

Abstract
UDP-GalNAc:polypeptide α-N-Acetylgalactosaminyltransferases (ppGalNAcTs), a family (EC 2.4.1.41) of enzymes that initiate mucin-type O-glycosylation, are structurally composed of a catalytic domain and a lectin domain. Previous studies have suggested that the lectin domain modulates the glycosylation of glycopeptide substrates and may underlie the strict glycopeptide specificity of some isoforms (ppGalNAcT-7 and -10). Using a set of synthetic peptides and glycopeptides based upon the sequence of the mucin, MUC5AC, we have examined the activity and glycosylation site preference of lectin domain deletion and exchange constructs of the peptide/glycopeptide transferase ppGalNAcT-2 (hT2) and the glycopeptide transferase ppGalNAcT-10 (hT10). We demonstrate that the lectin domain of hT2 directs glycosylation site selection for glycopeptide substrates. Pre-steady-state kinetic measurements show that this effect is attributable to two mechanisms, either lectin domain-aided substrate binding or lectin domain-aided product release following glycosylation. We find that glycosylation of peptide substrates by hT10 requires binding of existing GalNAcs on the substrate to either its catalytic or lectin domain, thereby resulting in its apparent strict glycopeptide specificity. These results highlight the existence of two modes of site selection used by these ppGalNAcTs: local sequence recognition by the catalytic domain and the concerted recognition of distal sites of prior glycosylation together with local sequence binding mediated, respectively, by the lectin and catalytic domains. The latter mode may facilitate the glycosylation of serine or threonine residues, which occur in sequence contexts that would not be efficiently glycosylated by the catalytic domain alone. Local sequence recognition by the catalytic domain differs between hT2 and hT10 in that hT10 requires a pre-existing GalNAc residue while hT2 does not.
The first committed step in mucin-type O-glycan biosynthesis is the transfer of GalNAc from UDP-GalNAc to Ser/Thr residues of proteins catalyzed by a large family of evolutionarily conserved enzymes, the ppGalNAcTs2 (1). The collective substrate specificity of the individual family members defines the mucin-type glycome, i.e. the Ser/Thr residues on a protein that acquire mucin-type O-glycans. Despite observations of biases in amino acid composition flanking known sites of O-glycosylation (2–5), no rigid consensus sequence for the prediction of sites has emerged. Consequently, current predictive methods are limited in their accuracy (6–8). In vitro studies using peptide acceptors have demonstrated that ppGalNAcT isoforms have distinct but overlapping substrate specificities (2, 9–12). Although a majority of these isoforms can glycosylate both naked peptides and glycopeptides (9–11), a subset, notably isoforms ppGalNAcT-7 (13) and ppGalNAcT-10 (14), has an apparent strict requirement for prior glycosylation of their substrates to add additional GalNAc.
ppGalNAcTs are type II transmembrane proteins with a short cytoplasmic tail, a single transmembrane region, a stem region of variable length followed by the catalytic and lectin domains. The crystal structures of soluble, transmembrane region-deleted forms of three isoforms, ppGalNAcT-1, -2, and -10 (15–17), have been determined. These structures show that the enzyme folds into two distinct domains, the catalytic domain containing a glycosyltransferase A-type fold and a lectin domain with a β-trefoil fold. However, the relative orientation of the two domains in these structures varies significantly. The interaction surface area between the two domains is relatively large in ppGalNAcT-1 (∼645 Å2 (15)) and -10 (17). This area is reduced to ∼325 Å2 in the hT2/UDP binary complex and almost absent in the hT2-UDP-acceptor peptide ternary complex (16). Studies of ppGalNAcT-1, -2, and -4 have demonstrated that mutations within the lectin domain specifically compromise transfer of GalNAc to partially glycosylated peptides (16, 18–20) and that glycopeptide activity is also selectively inhibited by high concentrations of free GalNAc (19, 20). Recently, Wandall et al. (19) demonstrated that the lectin domains of ppGalNAcT-2 and -4 specifically bind GalNAc. They also showed that the GalNAc density of peptide substrates with multiple acceptor sites after terminal glycosylation by hT2 and hT4 was reduced by mutational inactivation of the lectin domain binding. The lectin domain has been suggested to improve the kinetic properties of the enzymes toward partially glycosylated substrates, perhaps through simultaneous interactions of both domains with the substrates. These observations have led to the speculation that the apparent restriction of activity of isoforms ppGalNAcT-7 and -10 to glycopeptides is a property of the lectin domain of these enzymes (19).
We have directly examined the role of the lectin domain in glycosylation site selection by hT2 (a peptide/glycopeptide transferase) and compared it to that of hT10 (a glycopeptide transferase). Comparison of the preferred glycosylation sites of hT2 with and without its lectin domain shows that the lectin domain determines the site of glycosylation relative to the site of extant glycosylation. Binding and pre-steady-state kinetic measurements of hT2 and its lectin domain-deleted form support the view that the lectin domain assists with both substrate binding and product release of glycopeptide substrates. For hT10, the data demonstrate that the catalytic domain directs glycosylation to residues immediately N-terminal to extant GalNAcs while the lectin domain directs glycosylation to sites distal from extant GalNAcs. Thus, the strict glycopeptide specificity of hT10 arises from a requirement for recognition of pre-existing GalNAc(s) on acceptor substrates either by its catalytic domain or its lectin domain.
