

Abstract
The reversible oxidation of the active site cysteine in typical 2-Cys
peroxiredoxins (Prx) to sulfinic acid during oxidative stress plays an
important role in peroxide-mediated cell signaling. The catalytic
retroreduction of
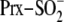 by
sulfiredoxin (Srx) has been proposed to proceed through two novel reaction
intermediates, a sulfinic phosphoryl ester and protein-based thiosulfinate.
Two scenarios for the repair mechanism have been suggested that differ in the
second step of the reaction. The attack of Srx or GSH on the
by
sulfiredoxin (Srx) has been proposed to proceed through two novel reaction
intermediates, a sulfinic phosphoryl ester and protein-based thiosulfinate.
Two scenarios for the repair mechanism have been suggested that differ in the
second step of the reaction. The attack of Srx or GSH on the
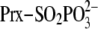 intermediate would result in either the formation of
Prx-Cys-S(=O)–S-Cys-Srx or the formation of Prx-Cys-S(=O)–S-G
thiosulfinates, respectively. To elucidate the mechanism of Prx repair, we
monitored the reduction of human
intermediate would result in either the formation of
Prx-Cys-S(=O)–S-Cys-Srx or the formation of Prx-Cys-S(=O)–S-G
thiosulfinates, respectively. To elucidate the mechanism of Prx repair, we
monitored the reduction of human
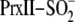 using rapid
chemical quench methodology and electrospray ionization time-of-flight mass
spectrometry. An 18O exchange study revealed that the Prx sulfinic
acid phosphoryl ester is rapidly formed and hydrolyzed (k = 0.35
min–1). Furthermore, we observed the exclusive formation of a
thiosulfinate linkage between Prx and Srx (k = 1.4
min–1) that collapses to the disulfide-bonded Srx-Prx species
(k = 0.14 min–1). Thus, the kinetic and chemical
competences of the first two steps in the Srx reaction have been demonstrated.
It is clear, however, that GSH may influence thiosulfinate formation and that
GSH and Srx may play additional roles in the resolution of the thiosulfinate
intermediate.
using rapid
chemical quench methodology and electrospray ionization time-of-flight mass
spectrometry. An 18O exchange study revealed that the Prx sulfinic
acid phosphoryl ester is rapidly formed and hydrolyzed (k = 0.35
min–1). Furthermore, we observed the exclusive formation of a
thiosulfinate linkage between Prx and Srx (k = 1.4
min–1) that collapses to the disulfide-bonded Srx-Prx species
(k = 0.14 min–1). Thus, the kinetic and chemical
competences of the first two steps in the Srx reaction have been demonstrated.
It is clear, however, that GSH may influence thiosulfinate formation and that
GSH and Srx may play additional roles in the resolution of the thiosulfinate
intermediate.
Proteineous Cys residues play a key role in the redox regulation of
biological systems due to the wide range of oxidation states that sulfur can
occupy (–2 to +4) (1,
2). Most Cys undergo only
small, reversible changes in oxidation state as exemplified by thiol-disulfide
exchange reactions (–2 to –1)
(3,
4). Remarkably, one Cys of the
eukaryotic, typical 2-Cys peroxiredoxins
(Prx)3 has been shown
to transition through five oxidation states. During the catalytic reduction of
H2O2, peroxynitrite, and alkyl hydroperoxides, the
peroxidatic Cys (Cys-SPH, –2) is oxidized to the sulfenic
acid (Cys-SPOH, 0)
(5). This activated moiety
subsequently reacts with the resolving Cys (Cys-SRH) of the other
Prx subunit within the homodimer to form an intermolecular disulfide bond
(Prx-Cys-SP–SR-Cys-Prx, –1). This disulfide
bond is readily reduced by an exogenous reductant such as thioredoxin (Trx)
(6). During oxidative stress,
however, a burst of peroxide can overwhelm this system and hyperoxidize the
sulfenic acid intermediate to form a stable sulfinic acid
(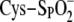 ,
+2). The resulting diminished peroxidase capacity of the cell has been thought
to locally increase peroxide levels and mediate signaling events
(7). In this context, human
PrxII inactivation promotes cell cycle arrest, which is resumed once the Prx
molecule is returned to the reduced state
(8). The unique reduction of
the sulfinic moiety by the enzyme sulfiredoxin (Srx) has been shown to play an
important regulatory role in peroxide-mediated transcriptional activation in
Schizosaccharomyces pombe
(9,
10).
