

Abstract
Heterogeneities in cell membranes due to the ordering of lipids and proteins are thought to play an important role in enabling protein and lipid trafficking throughout the secretory pathway and in maintaining cell polarization. Protein-coated vesicles provide a major mechanism for intracellular transport of select cargo, which may be sorted into lipid microdomains; however, the mechanisms and physical constraints for lipid sorting by protein coats are relatively unexplored. We studied the influence of membrane-tethered protein coats on the sorting, morphology, and phase behavior of liquid-ordered lipid domains in a model system of giant unilamellar vesicles composed of dioleoylphosphatidylcholine, sphingomyelin, and cholesterol. We created protein-coated membranes by forming giant unilamellar vesicles containing a small amount of biotinylated lipid, thereby creating binding sites for streptavidin and avidin proteins in solution. We found that individual tethered proteins colocalize with the liquid-disordered phase, whereas ordered protein domains on the membrane surface colocalize with the liquid-ordered phase. These observations may be explained by considering the thermodynamics of this coupled system, which maximizes its entropy by cosegregating ordered protein and lipid domains. In addition, protein ordering inhibits lipid domain rearrangement and modifies the morphology and miscibility transition temperature of the membrane, most dramatically near the critical point in the membrane phase diagram. This observation suggests that liquid-ordered domains are stabilized by contact with ordered protein domains; it also hints at an approach to the stabilization of lipid microdomains by cross-linked protein clusters or ordered protein coats.
INTRODUCTION
Cell membranes are complex materials, with hundreds of species of lipids and proteins forming heterogeneous and dynamic barriers for the cell and its organelles. Among the important features of these membranes is their ability to maintain distinct resident lipid and protein compositions, whereas molecules are continuously trafficked via protein-coated vesicles between the endoplasmic reticulum, the Golgi complex, and the plasma membrane. For example, Golgi enzymes must modify cargo proteins in transit to the plasma membrane. These enzymes, however, are retained in the Golgi, which has a lipid composition distinct from that of the plasma membrane. Thus, a mechanism is required to selectively sort lipids and proteins based on their destinations. One possibility is that protein coats, such as COPI, COPII, caveolae, and clathrin (1), cooperatively sort with lipid microdomains or lipid rafts, which are lateral domains enriched in sphingolipids, cholesterol, and signaling proteins (2). Model lipid bilayer membranes composed of saturated sphingolipids, unsaturated lipids, and cholesterol mimic some key properties of cell membranes, including the formation of liquid-ordered phases enriched in sphingolipids and cholesterol that are similar to lipid rafts (3,4). Although lipid rafts are difficult to observe in cell membranes due to their small size (10–100 nm) (5), larger domains form in model membranes such as supported lipid bilayers and vesicles (3,6,7). Giant unilamellar vesicles (GUVs) are attractive for studying the impact of membrane composition on the physical properties of domains via direct visualization of membrane phases and curvature (6,8). The physical chemistry of model membranes has provided insight into mechanisms for segregating and sorting lipids, and model membranes with controllable lipid composition remain an important testing ground for biologic functions that require membrane heterogeneity.
Lipid rafts are believed to play a critical role in sorting and concentrating proteins for cellular signaling and intracellular trafficking because select proteins have higher affinities for certain membrane environments (9). The physical characteristics that determine protein association with lipid environments may be based on the compatibility of their hydrophobic domains. This compatibility can be derived from matching between membrane thickness and transmembrane protein length (10,11), the shared physicochemical characteristics of lipids and lipidated protein tails (12), preferred curvatures (13), or chemical affinity (14). However, the influence of the cross-linking or complexation between membrane-associated proteins on membrane structure remains relatively unexplored (3,15), although it has been hypothesized that protein-lipid interactions, rather than lipid-lipid interactions, may control the organization of lipid rafts (16). Cross-linking in the plasma membrane occurs during receptor-ligand binding, the first step in the signaling cascade, and has the potential to dramatically influence membrane structure, perhaps stabilizing or enlarging protein-associated membrane domains (10,16–18). It has recently been demonstrated that the actin cytoskeleton can control the formation of membrane domains (19). Despite the likely importance of protein coats and complexes in determining membrane function, there is a dearth of experimental evidence from model protein-lipid bilayer membrane systems with which to explore the reciprocal influences of protein and lipid organization on molecular trafficking, phase behavior, and domain morphology.