EXPERIMENTAL PROCEDURES
Construction and Purification of Lectin Domain Deletion and Domain Swap Constructs—The various constructs used in this study are shown in Fig. 1a. All constructs were cloned into the Mlu1 and Age1 sites of the Pichia expression vector, pKN55-N6His-TEV and expressed with a Tobacco etch virus (TEV) protease-cleavable His6 fusion tag. Residues 75–571 of human ppGalNAc-T2 encoding a portion of the stem region and the entire catalytic and lectin domains (hT2) and catalytic domain of hT2 (residues 75–440, hT2CD) were expressed and purified as described previously (16). Residues 71–603 of human ppGal-NAc-T10 encoding a portion of the stem region and the entire catalytic and lectin domains (hT10) were PCR-amplified from RNA isolated from HEK cells (ATCC) using a NucleoSpin (BD Biosciences) kit. PCR amplification was done using a Superscript III one-step reverse transcription-PCR kit (Stratagene) and the primers 5′-ACCACGCGTTGAAAGTACGGTGGCCAGACTTT and 5′-ACCACCGGTCTACTGCTGCAGGTTGAGCGTGAA. The PCR product was cloned between the MluI/AgeI sites of pKN55-N6His-TEV. The catalytic domain of hT10 (residues 71–455, hT10CD) was PCR-amplified using the primers 5′-ACGACGCGTTGAAGGACTGGCATGACAAGGAGGCCATC and 5′-ACCACCGGTCTAGGGTGGGTAGAATTTGGGCAG and cloned between the MluI/AgeI sites of pKN55-N6His-TEV. For creation of the domain swap construct composed of the hT2 catalytic domain and the hT10 lectin domain, the hT2 catalytic domain (residues 75–440) was amplified using the primers ACCACGCGTTGAAAGTACGGTGGCCAGACTTT and TGCGGCCGGGGGCTCCACGGGTGGAACCCTTAACTCTGG (the primer encoding residues 435–440 of hT2 and residues 455–460 of the hT10 lectin domain). The lectin domain of hT10 (residues 455–603) was amplified using the primers CCAGAGTTAAGGGTTCCACCCGTGGAGCCCCCGGCCGCA (the primer encoding residues 435–440 of hT2 and residues 455–460 of hT10) and ACCACCGGTTCAGTTCCTATGAATTTTTCCAAGACTGT. These fragments were purified and mixed, and the full-length domain swap gene encoding residues 71–440 of hT2 fused with residues 455–603 of hT10 was generated by overlap extension PCR. The overlap PCR extension product encoding the domain swap gene was then amplified with the primers ACCACGCGTTGAAAGTACGGTGGCCAGACTTT and ACCACCGGTTCAGTTCCTATTGAATTTTTCCAAGACTGT and cloned into the Mlu1 and Age1 sites of the plasmid pKN55-N6His-TEV. The domain swap construct with the hT10 catalytic domain and the hT2 lectin domain was generated in a similar way. The primers used to amplify the hT10 catalytic domain were ACGACGCGTTGAAGGACTGGCATGACAAGGAGGCCATC and AGCTATATCCTGATGGTCCACGGGTGGGTAGAATTTGGG, and the primers used to amplify the hT2 lectin domain were CCCAAATTCTACCCACCCGTGGACCATCAGGATATAGCT and ACCACCGGTCTACTGCTGCAGGTTGAGCGTGAA. The plasmids were linearized and electroporated into Pichia pastoris strain SMD1168 to create stable transformants, and proteins were expressed in P. pastoris and purified as described (16).
FIGURE 1.

a, schematic representation of ppGalNAcT-2 and -10 constructs used in this study. Residues 1–74 of human ppGalNAcT-2 and residues 1–70 of human ppGalNAcT10 representing the transmembrane region and part of the stem region were deleted in the constructs. The catalytic domain is shown as clear bars and the lectin domain as hashed bars. b, peptide/glycopeptide substrates used. The sites of attachment of the extant GalNAc are highlighted.
Peptide Synthesis—The MUC5AC peptide used for this study was synthesized by the Facility for Biotechnology Resources, Center for Biologics Evaluation and Research at the National Institutes of Health, and glycopeptides MUC5AC-3, -13, and -3,13 were synthesized by Anaspec, San Jose, CA. The MUC5AC-9 glycopeptide was synthesized using Fmoc-Pro-NovaSyn® TGT (Novabiochem) resin, initial loading 0.20 mmol/g, to minimize problems apparently arising from dike-topiperizine formation, and with the glycopeptide synthesis following a protocol similar to earlier work.(21). All the amino acid derivatives used were commercially available α-amino Fmoc-protected acids or pentafluorophenyl esters (in the case of Thr) with t-butyl protection on Ser and Thr side chains and Fmoc-Thr(Ac3-α-d-GalNAc)-OH for the glycosylated residue. The TGT resin (125 mg) was placed in a glass vessel (8.5 ml) containing a porous polypropylene frit for manual synthesis. Fmoc removal was achieved with piperidine-dimethylformamide (DMF) (1:4) for 20 min, followed by 2-h couplings of corresponding amino acid derivatives (4 eq), which were mediated by 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (4 eq)/N-hydroxybenzotriazole (4 eq)/diisopropylethylamine (5 eq) in DMF (2 ml) at 25 °C. Double couplings were carried out on Fmoc-Thr(Ac3-α-d-Gal-NAc)-OH (2 eq, overnight and 0.5 eq, 2 h, respectively) as well as for the following two amino acid derivatives Fmoc-Pro-OH and Fmoc-Val-OH. Washings between reactions were carried out with DMF and CH2Cl2, then DMF again, and no intermediate capping steps were done. After chain assembly was completed and the N-terminal Fmoc group removed, the resin was washed with DMF and CH2Cl2 and dried in a desiccator overnight. Half of the glycopeptide resin was treated with trifluoroacetic acid/H2O (95/5) for 2 h to remove amino acid protecting groups. Separation and purification were carried out on semi-preparative reversed-phase high-performance liquid chromatography, followed by treatment with NaOMe/MeOH (∼pH 9 as detected by pH paper) for 3 h to remove acetyl protecting groups on the GalNAc and lyophilization to give the final fully deprotected glycopeptide, confirmed by matrix-assisted laser desorption ionization time-of-flight (observed: [M+H]+, 1704.4; [M+2H]2+, 852.7; calculated: 1703.8). The yield was 22% (14.7 mg) based on the initial loading of the resin.
Enzyme Assays and Determination of Kinetic Constants—(UDP-[1-14C]GalNAc was from PerkinElmer Life Sciences with a specific activity of 1.5 μCi/mmol). Reactions (25 μl) contained 10 mm MnCl2, 40 mm sodium cacodylate, 40 mm β-mercaptoethanol, and 0.1% Triton X-100 (pH 6.6) as described (11). The reactions were initiated by adding 0.05–0.2 pmol of enzyme, and incubation times were such that not more than 10% of the limiting substrate was converted to product. MUC5AC-3,13 was varied from 46.8 μm to 3 mm, MUC5AC and MUC5AC-3 were varied from 3 μm to 200 μm, and MUC5AC-13 was varied between 7.8 μm and 1.0 mm with UDP-GalNAc at 115 μm (0.18 μCi/mmol) for reactions with hT2CD-derived constructs. For reactions with hT10CD-derived constructs, MUC5AC-3 was varied between 18.5 μm and 300 μm and, MUC5AC-13 and MUC5AC-3,13 were varied between 15 μm and 1.0 mm. Reactions with the constructs with the hT2 catalytic domain were stopped by addition of 75 μl of 30 mm EDTA and worked up as described (11). GalNAc transfer reactions using all constructs containing the hT10 catalytic domain were accompanied by a high rate of UDP-GalNAc hydrolysis in addition to transfer. This hydrolysis precluded the use of anion exchange to eliminate unincorporated GalNAc so these reactions were terminated by the addition of 75 μl of 0.05% trifluoroacetic acid and glycopeptide products were purified using Sep-Pak reversedphase columns as described (22). Radioactive GalNAc incorporation into substrate peptide, purified by either of the two methods, was determined by liquid scintillation. Pseudo first-order kinetic constants were determined by non-linear regression fitting to the Michaelis-Menten equation using the program GraphPad, and the initial velocities were determined from duplicate measurements.