,
+2). The resulting diminished peroxidase capacity of the cell has been thought
to locally increase peroxide levels and mediate signaling events
(7). In this context, human
PrxII inactivation promotes cell cycle arrest, which is resumed once the Prx
molecule is returned to the reduced state
(8). The unique reduction of
the sulfinic moiety by the enzyme sulfiredoxin (Srx) has been shown to play an
important regulatory role in peroxide-mediated transcriptional activation in
Schizosaccharomyces pombe
(9,
10).
The Srx retroreduction reaction requires ATP, Mg2+, a conserved
active site Cys (Cys-99 in human Srx, hSrx), and an exogenous thiol reductant,
such as GSH or Trx. The crystal structure of the
Srx-ATP·Mg2+ complex (PDB 3CYI) coupled with an analysis of
the phosphorylation status of Srx and Prx variants supports that the first
step of the reaction (Fig. 1)
involves the direct attack of the Cys-sulfinic acid moiety onto the
γ-phosphate of ATP (11)
rather than an intervening transfer step from Srx
(12). The resulting sulfinic
phosphoryl ester
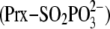 is transient and thought to lead to an intermolecular thiosulfinate
intermediate (i.e. an disulfide S-monoxide with +1, –1
oxidation states; step 2) either with Cys-99 of Srx
(Prx-Cys-SP(=O)–S-Cys-Srx; path 1) or with GSH
(Prx-Cys-SP(=O)–S-G; path 2)
(12,
13). These thiosulfinates
could then be reduced by GSH to release Prx-Cys-SPOH and either
Srx-S–S-G or G-S–S-G (step 3).
is transient and thought to lead to an intermolecular thiosulfinate
intermediate (i.e. an disulfide S-monoxide with +1, –1
oxidation states; step 2) either with Cys-99 of Srx
(Prx-Cys-SP(=O)–S-Cys-Srx; path 1) or with GSH
(Prx-Cys-SP(=O)–S-G; path 2)
(12,
13). These thiosulfinates
could then be reduced by GSH to release Prx-Cys-SPOH and either
Srx-S–S-G or G-S–S-G (step 3).
FIGURE 1.

Comparison of the proposed sulfinic acid reduction mechanisms of Srx. Path 1 represents the mechanism originally proposed by Biteau et al. (13). Path 2 incorporates modifications to the reaction pathway as suggested by Jeong et al. (12). Step 1 involves the formation of the sulfinic acid phosphoryl ester intermediate. In Step 2 of the reaction, the addition of a thiol group leads to the formation of different thiosulfinate intermediates. This intermediate is subsequently resolved by GSH in Step 3. The resulting sulfenic acid form of Prx could then go on to react with Srx, GSH, and the resolving Cys of the adjacent Prx molecule to form Prx-SP–S-Srx, Prx-SP–S-G, Prx-SP–SR-Prx species.
Over the last years, mass spectrometry (MS) has emerged as an important tool for the monitoring of both chemical and enzymatic reactions. These methods rely on either continuous flow and quenching through desolvation (time-resolved MS) or chemical quenching followed by direct injection of the quenched reaction mixture into the mass spectrometer (14–20). In this report, we investigated the human Srx reaction mechanism using rapid chemical quench methodology and electrospray ionization time-of-flight MS (ESI-TOF MS). We found that Prx sulfinic phosphoryl ester formation was reversible as indicated by 18O exchange. A thiosulfinate linkage between Prx and Srx was readily formed, and upon collapse of this intermediate, an increase in the disulfide-bonded Srx-Prx species was observed, proving the kinetic competency of this species. These results clarify the initial steps of the reaction mechanism but also raise questions as to the role of GSH, particularly within the cellular context.
EXPERIMENTAL PROCEDURES
Protein Preparation and Modification—Truncated versions of human Srx, the wild-type engineered truncation of hSrx (ET-Srx, residues 32–137) and the C99A mutant, were expressed in C41(DE3) Escherichia coli cells from a pET19 vector (Novagen) derivative containing a PreScission protease (GE Healthcare) cleavage site between Srx and the N-terminal His tag. The proteins were purified using nickel affinity and size-exclusion chromatography after the removal of the His tag (21). His-tagged human PrxII containing two point mutations (C70S and C172S, hPrxII-C2S) was also expressed in C41(DE3) cells and purified in the presence of 5 mm β-mercaptoethanol and 2 mm dithiothreitol (DTT) during the nickel affinity and size exclusion steps, respectively. The sulfinic acid form of hPrxII was generated by the addition of 2 or 5 mm H2O2 (or in some instances, isotopically labeled H 182O2, Icon Isotopes, Summit, NJ) to 130 μm PrxII-C2S in the presence of 50 mm DTT, 20 mm HEPES, pH 7.5, and 100 mm NaCl for 30 min at room temperature. Bio-Gel P6 spin columns (Bio-Rad) pre-equilibrated with 20 mm HEPES, pH 7.5, 100 mm NaCl were used to remove excess DTT and H2O2. The extent of overoxidation was determined by mass spectrometry. In some cases where incomplete overoxidation was observed, the remaining Cys were reduced with 2 mm DTT for 10 min at 37 °C and then alkylated by the addition of 50 mm iodoacetamide for 30 min. Approximately 5–10% of the protein was estimated to be alkylated.