In this article, we used model lipid bilayer membranes to investigate the sorting behavior of membrane-tethered protein coats and the influence of protein complexes on membrane phases. We synthesized GUVs containing a small amount of biotinylated lipid, which acts as an anchor for avidin and streptavidin proteins. Although unlabeled streptavidin interacts laterally to form two-dimensional ordered protein domains, avidin and labeled streptavidin do not form domains (20). This finding provides a physical model for other laterally interacting membrane-anchored proteins, such as glycosylphosphatidylinisotol (GPI)-anchored proteins, which are known to associate with lipid rafts (2). By using fluorescent microscopy imaging, we found that avidin and labeled streptavidin preferentially partition into the liquid-disordered (Ld) phase; however, ordered streptavidin protein domains colocalize with the “raft-like”, liquid-ordered (Lo) phase. In addition, protein ordering inhibits lipid domain rearrangement and modifies the morphology and miscibility transition temperatures of the membrane phases. These observations suggest that the thermodynamics of this coupled system can play an important role in determining its phase behavior and structure. Thus, we suggest an additional physical mechanism for protein sorting and further underline the essential role of proteins in determining domain morphology in cellular membranes.
MATERIALS AND METHODS
We prepared vesicles from a lipid mixture of cholesterol, sphingomyelin (SM), dioleoylphosphatidylcholine (DOPC), Texas Red dihexanoylphosphoethanolamine (TR-DHPE), and either biotinyl-dioleoylphosphatidylethanolamine (DOPE) or biotin-X-dihexanoylphosphoethanolamine (DHPE). Cholesterol, egg SM, brain SM (BSM), DOPC, and biotinyl-DOPE were purchased from Avanti Polar Lipids (Alabster, AL). TR-DHPE and biotin-X-DHPE were purchased from Invitrogen (Carlsbad, CA). All images and data accompanying this article are from mixtures containing BSM, cholesterol, and DOPC. In addition, we confirmed our protein partitioning results for the data points shown in Fig. 5 B with mixtures containing egg SM, cholesterol, and DOPC.
FIGURE 5.

(A) Effect of protein crystals on miscibility transition temperatures for membrane compositions in the ternary phase diagram. (B) Transition temperatures for a constant SM/cholesterol ratio of 1.5:1 indicated by the orange line in A, and (C) for a constant SM/DOPC ratio of 1:1 indicated by the blue line in A. Squares indicate liquid-liquid miscibility for bare vesicles. Circles indicate liquid gel miscibility for bare vesicles. Triangles indicate miscibility for protein-coated vesicles. The crosshatched region indicates no phase separation observed for protein-coated vesicles down to 10°C. The striped region indicates no phase melting observed for protein-coated vesicles up to 50°C. Error bars reflect the range of temperatures over which a transition is observed. (D) Images of membrane under Texas Red lipid probe illumination (left, red) and FITC-avidin protein probe illumination (right, green) in striped region at 30 mol% cholesterol, 40°C. Scale bar is 5 μm.
Membrane compositions were selected to lie along two lines in the ternary phase space defined by cholesterol, SM, and DOPC. One set of compositions was positioned approximately along a tie line defined by a 1.5:1 molar ratio of cholesterol/SM, and the other was approximately along a critical line defined by a 1:1 molar ratio of DOPC/SM (21,22). Concentrations of TR-DHPE and biotin-X-DHPE were maintained at 0.1 mol% and 5 mol%, respectively. All lipids were diluted in chloroform to a concentration of 5 mg/ml. All compositions cited refer to lipid solutions used to form the GUVs; the actual composition probably varies slightly between vesicles (23).
GUVs were prepared from a dried lipid film by electroformation (24,25). Lipid solutions were spread onto two indium tin oxide coated glass slides and then dried under vacuum for at least 30 min. The glass slides were assembled in parallel in a Teflon holder, filled with a 600 mM sucrose solution, and immersed in a circulating water bath at 50°C. GUVs were formed in an alternating current electric field at 10 Hz at 1 V for 1–2 h and then diluted at room temperature in a 600 mM glucose solution to induce the vesicles to sediment.
Streptavidin in lyophilized form and FITC-avidin in phosphate-buffered solution were purchased from Zymed (now Invitrogen). To examine the lipid phase preference of proteins, streptavidin and avidin were added to vesicles in a 1:1 molar ratio for a final concentration of 25 μg/ml, in excess of the amount required to fully coat all vesicles. For control experiments to test the partitioning of avidin alone, FITC-avidin was added to vesicle solutions for a final concentration of 25 μg/ml.