Pre-steady-state Kinetics—Reactions containing 500 μm peptide and 114 μm UDP-[1-14C]GalNAc (0.18 μCi/mmol) were initiated by the rapid addition of enzyme to a final concentration of 0.7–6 μm and terminated by the addition of 80 μl of 30 mm EDTA at 0-, 2-, 4-, 6-, 8-, 30-, 60-, and 120-s intervals. Glycopeptide product was quantitated as described (11). Data were evaluated for fit to both a linear and biphasic equation, and the best fit model in each case was used to determine kinetic parameters. Biphasic curves were fit by non-linear regression to the equation,
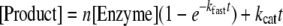 |
(Eq. 1) |
where, kfast and kcat represent the initial burst phase and the subsequent steady-state rate constants, respectively, and n represents the burst of glycopeptide formation per mole of enzyme present in the reaction (23).
Fluorescence Measurements—Spectra of enzyme tryptophan were recorded on a HORIBA Jobin Yvon FluoroLog-3 spectrofluorometer. Enzymes were diluted to 0.5 μm final concentration into buffer containing 10 mm MnCl2, 40 mm sodium cacodylate, 100 μm UDP, and various concentrations (500 nm to 300 μm) of peptide or glycopeptide and allowed to equilibrate for 5 min at room temperature. Emission spectra were recorded at 25 °C using a 3-mm/3-mm path length cuvette with excitation at 280 nm. Excitation and emission slit widths were set at 5 nm. Peptide concentration-dependent increase in fluorescence intensity at the emission maxima (330 nm for hT2 and 336 nm for hT2CD) at various concentrations of peptide (Fc) compared with the intensity in the absence of peptide (Fo) was used to monitor peptide binding. Dissociation constants were derived by fitting (Fc – Fo)/Fo versus [peptide] plots to a single site binding model by non-linear regression on GraphPad Prism (24).
SELDI-TOF-MS—Mass spectra were collected on a SELDI-TOF-MS PBS-II instrument using H4-RP assay chips and calibrated using the All-in-One Peptide Standard (Ciphergen). Spots were pre-treated with 5% acetonitrile, and 2 μl from reaction mixtures was spotted, allowed to dry, and washed three times with 5% acetonitrile. 1 μl of a 10 mg/ml 2,5-dihydroxybenzoic acid solution in 50% acetonitrile, 0.5% trifluoroacetic acid was applied to the spots, allowed to dry, and then used for data collection.
Edman Sequencing—Reactions for identification of glycosylation sites contained 1 mm each of the peptide and UDP-Gal-NAc and, 4 milliunits/ml of the enzyme where 1 unit of activity is the amount of enzyme the catalyzes the formation of 1 μmol of GalNAc transfer in 1 min for each enzyme substrate pair. MUC5AC-9 peptide glycosylations with hT10 were carried out at a peptide concentration of 500 μm. Reactions were allowed to proceed overnight at 37 °C. GalNAc glycosylation site analysis was performed on a Procise 494 Edman amino acid sequencer as described (25, 26). Prior to sequencing, peptides were chromatographed on Sephadex G-10 (run in 50 mm acetic acid, pH 4.5, with NH4OH) to remove buffer and contaminants (2). Proline sequence cycles were employed at each of the Pro positions in the MUC5AC peptide to reduce sequence lag. Integrated peaks for the glycosylated and non-glycosylated Ser and Thr residues were further corrected for lag and preview as described (25, 26). After such processing the Edman sequencing-derived percent glycosylation at all initially glycosylated sites of the glycosylated MUC5AC substrates was >80%.
RESULTS
The Lectin Domain of hT2 Directs the Site of GalNAc Addition to Glycopeptide Substrates—We showed previously that deleting the hT2 lectin domain compromised catalytic activity toward glycopeptide but not peptide substrates (15). To further investigate ppGalNAcT lectin domain function, we mapped the sites of GalNAc addition catalyzed by hT2 with or without its lectin domain to a set of glycopeptides, derived from the sequences in MUC5AC mucin (Fig. 1b). As a reference, we also mapped the sites used by the same hT2 constructs on the non-glycosylated MUC5AC peptide.
Edman sequencing of glycopeptide products obtained following the reaction of the MUC5AC peptide with hT2 and hT2CD showed that the preferred site of glycosylation on the MUC5AC peptide by these two forms of hT2 is Thr-9 (Fig. 2, a and e). Thr-3, -10, and -13 are glycosylated to a lesser extent. Substitution of the hT2 lectin domain with that from hT10 did not change the preference for Thr-9 (Fig. 2i). Thus, site selection on the MUC5AC naked peptide by hT2 is driven by local sequence recognition by the catalytic domain alone.
FIGURE 2.

Site of glycosylation of MUC5AC-derived (glyco)peptides by hT2CD-derived constructs determined by Edman sequencing. Data are presented normalized to the preferred site of glycosylation. Arrows indicate extant glycosylation. hT2, hT2CD, and hT2CD-hT10LD refer to full-length hT2, the catalytic domain of hT2, and the fusion of the catalytic domain of hT2 with the lectin domain of hT10, respectively.
In contrast, the lectin domain actively dictates site selection on the MUC5AC glycopeptides. As with the naked MUC5AC peptide, Thr-9 is the dominant site of GalNAc addition to the MUC5AC glycopeptides by hT2 in the absence of its lectin domain (Fig. 2, f–h). In the presence of the lectin domain, the site of glycosylation varied depending upon the location of the pre-existing GalNAc. For the MUC5AC-3 glycopeptide the residue most heavily glycosylated by hT2 was Thr-13 (Fig. 2b). Thus, the GalNAc on Thr-3 directed subsequent GalNAc addition to a residue 10 amino acids C-terminal to the extant glycosylation. The hT2 lectin domain targeted glycosylation to Thr-3, 10 residues N-terminal to the extant GalNAc (Fig. 2c), in the MUC5AC-13 glycopeptide. The most frequently glycosylated residue in the MUC5AC-3,13 diglycopeptide was Ser-5 (Fig. 2d). Thr-9 was not the preferred site of glycosylation by full-length hT2 for any of the MUC5AC glycopeptides, despite its availability, and in contrast to what was observed when the MUC5AC peptide was used as a substrate. The presence of the hT10 lectin domain was able to partially restore glycosylation to Thr-3 on the MUC5AC-13 glycopeptide (Fig. 2k) but not to Thr-13 and Ser-5 in the case of the MUC5AC-3 and MUC5AC-3,13 glycopeptides, respectively (Fig. 2, j and i).