Kinetic Assays—Reactions (50 μl) containing 60
μm
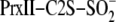 or
or
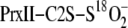 ,
1 mm ATP, 2 mm MgCl2, 20 mm HEPES,
pH 7.5, 100 mm NaCl were initiated by the addition of 40
μm WT or C99A Srx while stirring at 500 rpm at 37 °C. At the
appropriate incubation time, each sample was applied to a BioGel P6 spin
column to remove small molecules and buffer exchange into 50 mm
ammonium acetate, pH 3.0 or pH 5.0 (isotope exchange assay). For time points
shorter than 30 s, rapid quench experiments were performed using a Kintek
RFQ-3 rapid chemical quench instrument (Kintek Instruments, Austin, TX). The
quench-flow reactions were initiated by mixing 120 μm
,
1 mm ATP, 2 mm MgCl2, 20 mm HEPES,
pH 7.5, 100 mm NaCl were initiated by the addition of 40
μm WT or C99A Srx while stirring at 500 rpm at 37 °C. At the
appropriate incubation time, each sample was applied to a BioGel P6 spin
column to remove small molecules and buffer exchange into 50 mm
ammonium acetate, pH 3.0 or pH 5.0 (isotope exchange assay). For time points
shorter than 30 s, rapid quench experiments were performed using a Kintek
RFQ-3 rapid chemical quench instrument (Kintek Instruments, Austin, TX). The
quench-flow reactions were initiated by mixing 120 μm
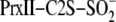 ,
2 mm ATP, and 4 mm Mg2+ in one loop (15
μl) with 80 μm SrxET in the other loop (15 μl) resulting
in concentrations identical to that described above. Each reaction was
quenched with 50 mm ammonium acetate, pH 3.0, and desalted prior to
mass analysis. SDS-PAGE analysis of the reactions utilized 8–16%
gradient gels (Bio-Rad) with non-reducing sample loading buffer and 20
mm N-ethylmaleimide.
,
2 mm ATP, and 4 mm Mg2+ in one loop (15
μl) with 80 μm SrxET in the other loop (15 μl) resulting
in concentrations identical to that described above. Each reaction was
quenched with 50 mm ammonium acetate, pH 3.0, and desalted prior to
mass analysis. SDS-PAGE analysis of the reactions utilized 8–16%
gradient gels (Bio-Rad) with non-reducing sample loading buffer and 20
mm N-ethylmaleimide.
Data Collection and Analysis—All ESI-TOF MS data were
collected on an Agilent MSD TOF system. The operating conditions for MS
analysis were as follows: positive ion mode, capillary voltage 3500 V,
nebulizer gas 30 p.s.i.g., drying gas 5.0 liter/min; fragmentor 140 V, gas
temperature 325 °C. The samples were injected into the ion source using a
syringe pump (KD Scientific) and a 250-μl Hamilton syringe connected to the
ion probe with a 50-μm inner diameter fused silica capillary. The injection
flow rate was 10 μl/min. The averaged MS spectra were deconvoluted using
the Agilent MassHunter work station software Version B.01.03. Data from the
isotope exchange reactions were quantified based upon the relative mass shift
(Δ mass) in the PrxII-C2S-S18O–2 species as
result of 18O/16O exchange. The Δ mass was fit to
a single exponential equation y = a(1 –
e–kt) to determine the rate
constant for the 18O/16O exchange. The formation and
decay of the intermolecular thiosulfinate and disulfide species were expressed
as a ratio of peak intensity of the respective species relative to the
constant amount of catalytically inactive iodoacetamide-labeled PrxII-C2S.
Fits of data were carried out using Sigma-Plot 10.0 and KinTekSim based on a
simple A ↔ B ↔ C ↔ D mechanism where A is the
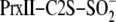 substrate, B is the Prx sulfinic phosphoryl ester intermediate, C is the
thiosulfinate intermediate ((Prx-Cys-S(=O)–S-Cys-Srx), and D is the
Srx-Prx covalent heterodimer.
substrate, B is the Prx sulfinic phosphoryl ester intermediate, C is the
thiosulfinate intermediate ((Prx-Cys-S(=O)–S-Cys-Srx), and D is the
Srx-Prx covalent heterodimer.