Fluorescence imaging
We used an inverted fluorescence microscope (Nikon Diaphot; Nikon, Melville, NY) with a 100× oil objective, equipped with a digital camera (Sensicam QE; Cooke, Romulus, MI), and an inverted confocal microscope (Zeiss 510; Carl Zeiss, Thornwood, NY) with a 60× oil objective, for imaging vesicles. Samples were sealed between a coverslip and a microwell with a 0.5 mm spacer (PC1R-0.5; Grace Bio-Labs, Bend, OR). Membrane and protein phases were visualized using two fluorescent probes with distinct excitation and emission spectra. The fluorescent lipid probe, TR-DHPE, was excited at 547 nm, and its emission was collected with a high-pass filter at 587 nm. It has been widely demonstrated that, over our range of lipid compositions, this probe preferentially partitions into the Ld phase, leaving the Lo phase dark (26). Streptavidin crystallizes to the exclusion of avidin and thus can be visualized as dark regions in contrast to fluorescently labeled FITC-avidin (20), excited with a bandpass filter (465–495 nm) with emission collected using a bandpass filter (515–555 nm).
Miscibility transition temperature measurements
Samples were placed into a temperature-controlled microscope stage; in addition, a collar heater was used to maintain the temperature of the objective. Beginning at a temperature of 25°C, the temperature was raised 5°C every 10 min until all vesicles appeared to be uniform. The temperature was then lowered 2°C every 10 min until vesicles showed the onset of phase separation; this defined the miscibility transition temperature Tm. Sample sizes of least 20 vesicles were used, and error bars reflect the range of temperatures over which a transition was observed. Temperatures probed were maintained between 10°C and 50°C to prevent the samples from freezing or the proteins from denaturing. Temperatures were calibrated from separate measurements using a thermocouple probe on an aqueous sample.
RESULTS AND DISCUSSION
Phase partitioning of biotinylated lipids
To investigate the effects of membrane-tethered proteins on membrane phase morphology and partitioning, we prepared vesicles composed of cholesterol, SM, DOPC, and 5 mol% biotin-X-DHPE or biotinyl-DOPE. Domains in the Lo phase enriched in SM and cholesterol appeared dark, whereas the Ld phase enriched in DOPC appeared bright (26). Vesicles displayed round domains as shown in Fig. 1, upper left, and Fig. 3, A–C. The area fraction of the Ld phase increased as the relative amount of DOPC was increased. Miscibility transition temperatures and domain morphology were similar to those of vesicles prepared from a mixture of cholesterol, SM, and DOPC without biotinylated lipid (22), confirming that the biotinylated lipid does not strongly perturb the phase characteristics of this model system.
FIGURE 1.

Phase-separated membrane (left) composed of DOPC/BSM/cholesterol in ratios of 70:12:8. Ld phase is indicated by lipid probe. Biotinylated lipid binds FITC-avidin (right), and partitions into the Ld phase. Scale bar is 10 μm.
FIGURE 3.

Effect of protein crystals on morphology of lipid phases for different lipid compositions in the following ratios of DOPC/BSM/cholesterol: (A, D, and G) 30:42:28, (B, E, and H) 50:30:20, (C, F, and I) 70:18:12. The first row (A–C) shows bare vesicles under Texas Red lipid probe illumination with characteristically round lipid domains. The middle row (D–F) shows the excitation of FITC-avidin, highlighting the streptavidin domains (dark) on the same vesicles shown under Texas Red lipid probe illumination in the right column (G–I). Scale bars are 20 μm.
Biotin strongly binds avidin and streptavidin; thus, these proteins form a surface coat when added to a solution containing GUVs. In the case of biotin-X-DHPE, biotin is attached to the same saturated lipid as the fluorescent probe TR-DHPE and similarly creates a steric repulsion due to its biotin-conjugated headgroup. Therefore, we can expect that it should preferentially partition into the Ld phase. In the case of biotinyl-DOPE, biotin is attached to an unsaturated lipid with no spacer and should partition into the Ld phase (23). Indeed, vesicles coated with either 100% fluorescently labeled avidin or 100% fluorescently labeled streptavidin (data not shown), neither of which forms two-dimensional crystals, confirmed this preference, as shown in Fig. 1 for FITC-avidin. We estimated a partition coefficient for biotin-X-DHPE by assuming that the fluorescence intensity of FITC-avidin, corrected by subtracting the background intensity, is directly proportional to the concentration of biotinylated lipid in each phase. This yields a partition coefficient of K = 0.17 ± 0.043, corresponding to a free energy of transfer from the Ld to the Lo phase of ɛ =1.78 ± 0.28 kBT per molecule, or 1.05 ± 0.15 kcal/mol. This is comparable to values found for DOPC (27).