The Catalytic and Lectin Domains of hT10 Direct Glycosylation Site Selection—We next examined the roles of the catalytic and lectin domains in determining the sites of glycosylation for hT10, shown previously to have a strict specificity for glycopeptide substrates (12, 13). It has been suggested that the glycopeptide specificity of hT10 is conferred by its lectin domain (18), and thus, deletion of the lectin domain would be predicted to abolish enzyme activity. Mass spectrometric analysis of the products obtained after overnight glycosylation by hT10 showed the transfer of a single GalNAc to MUC5AC-3 and MUC5AC-13 peptides (data not shown). With the MUC5AC-3,13 diglycopeptide, the predominant species formed also contained a single additional GalNAc, although a small amount of material with two GalNAcs added was also observed. The deletion of the hT10 lectin domain or exchange with the hT2 lectin domain did not alter the mass spectrometry profile compared with full-length hT10 (data not shown) indicating that the lectin domain is not required for catalysis by hT10. This is in agreement with the observations made for human ppGalNAcT1 and -T4 (20, 27).
Edman sequencing of the reaction products catalyzed by hT10 with and without its lectin domain using the MUC5AC glycopeptide substrates showed that the products were virtually identical (compare Fig. 3, a–c and d–f). In each instance the preferred site of glycosylation was immediately N-terminal to the GalNAc already present on the glycopeptide as shown previously in studies of full-length hT10. Thus, it is the hT10 catalytic domain that directs the site of glycosylation on these substrates. Exchange of the hT10 lectin domain with that from hT2 did not alter the site selection compared with the full-length hT10 (Fig. 3, compare a and g, b and h, and c and i).
FIGURE 3.

Site of glycosylation of MUC5AC derived glycopeptides (horizontal) by hT10CD-derived constructs (vertical) determined by Edman sequencing. Data are presented normalized to the preferred site of glycosylation. Arrows indicate extant glycosylation.
It is not surprising that the catalytic domain is involved in site selection on the above glycopeptides given that the site of glycosylation is adjacent to the existing GalNAc. We therefore examined the activity of the hT10CD-derived constructs on the MUC5AC-9 glycopeptide, because a previous study had suggested that mouse T10 glycosylates this glycopeptide on Thr-3, five residues N-terminal of the existing GalNAc(28). Fig. 4 shows the mass spectrometry profiles of the products obtained by glycosylation of the MUC5AC-9 glycopeptide by various hT10 catalytic domain-derived constructs. In this case, deletion of the lectin domain resulted in a loss of activity that could not be restored by the hT2 lectin domain. Edman sequencing of the product obtained on glycosylation of MUC5AC-9 by hT10 showed the preferred site of glycosylation to be Thr-2 and not Thr-3 as previously reported for mouse T10 (28). Thus, as in the case of hT2, the hT10 lectin domain can direct glycosylation to sites distal from existing GalNAc residues. hT10 is unable to recognize naked peptides and thus differs from hT2 in that it must recognize existing GalNAc via either its catalytic domain or lectin domain.
FIGURE 4.

a, mass spectrometry (SELDI) profiles for the glycosylation of MUC5AC-9 by hT10CD-derived constructs. The three peaks (0, 1, and 2) within the regions marked as the mono- and diglycosylated peptides represent the naked, mono-, and di-sodiated species. hT10 with its native lectin domain alone is able to transfer a GalNAc to the MUC5AC-9 peptide. Deletion or substitution of its lectin domain leads to loss of this activity.
Table 1 summarizes the steady-state kinetic constants derived from initial-rate glycosylation assays by the hT10 constructs. The kinetic parameters for the MUC5AC-3/13 glycopeptides for the different constructs are comparable. This supports the above data showing that site selection on these two substrates is determined by the catalytic domain alone. Compared with MUC5AC-3 glycopeptide, the rate of glycosylation of the MUC5AC-9 glycopeptide by hT10 is significantly slower with a corresponding higher Km even though the site of GalNAc addition is the same. This suggests that, although the binding of the GalNAc at Thr-9 to the lectin domain is able to compensate for the absence of interactions of GalNAc with the catalytic domain, this lectin domain mediated binding weaker than that to the catalytic domain.
TABLE 1.
Kinetic parameters for glycosylation by hT10 catalytic domain-derived constructs
|
Peptide
|
Site
|
hT10
|
hT10CD
|
hT10CD-hT2LD
|
|||
|---|---|---|---|---|---|---|---|
| kcat | Km | kcat | Km | kcat | Km | ||
| min-1 | μm | min-1 | μm | min-1 | μm | ||
| MUC5AC-3 | Thr-2 | 19.8 ± 0.6 | 96 ± 6.8 | 14.4 ± 1.2 | 50 ± 6.2 | 9.0 ± 0.6 | 107 ± 13.4 |
| MUC5AC-13 | Thr-12 | 24.0 ± 1.2 | 530 ± 49 | 18.6 ± 1.2 | 770 ± 73 | 17.4 ± 1.2 | 640 ± 60 |
| MUC5AC-3,13 | Thr-2, -12 | 84.0 ± 7.8 | 440 ± 35 | 45.6 ± 7.8 | 1000 ± 150 | 31.2 ± 1.8 | 570 ± 58 |
| MUC5AC-9 | Thr-2 | 7.6 ± 0.3a | 2400 ± 150 | NDb | NDb | ||
Activity at 1 mm peptide concentration.
ND, no activity detected.