RESULTS AND DISCUSSION
Reversibility of the Phosphorylated Prx Intermediate—The
mechanism of Prx sulfinic acid reduction by Srx has been proposed to proceed
through two novel protein intermediates, sulfinic phosphoryl ester and
thiosulfinate (Fig. 1).
Previous efforts to identify these intermediates using wild-type proteins have
been unsuccessful
(11–13).
Recently, we were able to trap a phosphorylated form of Prx where the
Cys-SP residue was mutated to Asp
 (11). In the study described
here, the reaction mechanism of
(11). In the study described
here, the reaction mechanism of
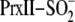 reduction
by Srx was investigated using a combination of rapid chemical quench and
ESI-TOF MS. Given the facile interchange between activated sulfur species,
which could potentially complicate the interpretation of experimental data, a
decision was made to remove all thiols that were not necessary for reduction
of the Prx molecule. This was accomplished by using the hPrxII-C2S variant
(C70S, C172S) and performing the reactions in the absence of GSH. Under these
conditions, Srx, ATP, and Mg2+ are sufficient to repair the
reduction
by Srx was investigated using a combination of rapid chemical quench and
ESI-TOF MS. Given the facile interchange between activated sulfur species,
which could potentially complicate the interpretation of experimental data, a
decision was made to remove all thiols that were not necessary for reduction
of the Prx molecule. This was accomplished by using the hPrxII-C2S variant
(C70S, C172S) and performing the reactions in the absence of GSH. Under these
conditions, Srx, ATP, and Mg2+ are sufficient to repair the
 moiety.
moiety.
The initial control reactions were quenched at different time points by
passing the samples through spin columns equilibrated with ammonium acetate
(pH< 5) prior to MS analysis. A molecular peak corresponding to the mass of
the thiosulfinate intermediate was consistently observed along with the Srx,
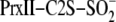 ,
and Srx-Prx species; however, there was no evidence for the presence of the
Prx sulfinic phosphoryl ester intermediate. In an effort to determine the Srx
requirement for the formation of the Prx phosphoryl intermediate, the
isotopically labeled
,
and Srx-Prx species; however, there was no evidence for the presence of the
Prx sulfinic phosphoryl ester intermediate. In an effort to determine the Srx
requirement for the formation of the Prx phosphoryl intermediate, the
isotopically labeled
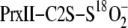 was incubated with Srx and excess ATP and Mg2+ at pH 7.5, and the
18O/16O exchange reaction was monitored at different
time points over a 15 min period (Fig.
2A). A mass decrease of 2 Da was observed in the
was incubated with Srx and excess ATP and Mg2+ at pH 7.5, and the
18O/16O exchange reaction was monitored at different
time points over a 15 min period (Fig.
2A). A mass decrease of 2 Da was observed in the
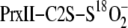 molecule with the rate of mass exchange consistent with a first order rate
constant of 0.35 min–1
(Fig. 2B). A full
exchange of the 18O for 16O, i.e. a mass
decrease of 4 Da, was not observed. The 18O/16O exchange
reaction was not observed when the reaction was performed at pH 5, ATP and
Mg2+ were omitted, and the Srx C99A mutant was used
(Fig. 2A). ATP
hydrolysis in the absence of an exogenous thiol (i.e. GSH) is
consistent with the increased level of 32Pi release over
the amount of sulfinic acid substrate observed by Jeong et al.
(12). Similar to the findings
described here, the use of the C99A Srx variant prevented ATP hydrolysis.
molecule with the rate of mass exchange consistent with a first order rate
constant of 0.35 min–1
(Fig. 2B). A full
exchange of the 18O for 16O, i.e. a mass
decrease of 4 Da, was not observed. The 18O/16O exchange
reaction was not observed when the reaction was performed at pH 5, ATP and
Mg2+ were omitted, and the Srx C99A mutant was used
(Fig. 2A). ATP
hydrolysis in the absence of an exogenous thiol (i.e. GSH) is
consistent with the increased level of 32Pi release over
the amount of sulfinic acid substrate observed by Jeong et al.
(12). Similar to the findings
described here, the use of the C99A Srx variant prevented ATP hydrolysis.
FIGURE 2.

Srx-dependent incorporation of 16O in
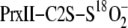 supports the reversible formation of PrxII sulfinic phosphoryl
intermediate. A, deconvoluted ESI-TOF MS spectra of
supports the reversible formation of PrxII sulfinic phosphoryl
intermediate. A, deconvoluted ESI-TOF MS spectra of
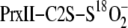 mass peak at increasing reaction times. A negative mass shift is observed in
the presence of WT Srx (left panel) due to PrxII sulfinic phosphoryl
intermediate hydrolysis in H 162O-based reaction buffer.
