B., Borges, M., and Guilvard, E. (2000) Gene (Amst.) 253 271–280 [DOI] [PubMed] [Google Scholar]
- 15.Panigrahi, A. K., Zíková, A., Dalley, R. A., Acestor, N., Ogata, Y., Anupama, A., Myler, P. J., and Stuart, K. D. (2008) Mol. Cell Proteomics, 7 534–545 [DOI] [PubMed] [Google Scholar]
- 16.Vanhamme, L., Perez-Morga, D., Marchal, C., Speijer, D., Lambert, L., Geuskens, M., Alexandre, S., Ismaili, N., Goringer, U., Benne, R., and Pays, E. (1998) J. Biol. Chem. 273 21825–21833 [DOI] [PubMed] [Google Scholar]
- 17.Pelletier, M., Pasternack, D. A., and Read, L. K. (2005) Mol. Biochem. Parasitol. 144 206–217 [DOI] [PubMed] [Google Scholar]
- 18.Madison-Antenucci, S., and Hajduk, S. L. (2001) Mol. Cell 7 879–886 [DOI] [PubMed] [Google Scholar]
- 19.Madison-Antenucci, S., Sabatini, R. S., Pollard, V. W., and Hajduk, S. L. (1998) EMBO J. 17 6368–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riley, G. R., Corell, R. A., and Stuart, K. (1994) J. Biol. Chem. 269 6101–6108 [PubMed] [Google Scholar]
- 21.Koslowsky, D. J., Goringer, H. U., Morales, T. H., and Stuart, K. (1992) Nature 356 807–809 [DOI] [PubMed] [Google Scholar]
- 22.Goulah, C. C., Pelletier, M., and Read, L. K. (2006) RNA (N. Y.) 12 1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goulah, C. C., and Read, L. K. (2007) J. Biol. Chem. 282 7181–7190 [DOI] [PubMed] [Google Scholar]
- 24.Hajduk, S. L., Adler, B., Madison-Antenucci, S., McManus, M., and Sabatini, R. (1997) Nucleic Acids Symp. Ser. 36 15–18 [PubMed] [Google Scholar]
- 25.Kiledjian, M., and Dreyfuss, G. (1992) EMBO J. 11 2655–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghisolfi, L., Joseph, G., Amalric, F., and Erard, M. (1992) J. Biol. Chem. 267 2955–2959 [PubMed] [Google Scholar]
- 27.Yamaguchi, Y., Narita, T., Inukai, N., Wada, T., and Handa, H. (2001) J. Biochem. (Tokyo) 129 185–191 [DOI] [PubMed] [Google Scholar]
- 28.Ruiz, F. H., Silva, E., and Inestrosa, N. C. (2000) Biochem. Biophys. Res. Commun. 269 491–495 [DOI] [PubMed] [Google Scholar]
- 29.El-Shami, M., Pontier, D., Lahmy, S., Braun, L., Picart, C., Vega, D., Hakimi, M.-A., Jacobsen, S. E., Cooke, R., and Lagrange, T. (2007) Genes Dev. 21 2539–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shichijo, S., Azuma, K., Komatsu, N., Kawamoto, N., Takedatsu, H., Shomura, H., Sawamizu, H., Maeda, Y., Ito, M., and Itoh, K. (2003) Int. J. Mol. Med. 12 895–902 [PubMed] [Google Scholar]
- 31.Zeiner, G. M., Sturm, N. R., and Campbell, D. A. (2003) Eukaryot. Cell 2 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris, M. E., Moore, D. R., and Hajduk, S. L. (1990) J. Biol. Chem. 265 11368–11376 [PubMed] [Google Scholar]
- 33.Panigrahi, A. K., Schnaufer, A., Ernst, N. L., Wang, B., Carmean, N., Salavati, R., and Stuart, K. (2003) RNA (N. Y.) 9 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickstead, B., Ersfeld, K., and Gull, K. (2002) Mol. Biochem. Parasitol. 125 211–216 [DOI] [PubMed] [Google Scholar]
- 35.Salavati, R., Ernst, N. L., O'Rear, J., Gilliam, T., Tarun, S., Jr., and Stuart, K. (2006) RNA (N. Y.) 12 819–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang, C. E., O'Hearn, S. F., and Sollner-Webb, B. (2002) Mol. Cell. Biol. 22 3194–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rusche, L. N., Huang, C. E., Piller, K. J., Hemann, M., Wirtz, E., and Sollner-Webb, B. (2001) Mol. Cell. Biol. 21 979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carnes, J. T. J., Peltan, A., Fleck, M., and Stuart, K. (2008) Mol. Cell. Biol. 28 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert, L., Muller, U. F., Souza, A. E., and Goringer, H. U. (1999) Nucleic Acids Res. 27 1429–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panigrahi, A. K., Ernst, N. L., Domingo, G. J., Fleck, M., Salavati, R., and Stuart, K. D. (2006) RNA (N. Y.) 12 1038–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aphasizhev, R., Aphasizheva, I., Nelson, R. E., and Simpson, L. (2003) RNA (N. Y.) 9 62–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang, Q., and Jankowsky, E. (2005) Biochemistry 44 13591–13601 [DOI] [PubMed] [Google Scholar]
- 43.Mayer, O., Rajkowitsch, L., Lorenz, C., Konrat, R., and Schroeder, R. (2007) Nucleic Acids Res. 35 1257–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hitti, E., Neunteufl, A., and Jantsch, M. F. (1998) Nucleic Acids Res. 26 4382–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buvoli, M., Cobianchi, F., and Riva, S. (1992) Nucleic Acids Res. 20 5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ammerman, M. A., Fisk, J. C., and Read, L. K. (2008) RNA (N. Y.) 14 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]