The Binding of MUC5AC-13 but Not MUC5AC-3 to ppGalNAcT-2 Is Lectin-mediated—The data presented above in Fig. 2 establish a role for the hT2 lectin domain in determining the site of glycosylation of peptide sequences with multiple acceptor sites. In particular, the lectin domain directs glycosylation 10 residues N- or C-terminal to an extant GalNAc for the MUC5AC-13 or MUC5AC-3 glycopeptides, respectively (Fig. 2, b and c). One explanation is that the lectin domain binds to the existing GalNAc and positions the acceptor Thr in the active site of the enzyme. This model predicts that removal of the lectin domain should reduce the binding of the MUC5AC-3 and -13 glycopeptides to hT2. We therefore measured the binding of these two glycopeptides and the MUC5AC peptide to hT2 with and without its lectin domain. Dissociation constants were measured by exploiting the peptide-concentration-dependent increase in intrinsic tryptophan fluorescence of both hT2 and hT2CD. Fluorescence spectra were recorded in the presence of Mn2+ and UDP to closely mimic peptide binding in the presence of UDP-GalNAc without actual catalysis occurring during the study. Crystal structures of hT2 in the presence of an acceptor peptide and UDP show that the acceptor Thr hydroxyl is ideally placed to be the GalNAc acceptor and hydrogen bonds with β-phosphate oxygen of UDP (16). However, using short acceptor peptides, the catalytic mechanism of mouse ppGalNAcT-1 has been shown to be random sequential (29). Dissociation constants measured under these conditions, albeit of a non-productive complex, are likely to closely reflect peptide affinities during catalysis. The fluorescence spectra of the lectin domain alone were unaffected by the peptides, suggesting that the change in emission intensity was a result of binding to the catalytic domain. The dissociation constants presumably reflect the binding of the peptide in an orientation that positions the preferred site of glycosylation within the active site.
The binding of the MUC5AC-13 glycopeptide is reduced 4.9-fold in the absence of the lectin domain suggesting that lectin-assisted substrate binding contributes to site selection in this instance (Fig. 5, 19 μm for hT2 versus 93 μm for hT2CD). Surprisingly, the affinities of both hT2 and hT2CD for the MUC5AC-3 glycopeptide are comparable (11 μm versus 10 μm, respectively) suggesting that the lectin domain does not contribute to the binding of this glycopeptide. These data suggest that the lectin domain aids binding when the potential site of glycosylation is N-terminal to the site of extant glycosylation, but when the site of glycosylation is C-terminal to the existing glycosylation the lectin domain influences site selection by a means not directly related to GalNAc binding.
FIGURE 5.

Determination of dissociation constants for peptides using intrinsic tryptophan fluorescence enhancement. The plots of the fractional change in intensities at emission maxima at different peptide concentrations (Fc – Fo/Fo) as a function of peptide concentration were fit to a single site binding model. Fc and Fo represent the intensity at emission maxima at different peptide concentrations and in the absence of peptide, respectively. Table b shows the dissociation constants derived from the fits in a for hT2 and hT2CD.
If the hT2 lectin domain does not contribute to the binding of the MUC5AC-3 glycopeptide, how is it able to direct glycosylation to Thr-13 versus, for example, Thr-9? One explanation is that the turnover of the enzyme is greater when Thr-13 of MUC5AC-3 is glycosylated compared with when Thr-9 is glycosylated. We therefore measured the steady-state kinetic parameters of hT2 with and without its lectin domain for the MUC5AC peptide and glycopeptides. The parameters derived from initial rate determinations at room temperature are presented in Table 2. In agreement with the binding constants, the Km for MUC5AC-3 is not altered on deletion of the lectin domain. Rather, site selection is driven by a 3.5-fold increase in turnover when Thr-13 of MUC5AC-3 is glycosylated compared with when Thr-9 is glycosylated (Table 2). For the MUC5AC-13 glycopeptide, the increase in Kd for hT2 compared with hT2CD is also reflected in an increased Km and is the decisive factor for glycosylation site selection.
TABLE 2.
Kinetic parameters for glycosylation by hT2 catalytic domain-derived constructs
Reactions were carried out at 25 °C.
|
Peptide
|
Site
|
hT2CD
|
Site
|
hT2CD
|
||
|---|---|---|---|---|---|---|
| kcat | Km | kcat | Km | |||
| min-1 | μm | min-1 | μm | |||
| MUC5AC | Thr-9 | 3.1 ± 0.1 | 20 ± 2.9 | Thr-9 | 3.0 ± 0.2 | 18 ± 1.1 |
| MUC5AC-3 | Thr-13 | 13.1 ± 0.4 | 33 ± 3.2 | Thr-9 | 3.5 ± 0.1 | 36 ± 3.6 |
| MUC5AC-13 | Thr-13 | 6.2 ± 0.3 | 18 ± 1.0 | Thr-9 | 4.0 ± 0.3 | 170 ± 3.1 |
Pre-steady-state Kinetics: The Lectin Domain Can Aid Glycopeptide Product Release—The kinetic data presented in Table 2 are steady-state values in which kcat reflects the rate-limiting step for sugar transfer but the identity of the rate-determining step is not identified by these measurements. We exploited the fact that the steady-state rates of hT2 catalysis are on the order of <1 reaction per second at room temperature (Table 2) and studied pre-steady-state reaction kinetics on the second time scale to identify the rate-limiting step of the reaction. Comparison of these rates for the lectin domain deletion construct and the full-length enzyme would identify the specific step in which the lectin domain contributes during the catalytic cycle.
Representative plots of pre-steady-state measurements carried out in the presence of various concentrations of the enzyme with the peptide substrate MUC5AC and the monoglycosylated peptides, MUC5AC-3, are shown in Fig. 6. Data were analyzed as described under “Experimental Procedures.” The biphasic time courses (Fig. 6a) indicated a relatively fast formation of the enzyme-product (glycopeptide) complex followed by a slower rate-limiting step, most likely product release. The linear time courses (Fig. 6b) suggest that the rate-limiting step is either catalysis or the preceding substrate binding step. Values of n for the various fits were between 0.75 and 0.8 indicating the formation of ∼1 molecule of product per enzyme molecule during the burst phase consistent with a single turnover event. The rate constants obtained from the linear region of both categories were comparable to the kcat values from steady-state measurements at room temperature listed in Table 2.
FIGURE 6.

Representative pre-steady state product formation plots for hT2 with MUC5AC peptide at the indicated concentrations of enzyme (biphasic) (a) and hT2 with MUC5AC-3 (linear) (b). Lines in a represent fits to a single exponential equation with a linear component as described under “Experimental Procedures.”
Time courses for glycosylation of MUC5AC (Fig. 6a) and MUC5AC-13 (data not shown) by hT2 were biphasic, whereas those for transfer to the MUC5AC-3 glycopeptide were linear (Fig. 6b). For hT2CD, burst phases were observed for transfer to the MUC5AC peptide and to the MUC5AC-3 glycopeptide, whereas a linear slope was observed for transfer to the MUC5AC-13 peptide (data not shown) even though each of these events represent transfer to the same site (Thr-9) on the substrates.