The 18O/16O exchange reaction was not observed when C99A
Srx was used instead of WT Srx (right panel). amu, atomic
mass units. B, the mass shift determined at each reaction time point
was plotted against time and fit to a single exponential increase equation
with an amplitude of 1.96 ± 0.04 atomic mass units and a rate of 0.35
± 0.02 min–1.
mass peak at increasing reaction times. A negative mass shift is observed in
the presence of WT Srx (left panel) due to PrxII sulfinic phosphoryl
intermediate hydrolysis in H 162O-based reaction buffer.
The 18O/16O exchange reaction was not observed when C99A
Srx was used instead of WT Srx (right panel). amu, atomic
mass units. B, the mass shift determined at each reaction time point
was plotted against time and fit to a single exponential increase equation
with an amplitude of 1.96 ± 0.04 atomic mass units and a rate of 0.35
± 0.02 min–1.
Together, these observations illustrate the necessity of phosphorylation for 18O exchange and the reversibility of the first step of the reaction. These results are consistent with the rapid phosphorylation of the Prx sulfinic acid moiety. It is also apparent that the formation and slow hydrolysis of the sulfinic phosphoryl ester are both possibly dependent on Cys-99 of Srx. Cys-99 and the other surrounding active site residues may influence the correct positioning of the sulfinic acid moiety, associated loop residues, and water structure through hydrogen-bonding and hydrophobic interactions. Support for this notion comes from the apparent stereospecific exchange of 18O within the sulfinic acid and the geometric relationship of the γ-phosphate group of ATP within Srx that facilitates an inline attack by the Prx molecule and not Srx (11, 22). A crystal structure of the Srx-Prx-ATP·Mg2+ complex and future time-resolved ESI-TOF MS experiments performed without chemical quenching and on the millisecond time scale will hopefully clarify this issue (15, 19, 20).
Identification of an Intact Thiosulfinate Intermediate between Srx and
Prx—In the second step of the repair of
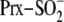 (Fig. 1), two different
thiosulfinate intermediates have been proposed. Although Biteau et
al. (13) proposed the
formation of a protein-protein thiosulfinate (Prx-Cys-S(=O)–S-Cys-Srx),
Jeong et al. (12)
proposed a protein-glutathione thiosulfinate (Prx-Cys-S(=O)–SG)
intermediate. In a similar approach to the isotope exchange study, we sought
to identify the thiosulfinate species using ESI-TOF MS.
(Fig. 1), two different
thiosulfinate intermediates have been proposed. Although Biteau et
al. (13) proposed the
formation of a protein-protein thiosulfinate (Prx-Cys-S(=O)–S-Cys-Srx),
Jeong et al. (12)
proposed a protein-glutathione thiosulfinate (Prx-Cys-S(=O)–SG)
intermediate. In a similar approach to the isotope exchange study, we sought
to identify the thiosulfinate species using ESI-TOF MS.
Although nothing is known about protein-based thiosulfinates, studies on
small molecule thiosulfinates have shown that they are very reactive with
other thiols and stabilized at low pH
(2,
23,
24). Therefore, a reaction
with the
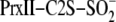 mutant in slight excess over Srx at pH 7.5 was rapidly quenched at different
time points by the addition of ammonium acetate, pH 3.0, followed by desalting
on a Bio-spin column equilibrated with the same buffer. Under these
conditions, we were able to observe two species with the average molecular
mass of 37,723.00 ± 0.14 and 37,739.70 ± 0.11 Da, which
correspond to Prx-S–S-Srx heterodimer (expected mass 37,723.35 Da) and
the thiosulfinate-linked complex between Srx and Prx (expected mass 37,739.35
Da) (Fig. 3,
A–C). As expected, the mass of the thiosulfinate
complex was 2 Da higher when the isotopically labeled
mutant in slight excess over Srx at pH 7.5 was rapidly quenched at different
time points by the addition of ammonium acetate, pH 3.0, followed by desalting
on a Bio-spin column equilibrated with the same buffer. Under these
conditions, we were able to observe two species with the average molecular
mass of 37,723.00 ± 0.14 and 37,739.70 ± 0.11 Da, which
correspond to Prx-S–S-Srx heterodimer (expected mass 37,723.35 Da) and
the thiosulfinate-linked complex between Srx and Prx (expected mass 37,739.35
Da) (Fig. 3,
A–C). As expected, the mass of the thiosulfinate
complex was 2 Da higher when the isotopically labeled
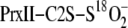 was used (data not shown). The disulfide and thiosulfinate species were absent
when the reaction was performed with the C99A mutant of Srx.
was used (data not shown). The disulfide and thiosulfinate species were absent
when the reaction was performed with the C99A mutant of Srx.