Effect of lipid phases on growth of two-dimensional streptavidin crystals
We have shown that, upon binding biotin, streptavidin can interact with neighboring molecules to crystallize in two dimensions on the surface of a GUV (20). We tested the effect of lipid domains on protein crystallization by adding streptavidin and FITC-avidin in a 1:1 ratio to phase-separated GUVs (cholesterol/SM/DOPC in ratios of 20:30:50) containing 5 mol% biotinyl-DOPE. Within 10 min, streptavidin-enriched domains were clearly visible as dark regions within the avidin-enriched Ld phase, whereas lipid phases appeared unperturbed, as shown in Fig. 2 A. We are unable to conclude from this data whether domains also form within the Lo phase. Surprisingly, after ∼1 hour, streptavidin domains were found on many GUVs at boundaries between the Lo and Ld phases. This led to a change in the initially circular morphology of the lipid phases, which became deformed and took on a ragged appearance with sharp edges, as shown in Fig. 2 B. At even later times, protein domains were no longer visible within the Ld phase on a subset of GUVs, but the phase boundaries remained perturbed, as shown in Fig. 2 C.
FIGURE 2.

Lipid partitioning as shown by a TR-DHPE probe (left) and protein organization as shown by FITC-avidin (middle) for different GUVs composed of DOPC/BSM/cholesterol in ratios of 50:30:20 and 5% biotinyl-DOPE (A) after ∼10 min, (B) after ∼60 min, and (C) after ∼90 min. Yellow arrows indicate protein crystals in the Ld phase. Scale bars are 10 μm.
The physicochemical characteristics of lipids strongly influence their intermolecular interactions and phase behavior (23). To probe these effects, we repeated the crystallization experiment using phase-separated GUVs (cholesterol/SM/DOPC 20:30:50) containing 5 mol% biotin-X-DHPE. Within the time required to mix the proteins with GUVs and begin imaging (∼10 min), the lipid phases had already rearranged to form irregular domains with sharp edges. This change in morphology was similar to the changes observed at later times in GUVs formed with biotinyl-DOPE in Fig. 2 C. The observations of the disappearance of streptavidin crystals accompanied by the change in lipid domain morphology suggest that the crystals preferentially form in or translocate into the Lo phase, a point we will demonstrate in greater detail below. Because the saturated lipid DHPE preferentially partitions into the Lo phase, we hypothesize that translocation of streptavidin crystals may occur more rapidly than in the case of the unsaturated DOPE, whose lipid tails should not show preferential partitioning into the Lo phase. Alternatively, if a large enough fraction of biotin-X-DHPE were to partition into the Lo phase, streptavidin crystals might be able to form there.
Streptavidin crystallization on GUVs of varied lipid composition (along an approximate tie line)
To explore the relationship between lipid and protein phases, we examined their morphologies as revealed by their fluorescent probes for GUVs composed of different lipid mixtures. We chose lipid compositions to lie approximately along a tie line, defined by a 1.5:1 molar ratio of cholesterol/SM, with 5 mol% biotin-X-DHPE to enable protein binding. As expected, the area fraction of the Ld phase increased as the relative amount of DOPC was increased. This is illustrated in Fig. 3, A–C, in a control experiment with no protein present.
Streptavidin and FITC-avidin were added in a 1:1 ratio to phase-separated GUVs. In Fig. 3 D, streptavidin crystals appear dark against the bright FITC-avidin and display an X-shaped morphology, which is characteristic of transport-limited growth (20). Both streptavidin and avidin are ∼5 nm in size, and each has four biotin-binding sites; however, only two are simultaneously available to the membrane surface (28). Thus, ∼2 of every 40 lipids were bound to a single protein, as shown schematically in Fig. 4. The protein should only interact directly with the two bound lipids because biotin is attached to lipid headgroups with a spacer, leaving a ∼1 nm gap between the bound protein and the lipid headgroups (Fig. 4, blue).