The steady-state and, where applicable, pre-steady-state rates for the glycosylation of the (glyco)peptides by the full-length and lectin domain deletions are summarized in Table 3. Also indicated are the Km values and the dissociation constants determined by fluorescence. The pre-steady-state behavior of hT2 and hT2CD with the MUC5AC peptide are similar. Catalysis is fast with a subsequent rate-limiting product release step that is not influenced by the lectin domain. The rate-limiting product release step is likely the rate of glycosylated peptide release rather than UDP release, because the latter would be similar across all peptides studied. This agrees with the fact that both these constructs transfer to Thr-9 of the peptide and that the steady-state kinetic parameters are comparable (Fig. 7, a and b).
TABLE 3.
Summary of steady-state and pre-steady-state kinetic constants for hT2 and hT2CD
All measurements were carried out at 25 °C.
| Enzyme construct | Site | Km | Kd | kfast (burst phase)a | kcat (steady state)a | ||
|---|---|---|---|---|---|---|---|
| μm | min-1 | ||||||
| MUC5AC | |||||||
| hT2 | Thr-9 | 20 ± 2.9 | ∼2 | 7.4 ± 0.5 | 2.4 ± 0.1 | ||
| hT2CD | Thr-9 | 18 ± 1.1 | ∼1 | 11.6 ± 1.8 | 3.2 ± 0.4 | ||
| MUC5AC-3 | |||||||
| hT2 | Thr-13 | 33 ± 3.2 | 11 ± 1.0 | Not observed | 13.2 ± 1.8 | ||
| hT2CD | Thr-9 | 36 ± 3.6 | 10 ± 1.1 | 8.2 ± 1.3 | 2.4 ± 0.1 | ||
| MUC5AC-13 | |||||||
| hT2 | Thr-3 | 18 ± 1.0 | 19 ± 3.1 | 10.2 ± 1.4 | 6.5 ± 0.3 | ||
| hT2CD | Thr-9 | 170 ± 3.1 | 93 ± 8.5 | Not observed | 4.5 ± 0.4 | ||
Derived from pre-steady-state experiments.
FIGURE 7.

Suggested mechanism for lectin domain directed site selection in hT2. Binding of the MUC5AC peptide is not LD dependent (a and b). Site selection for the MUC5AC-3 glycopeptide is largely a result of the faster release of the MUC5AC-3,13 product compared with the MUC5AC-3,9 glycopeptide product (c and d). The lectin domain directly binds the extant GalNAc on MUC5AC-13 directing site selection (e and f). Sites of pre-existing GalNAc are represented as open circles and sites of GalNAc addition by filled circles.
The steady-state rate for the glycosylation at Thr-13 on MUC5AC-3 by hT2 reflects the actual rate of catalysis. Product release is no longer rate-limiting. Even though the affinities for Thr-9 (hT2CD Kd = 10 μm) and Thr-13 (hT2 Kd = 11 μm, Fig. 5) are comparable, Thr-13 is the preferred site, because the MUC5AC-3,13 glycopeptide product is released faster than the MUC5AC-3,9 glycopeptide (Fig. 7d). In the absence of the lectin domain, product release once again becomes rate-limiting, and catalysis occurs preferentially at Thr-9 (Fig. 7c). These results suggest that the lectin domain plays a role in selectively enhancing the rate of release of the MUC5AC-3,13 product. This allows faster cycling of the enzyme when the product is MUC5AC-3,13 rather than MUC5AC-3,9, increasing the ratio of the former in the product formed (Fig. 7d). However, an additional contribution of lectin-mediated enhancement in binding with Thr-13 in the active site cannot be excluded.
The situation is reversed for the glycosylation of the MUC5AC-13 glycopeptide. Here, product release is rate-limiting for transfer to Thr-3 by the full-length enzyme, whereas transfer to Thr-9 in the absence of the lectin domain proceeds with a uniform rate (Table 3). This observation can be reconciled by examination of the affinity of the peptide (both Km and Kd values) for the two constructs. Evidently, the presence of the lectin domain creates a site of higher affinity (Thr-3) on this glycopeptide, which is otherwise not available to the catalytic domain. The higher affinity for Thr-3 recognition (Kd = 19 μm) versus that for Thr-9 recognition (Kd = 93 μm) directs glycosylation to Thr-3. Here, the lectin domain contributes directly to glycopeptide binding, thereby determining the site of glycosylation (Fig. 7, e and f). The direct contribution of the lectin domain in this case also accounts for the ability of the lectin domain of hT10 to partially compensate for the hT2 lectin domain function (Fig. 2k). The increased Km and Kd for glycosylation by hT2CD at Thr-9 on MUC5AC-13 compared with the same parameters for the MUC5AC and MUC5AC-3 peptides is likely a result of steric hindrance by the GalNAc on Thr-13.
DISCUSSION
The results demonstrate two modes of glycosylation site selection by ppGalNAcTs (Fig. 8). One level of selection is dictated by the peptide-binding groove of the catalytic domain and would be influenced only by the local sequence of residues immediately surrounding the Thr/Ser destined for glycosylation. The distinction between the peptide/glycopeptide transferase hT2 and the strict glycopeptide transferase hT10 arises from the ability of the catalytic domain of hT2 to bind naked peptides. The catalytic domain of hT10 however, has a minimal (if any) affinity for naked peptides. Local sequence recognition by its catalytic domain requires the presence of a GalNAc on the substrate immediately C-terminal to the residue being glycosylation. Current attempts to establish predictive methods for O-glycosylation and to compare the substrate preferences of different GalNAc transferases have focused only on this mode of site selection (7, 8, 30). Distinct preferences for certain amino acids flanking the glycosylation site have been demonstrated, underscoring the apparent significance of this mode of substrate selection (2).
FIGURE 8.

Schematic representation of the two modes of substrate recognition by ppGalNAcTs. a, local sequence recognition that differs between hT2 and hT10, with hT10 requiring binding of extant GalNAc to the catalytic domain (CD). b, concerted recognition of local sequence by the catalytic domain and preexisting GalNAc by the lectin domain (LD) that is common to both hT2 and hT10. Sites of GalNAc addition are shown as filled black circles and pre-existing GalNAc are shown as open circles. Note that the different relative orientations of the CD and LD are shown only highlight conformational flexibility suggested by available crystal structures and are not a true representation of actual data.
The second mode of selection is determined by the lectin domain, which directs glycosylation to sites distal form the extant glycosylation. Concerted recognition of the pre-existing glycosylation by the lectin domain and local sequence by the catalytic domain creates glycosylation sites that are otherwise not accessible to the catalytic domain alone due to low affinities (Fig. 8b).