FIGURE 3.

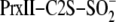 reduction by WT Srx monitored by ESI-TOF MS and SDS-PAGE. A and
B, deconvoluted ESI-TOF MS spectra of protein species present at
different reaction time points. The spectra are focused on the
reduction by WT Srx monitored by ESI-TOF MS and SDS-PAGE. A and
B, deconvoluted ESI-TOF MS spectra of protein species present at
different reaction time points. The spectra are focused on the
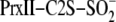 (25,736
mass peak), Prx-Cys-S–S-Cys-Srx (37,723 mass peak), and
Prx-Cys-S(=O)–S-Cys-Srx species (37,739 mass peak). amu, atomic
mass units. Panel B is a closeup of the Prx-Cys-S–S-Cys-Srx and
Prx-Cys-S(=O)–S-Cys-Srx species. C, SDS-PAGE analysis of the
reaction quenched at different time points (inset). Formation of
Prx-Cys-S–S-Cys-Srx was plotted and fit to a single exponential increase
equation with a rate of 0.3 ± 0.05 min–1. D,
global fit of kinetic data using KinTekSim. The plot shows the modeled kinetic
profile for the Prx sulfinic phosphoryl ester intermediate and the determined
kinetic profiles of thiosulfinate intermediate (Prx-Cys-S(=O)–S-Cys-Srx)
and product formation (Prx-Cys-S–S-Cys-Srx) (reaction scheme and rate
constants associated with the proposed kinetic steps are shown in
Fig. 4).
(25,736
mass peak), Prx-Cys-S–S-Cys-Srx (37,723 mass peak), and
Prx-Cys-S(=O)–S-Cys-Srx species (37,739 mass peak). amu, atomic
mass units. Panel B is a closeup of the Prx-Cys-S–S-Cys-Srx and
Prx-Cys-S(=O)–S-Cys-Srx species. C, SDS-PAGE analysis of the
reaction quenched at different time points (inset). Formation of
Prx-Cys-S–S-Cys-Srx was plotted and fit to a single exponential increase
equation with a rate of 0.3 ± 0.05 min–1. D,
global fit of kinetic data using KinTekSim. The plot shows the modeled kinetic
profile for the Prx sulfinic phosphoryl ester intermediate and the determined
kinetic profiles of thiosulfinate intermediate (Prx-Cys-S(=O)–S-Cys-Srx)
and product formation (Prx-Cys-S–S-Cys-Srx) (reaction scheme and rate
constants associated with the proposed kinetic steps are shown in
Fig. 4).
Reactions with up to 1 mm GSH were also analyzed, and no evidence was found for the formation of a Prx-GSH thiosulfinate intermediate (data not shown). Moreover, the addition of GSH resulted in the complete disappearance of the disulfide-bonded and thiosulfinate Srx-Prx species and formation of the glutathionylated adducts of Srx and Prx, 12,327.1 and 25,737.0 Da, respectively. Thus, under physiological conditions where GSH is present at 1–10 mm, there may be a competition between Srx and GSH for the Prx sulfinic phosphoryl ester to form the thiosulfinate species. Given local concentration effects and the close proximity of Cys-99 of Srx to the Prx sulfinic moiety (11, 22), however, the attack of Srx should be efficient as suggested by the rate analyses described below. Nonetheless, it is clear that additional time- and concentration-dependent ESI-TOF MS experiments will be required to deconvolute GSH contributions to the kinetics of the Srx-Prx thiosulfinate and the putative Prx-GSH thiosulfinate formation and resolution, although the latter species could not be experimentally observed. No matter which thiolsulfinate path is taken in step 2, the result is the same, a repaired Prx molecule with regained enzymatic activity by the action of Srx.
Given that complete repair of the Prx molecule is possible without the
addition of GSH, we modeled the Srx reaction using KinTekSim to validate the
reaction scheme, as described under “Experimental Procedures.” The
rate constants for the reaction steps were determined based on the relative
intensities of disulfide and thiosulfinate species
(Fig. 3, A and
B) and were as follows: 0.35 min–1 for
sulfinic phosphoryl ester hydrolysis, 1.4 min–1 for
thiosulfinate formation, and 0.14 min–1 for
Prx-Cys-S–S-Cys-Srx formation. The latter rate is comparable with the
rate of 0.3 min–1 determined by SDS-PAGE analysis
(Fig. 3C). The
resulting global fit shown in Fig.
3D is consistent with the rapid formation of the sulfinic
phosphoryl ester (>2 min–1) and the following mechanism
(Fig. 4). In the absence of
GSH,
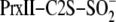 is rapidly phosphorylated to generate the sulfinic phosphoryl ester
intermediate. This reaction is reversible by the attack of hydroxide ions at a
rate of 0.35 min–1 at pH 7.5. This equilibrium is driven
forward by the thiol attack of Cys-99 in Srx at a rate of 1.4
min–1 to form a thiosulfinate intermediate between Srx and
Prx. One additional Srx can then attack the thiosulfinate bond to generate the
sulfenic acid form of Prx and Srx-S–S-Srx. Srx could also readily react
with Prx-Cys-SOH to generate the observed Prx-Cys-S–S-Cys-Srx disulfide
bond at a rate of 0.14 min–1.
is rapidly phosphorylated to generate the sulfinic phosphoryl ester
intermediate. This reaction is reversible by the attack of hydroxide ions at a
rate of 0.35 min–1 at pH 7.5. This equilibrium is driven
forward by the thiol attack of Cys-99 in Srx at a rate of 1.4
min–1 to form a thiosulfinate intermediate between Srx and
Prx. One additional Srx can then attack the thiosulfinate bond to generate the
sulfenic acid form of Prx and Srx-S–S-Srx. Srx could also readily react
with Prx-Cys-SOH to generate the observed Prx-Cys-S–S-Cys-Srx disulfide
bond at a rate of 0.14 min–1.
FIGURE 4.

Structural and kinetic intermediates along the human Srx reaction
coordinate. A, the
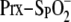 moiety (purple), Cys sulfinic acid 52 (Csd52), poised for an
inline attack of the γ-phosphate of ATP, based on the
ATP·Mg2+·Srx (blue) and
Srx-S–SP-PrxI crystal structures (Protein Data Bank numbers
3CYI and 2RII) (11,
22). The resulting
phosphorylated intermediate (not shown) can be readily hydrolyzed. B,
model of the thiosulfinate intermediate between Srx and Prx. C, the
crystallographically determined Prx-SP–S-Srx disulfide-bonded
complex (Protein Data Bank number 2RII). This complex could form through
several steps involving reactions between the sulfenic acid form of Prx and
Srx.
moiety (purple), Cys sulfinic acid 52 (Csd52), poised for an
inline attack of the γ-phosphate of ATP, based on the
ATP·Mg2+·Srx (blue) and
Srx-S–SP-PrxI crystal structures (Protein Data Bank numbers
3CYI and 2RII) (11,
22). The resulting
phosphorylated intermediate (not shown) can be readily hydrolyzed. B,
model of the thiosulfinate intermediate between Srx and Prx. C, the
crystallographically determined Prx-SP–S-Srx disulfide-bonded
complex (Protein Data Bank number 2RII). This complex could form through
several steps involving reactions between the sulfenic acid form of Prx and
Srx.
The studies described here prove the kinetic competence of the Prx sulfinic phosphoryl ester and Prx-Srx thiosulfinate intermediates and the chemical identity of the latter. Moreover, these data coupled with previous structural analyses support a unique reaction mechanism where Srx functions as an ATP carrier and facilitator of the unfolding of the Prx active site. This process enables the sulfinic acid moiety to attack the γ-phosphate of ATP, leading to the sequential formation of the sulfinic phosphoryl ester and thiosulfinate intermediate with Srx. Collapse of the thiosulfinate can be mediated by Srx, when GSH is not included in the reaction and the Cys-SP residue of Prx is mutated. Therefore, the contribution of these potentially important thiols to the Srx retroreduction reaction and hydrogen peroxide-mediated cell signaling will ultimately need to be evaluated. Combined kinetics and mass spectrometry methodologies are poised to answer these questions and those of other reactions with novel reaction intermediates and chemistry that have eluded conventional methods of analysis.
Note Added in Proof—Recent studies of the yeast Srx/Prx system have also led to the identification of the Prx-Cys-S(=O)-S-Cys-Srx thiosulfinate catalytic intermediate (25).
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM072866 (to W. T. L.). This work was also supported by American Heart Association Postdoctoral Fellowship 0725399U (to T. J. J.) and American Heart Association Grant SDG 0730069N (to C. M. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Prx, peroxiredoxin; Srx, sulfiredoxin; h, human; Csd, Cys sulfinic acid; MS, mass spectrometry; ESI-TOF-MS, electrospray ionization time-of-flight MS; DTT, dithiothreitol; WT, wild type.