These lectin domain-mediated effects are reminiscent of the roles of the carbohydrate binding modules of many glycoside hydrolases, which often bind sugars contained in the substrate of the cognate catalytic module (31). Notably, in some xylanases that also contain a family 13 carbohydrate binding module like the ppGalNAcTs, deletion of the carbohydrate binding module has been shown to affect activity on insoluble but not on soluble substrates (32, 33).
The lectin domain influences glycosylation site selection over a wide range of distances with respect to the existing glycosylation. As demonstrated here for hT2, depending on the peptide, the lectin domain is able to direct glycosylation to positions both N- and C-terminal to the extant glycosylation. The number of residues between the extant GalNAc and lectin-directed glycosylation site is also variable. This is also true of hT10. Glycosylation of MUC5AC-9 glycopeptide, shown here to be lectin-mediated, occurs on Thr-3 (28), five residues to the N-terminal of the extant glycosylation. Similarly, Kubota et al. (17) have shown that the glycosylation of Ser-3 by hT10 on an IgA hinge region-derived peptide already glycosylated on Ser-11 is lectin domain-mediated. This variability likely arises due to a combination of flexibility in the relative orientation of the lectin and catalytic domain and conformational variation in the glycopeptide substrate.
Lectin domain-mediated effects on glycopeptide glycosylation by ppGalNAcT1, -T2, and -T4 have been documented (20, 27, 34). However, the mechanism by which the lectin domain directs site selection is not clear. The pre-steady state kinetic results with hT2 presented here suggest that the lectin domain can modulate the site of glycosylation by directly aiding substrate binding and possibly by enhancing product release. How the lectin domain mediates these two different effects is not directly evident from an examination of the crystal structure of hT2. With the structures of the two complexes currently available (UDP-bound and peptide/UDP-bound), it is not possible to span the proposed GalNAc binding site(s) on the lectin domain and the site of catalysis with the glycopeptides used in this study. But the significantly different conformations of the lectin domain with respect to the catalytic domain between the two available structures of hT2 suggest the existence of considerable conformational flexibility (16). It is conceivable that these two domains could be in greater proximity in the presence of a glycopeptide substrate allowing the lectin domain to mediate site selection by direct binding. In addition, the lectin domain of ppGalNAcTs comprises three independent glycan binding sites. The alpha site of hT2 and the beta site of hT10 have been shown to have affinity for GalNAc. These three sites could play different roles in the binding of different glycopeptide substrates contributing to greater context-dependent complexity in substrate recognition.
The density of terminal glycosylation of peptides with multiple acceptor sites is diminished when lectin domain function is compromised by site-directed mutagenesis (19). Taken together with the observations presented here, it is likely that the elimination of lectin domain function would not only reduce glycosylation density but also result in peptides that are glycosylated at very different positions. Domains carrying mucin-type O-glycosylation are frequently composed of repeat sequences. The cellular glycosylation machinery requires the means not only to achieve high local densities within the repeats but also to space out the addition of the GalNAc across the many tandem repeats. The ability of the lectin domain to direct glycosylation to sites distal from the existing GalNAcs represents a mechanism that would permit spacing of the GalNAcs. High GalNAc density within each unit of the repeat could then be achieved by catalytic domain activity alone, especially in the case of transferases such as ppGalNAcT-10, which have a strict glycopeptide specificity.
The results presented here have direct implications for prediction methods for mucin-type O-glycosylation. It is evident that the final glycosylation state of a protein in vivo would be dependent of the repertoire of ppGalNAcTs that are expressed in that cell. An additional degree of complexity could arise from the order in which the protein substrate encounters each transferase isoform within the Golgi. Our understanding of the specific cellular location of native ppGalNAcTs within the Golgi (or endoplasmic reticulum) is incomplete (35, 36). If different ppGalNAcTs are colocalized within the same subcellular compartment, different molecules of the same protein could encounter the transferase isoforms in a different sequence. Heterogeneity in O-glycosylation would therefore result not only from incomplete glycosylation but also from an alteration of the specificity of latter transferases based on the activity of the early transferases. Prediction methods will also be confounded by the apparent lack of strict distance criteria for site selection with respect to the extant glycosylation. In vitro studies with random glycopeptides will help rationalize these distance criteria. Crystal structures of these isoforms in complex with different glycopeptide substrates will be necessary to understand the structural basis of lectin domain-mediated glycopeptide recognition and would perhaps help in framing rules for glycosylation site selection.
Acknowledgments
We thank Dr. James Hurley, NIDDK, NIH for the use of the spectrofluorometer. We thank Dr. Kelly Ten Hagen and Dr. John Hanover for helpful suggestions.
This work was supported, in whole or in part, by the National Institutes of Health, through funds of the intramural program of the NIDDK and Grants NCI-RO1 CA-78834 (to T. A. G.) and NIGMS-RO1 GM-066148 (to D. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ppGalNAcT, UDP-GalNAc:polypeptide α-N-acetylgalactosaminyltransferase; TEV, tobacco etch virus; hT2, human ppGalNAcT-2 residues 75–571; hT10, human ppGalNAcT-10 residues 71–603; CD, catalytic domain; LD, lectin domain; SELDI-TOF, surface-enhanced laser desorption ionization-time of flight; DMF, dimethylformamide; Fmoc, N-(9-fluorenyl)methoxycarbonyl.
References
- 1.Ten Hagen, K. G., Fritz, T. A., and Tabak, L. A. (2003) Glycobiology 13 1R–16R [DOI] [PubMed] [Google Scholar]
- 2.Gerken, T. A., Raman, J., Fritz, T. A., and Jamison, O. (2006) J. Biol. Chem. 281 32403–32416 [DOI] [PubMed] [Google Scholar]
- 3.Gerken, T. A., Zhang, J., Levine, J., and Elhammer, A. (2002) J. Biol. Chem. 277 49850–49862 [DOI] [PubMed] [Google Scholar]
- 4.O'Connell, B. C., Hagen, F. K., and Tabak, L. A. (1992) J. Biol. Chem. 267 25010–25018 [PubMed] [Google Scholar]
- 5.Nehrke, K., Ten Hagen, K. G., Hagen, F. K., and Tabak, L. A. (1997) Glycobiology 7 1053–1060 [DOI] [PubMed] [Google Scholar]
- 6.Sparrow, L. G., Gorman, J. J., Strike, P. M., Robinson, C. P., McKern, N. M., Epa, V. C., and Ward, C. W. (2007) Proteins 66 261–265 [DOI] [PubMed] [Google Scholar]
- 7.Hansen, J. E., Lund, O., Engelbrecht, J., Bohr, H., and Nielsen, J. O. (1995) Biochem. J. 308 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen, J. E., Lund, O., Tolstrup, N., Gooley, A. A., Williams, K. L., and Brunak, S. (1998) Glycoconj. J. 15 115–130 [DOI] [PubMed] [Google Scholar]
- 9.Nehrke, K., Hagen, F. K., and Tabak, L. A. (1998) Glycobiology 8 367–371 [DOI] [PubMed] [Google Scholar]
- 10.Wandall, H. H., Hassan, H., Mirgorodskaya, E., Kristensen, A. K., Roepstorff, P., Bennett, E. P., Nielsen, P. A., Hollingsworth, M. A., Burchell, J., Taylor-Papadimitriou, J., and Clausen, H. (1997) J. Biol. Chem. 272 23503–23514 [DOI] [PubMed] [Google Scholar]
- 11.Hagen, F. K., Ten Hagen, K. G., Beres, T. M., Balys, M. M., VanWuyckhuyse, B. C., and Tabak, L. A. (1997) J. Biol. Chem. 272 13843–13848 [DOI] [PubMed] [Google Scholar]
- 12.Nakagawa, T., Martinez, S. R., Goto, Y., Koyanagi, K., Kitago, M., Shingai, T., Elashoff, D. A., Ye, X., Singer, F. R., Giuliano, A. E., and Hoon, D. S. (2007) Clin. Cancer Res. 13 4105–4110 [DOI] [PubMed] [Google Scholar]
- 13.Bennett, E. P., Hassan, H., Hollingsworth, M. A., and Clausen, H. (1999) FEBS Lett. 460 226–230 [DOI] [PubMed] [Google Scholar]
- 14.Ten Hagen, K. G., Bedi, G. S., Tetaert, D., Kingsley, P. D., Hagen, F. K., Balys, M. M., Beres, T. M., Degand, P., and Tabak, L. A. (2001) J. Biol. Chem. 276 17395–17404 [DOI] [PubMed] [Google Scholar]
- 15.Fritz, T. A., Hurley, J. H., Trinh, L. B., Shiloach, J., and Tabak, L. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 15307–15312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fritz, T. A., Raman, J., and Tabak, L. A. (2006) J. Biol. Chem. 281 8613–8619 [DOI] [PubMed] [Google Scholar]
- 17.Kubota, T., Shiba, T., Sugioka, S., Furukawa, S., Sawaki, H., Kato, R., Wakatsuki, S., and Narimatsu, H. (2006) J. Mol. Biol. 359 708–727 [DOI] [PubMed] [Google Scholar]
- 18.Tenno, M., Saeki, A., Kezdy, F. J., Elhammer, A. P., and Kurosaka, A. (2002) J. Biol. Chem. 277 47088–47096 [DOI] [PubMed] [Google Scholar]
- 19.Wandall, H. H., Irazoqui, F., Tarp, M. A., Bennett, E. P., Mandel, U., Takeuchi, H., Kato, K., Irimura, T., Suryanarayanan, G., Hollingsworth, M. A., and Clausen, H. (2007) Glycobiology 17 374–387 [DOI] [PubMed] [Google Scholar]
- 20.Hassan, H., Reis, C. A., Bennett, E. P., Mirgorodskaya, E., Roepstorff, P., Hollingsworth, M. A., Burchell, J., Taylor-Papadimitriou, J., and Clausen, H. (2000) J. Biol. Chem. 275 38197–38205 [DOI] [PubMed] [Google Scholar]
- 21.Liu, M., Barany, G., and Live, D. (2005) Carbohydr. Res. 340 2111–2122 [DOI] [PubMed] [Google Scholar]
- 22.O'Connell, B. C., and Tabak, L. A. (1993) Anal. Biochem. 210 423–425 [DOI] [PubMed] [Google Scholar]
- 23.Xu, Y., Eads, J., Sacchettini, J. C., and Grubmeyer, C. (1997) Biochemistry 36 3700–3712 [DOI] [PubMed] [Google Scholar]
- 24.Bashor, C., Denu, J. M., Brennan, R. G., and Ullman, B. (2002) Biochemistry 41 4020–4031 [DOI] [PubMed] [Google Scholar]
- 25.Gerken, T. A., Owens, C. L., and Pasumarthy, M. (1998) J. Biol. Chem. 273 26580–26588 [DOI] [PubMed] [Google Scholar]
- 26.Gerken, T. A., Owens, C. L., and Pasumarthy, M. (1997) J. Biol. Chem. 272 9709–9719 [DOI] [PubMed] [Google Scholar]
- 27.Hagen, F. K., Hazes, B., Raffo, R., deSa, D., and Tabak, L. A. (1999) J. Biol. Chem. 274 6797–6803 [DOI] [PubMed] [Google Scholar]
- 28.Tetaert, D., Ten Hagen, K. G., Richet, C., Boersma, A., Gagnon, J., and Degand, P. (2001) Biochem. J. 357 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wragg, S., Hagen, F. K., and Tabak, L. A. (1995) J. Biol. Chem. 270 16947–16954 [DOI] [PubMed] [Google Scholar]
- 30.Julenius, K., Molgaard, A., Gupta, R., and Brunak, S. (2005) Glycobiology 15 153–164 [DOI] [PubMed] [Google Scholar]
- 31.Boraston, A. B., Bolam, D. N., Gilbert, H. J., and Davies, G. J. (2004) Biochem. J. 382 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boraston, A. B., Tomme, P., Amandoron, E. A., and Kilburn, D. G. (2000) Biochem. J. 350 933–941 [PMC free article] [PubMed] [Google Scholar]
- 33.Dupont, C., Roberge, M., Shareck, F., Morosoli, R., and Kluepfel, D. (1998) Biochem. J. 330 41–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanisch, F. G., Reis, C. A., Clausen, H., and Paulsen, H. (2001) Glycobiology 11 731–740 [DOI] [PubMed] [Google Scholar]
- 35.Roth, J., Wang, Y., Eckhardt, A. E., and Hill, R. L. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 8935–8939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rottger, S., White, J., Wandall, H. H., Olivo, J. C., Stark, A., Bennett, E. P., Whitehouse, C., Berger, E. G., Clausen, H., and Nilsson, T. (1998) J. Cell Sci. 111 45–60 [DOI] [PubMed] [Google Scholar]