References
- 1.Giles, G. I., and Jacob, C. (2002) Biol. Chem. 383 375–388 [DOI] [PubMed] [Google Scholar]
- 2.Jacob, C. (2006) Nat. Prod. Rep. 23 851–863 [DOI] [PubMed] [Google Scholar]
- 3.Sevier, C. S., and Kaiser, C. A. (2006) Antioxid. Redox Signal. 8 797–811 [DOI] [PubMed] [Google Scholar]
- 4.Toledano, M. B., Kumar, C., Le Moan, N., Spector, D., and Tacnet, F. (2007) FEBS Lett. 581 3598–3607 [DOI] [PubMed] [Google Scholar]
- 5.Ellis, H. R., and Poole, L. B. (1997) Biochemistry 36 15013–15018 [DOI] [PubMed] [Google Scholar]
- 6.Wood, Z. A., Schröder, E., Harris, J. R., and Poole, L. B. (2003) Trends Biochem. Sci. 28 32–40 [DOI] [PubMed] [Google Scholar]
- 7.Wood, Z. A., Poole, L. B., and Karplus, P. A. (2003) Science 300 650–653 [DOI] [PubMed] [Google Scholar]
- 8.Phalen, T. J., Weirather, K., Deming, P. B., Anathy, V., Howe, A. K., van der Vliet, A., Jönsson, T. J., Poole, L. B., and Heintz, N. H. (2006) J. Cell Biol. 175 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bozonet, S. M., Findlay, V. J., Day, A. M., Cameron, J., Veal, E. A., and Morgan, B. A. (2005) J. Biol. Chem. 280 23319–23327 [DOI] [PubMed] [Google Scholar]
- 10.Vivancos, A. P., Castillo, E. A., Biteau, B., Nicot, C., Ayte, J., Toledano, M. B., and Hidalgo, E. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 8875–8880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jönsson, T. J., Murray, M. S., Johnson, L. C., and Lowther, W. T. (2008) J. Biol. Chem. 10.1074/jbc.M803244200 [DOI] [PMC free article] [PubMed]
- 12.Jeong, W., Park, S. J., Chang, T. S., Lee, D. Y., and Rhee, S. G. (2006) J. Biol. Chem. 281 14400–14407 [DOI] [PubMed] [Google Scholar]
- 13.Biteau, B., Labarre, J., and Toledano, M. B. (2003) Nature 425 980–984 [DOI] [PubMed] [Google Scholar]
- 14.Bothner, B., Chavez, R., Wei, J., Strupp, C., Phung, Q., Schneemann, A., and Siuzdak, G. (2000) J. Biol. Chem. 275 13455–13459 [DOI] [PubMed] [Google Scholar]
- 15.Furdui, C. M., Lew, E., Schlessinger, J., and Anderson, K. S. (2006) Mol. Cell 17 1–7 [DOI] [PubMed] [Google Scholar]
- 16.Gross, J. W., Hegeman, A. D., Vestling, M. M., and Frey, P. A. (2000) Biochemistry 39 13633–13640 [DOI] [PubMed] [Google Scholar]
- 17.Konermann, L., Collings, B. A., and Douglas, D. J. (1997) Biochemistry 36 5554–5559 [DOI] [PubMed] [Google Scholar]
- 18.Paiva, A., Tilton, R. F., Crooks, G. C., Liang, P.-H., and Anderson, K. S. (1997) Biochemistry 36 15472–15476 [DOI] [PubMed] [Google Scholar]
- 19.Li, Z., Sau, A., Shen, S., Whitehouse, C., Baasov, T., and Anderson, K. S. (2003) J. Am. Chem. Soc. 125 9938–9939 [DOI] [PubMed] [Google Scholar]
- 20.Li, Z., Sau, A. K., Furdui, C. M., and Anderson, K. S. (2005) Anal. Biochem. 343 35–47 [DOI] [PubMed] [Google Scholar]
- 21.Jönsson, T. J., Murray, M. S., Johnson, L. C., Poole, L. B., and Lowther, W. T. (2005) Biochemistry 44 8634–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jönsson, T. J., Johnson, L. C., and Lowther, W. T. (2008) Nature 451 98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kice, J. L., and Rogers, T. E. (1974) J. Am. Chem. Soc. 96 8015–8019 [Google Scholar]
- 24.Shen, C., Xiao, H., and Parkin, K. L. (2002) J. Agric. Food Chem. 50 2644–2651 [DOI] [PubMed] [Google Scholar]
- 25.Roussel, X., Bechade, G., Kriznik, A., Van Dorsselaer, A., Sanglier-Cianferani, S., Branlant, G., and Rahuel-Clermont, S. (2008) J. Biol. Chem. 10.1074/jbc.M800493200. [DOI] [PubMed]