FIGURE 4.

Schematic illustration of the upper leaflet of a protein-coated lipid bilayer membrane. Green represents fluorescently labeled avidin; black represents crystallizable streptavidin; blue represents biotin groups; red represents DOPC; light gray represents SM; and dark gray represents cholesterol. There are ∼40 lipids in the area under a single streptavidin or avidin molecule. Scales are approximate. The lipid bilayer is ∼5 nm thick, whereas streptavidin and avidin are ∼4 nm in height. Partitioning of lipid is simplified in this schematic because lipid composition in coexisting phases is determined by the endpoints of tie lines for any initial composition. The degree of partitioning of protein depends on the relative area fraction of lipid and protein phases.
Surprisingly, this small degree of tethering can give rise to dramatic effects when coupled with the long-range ordering provided by streptavidin crystals. Bare vesicles displayed the characteristic round domain morphology, which has been shown to minimize the free energy of the system (6), as shown in Fig. 3, A–C. We found that streptavidin crystals colocalized with Lo domains in lipid membranes as illustrated in Fig. 3, where images are shown of the protein coat with dark crystals (D–F) and of the lipid domains in the same vesicles (G–I). These findings were in striking contrast to the preference of the noncrystalline protein for the Ld phase illustrated in Fig. 1. Furthermore, the morphologies of both the membrane domains and the crystals were modified. The morphology of the Lo domains were no longer round; instead they took on the shape of the streptavidin crystals, as seen most clearly in Fig. 3 G, and crystalline regions could reflect the original, round shape of membrane domains as shown in Fig. 3 E.
Possible mechanisms for the sorting of streptavidin crystals
Several mechanisms have been proposed for the sorting of molecules into lipid domains: 1), hydrophobic matching (10,11); 2), the shared physicochemical characteristics of lipids and lipidated protein tails (12); 3), curvature (13); and 4) chemical affinity (14). All of these mechanisms are based on a reduction of the free energy of the system as a result of molecular sorting. Hydrophobic matching is relevant for transmembrane proteins, which have a hydrophobic domain inserted into the membrane. If this hydrophobic length differs from the lipid bilayer thickness, the bilayer and/or protein must deform to accommodate the mismatch. As a result, proteins should partition into lipid environments that match their hydrophobic length, an expectation corroborated by many proteins in the secretory pathway (11). Because streptavidin is membrane-tethered, hydrophobic matching is unlikely to play an important role. Similarly, although it is likely that different lipid phases are characterized by distinct bending energies (29), it has been demonstrated that these differences are small in GUVs formed from model lipid mixtures, so that line energies are the determinant of membrane curvature (6). Finally, we used a biotinylated lipid that should partition preferentially into the Ld phase base on its physicochemical characteristics, and we demonstrated that noncrystallizing avidin bound to the biotinylated lipid remains in the Ld phase, thereby ruling out chemical affinity.
Because we observed sorting only for ordered streptavidin crystals and not individual avidin molecules, we hypothesize that ordering may be an important cue for colocalization. A similar observation has been made for the ganglioside GM1, which, in its monomeric form, partitions preferentially into the Ld phase. Upon binding cholera toxin subunit B, GM1 forms pentamers that repartition into the Lo phase (8,30). Similarly, at higher concentrations of GM1, where clusters can form even in the absence of cholera toxin subunit B, GM1 partitions preferentially into the Lo phase (31). This translocation is sensitive to the chemistry of the lipid tails, because a synthetic GM1 lacking a saturated tail remains in the Ld phase (8). This effect has been observed for a phospholipid analog system as well, where the cross-linking of a saturated phospholipid analog resulted in a fourfold increase in partitioning into the Lo phase, whereas cross-linking an unsaturated phospholipid analog had little effect (32). Sorting of GPI-anchored proteins in polarized Madin-Darby canine kidney cells has also been shown to depend on their oligomerization (33).
We propose that the sorting of protein into the Lo phase upon crystallization, as well as the change in domain morphology, can be described by simple thermodynamic considerations. In the case of a bare membrane, the partitioning of lipid into Lo and Ld phases is a result of a minimization of the free energy, which can be written as a sum of contributions from enthalpic and entropic terms. Enthalpic terms account for intermolecular interactions, whereas entropic terms account for lipid ordering and partitioning (34). A complete quantitative model is precluded by our lack of knowledge of the form of the interaction potential; however, we can estimate the difference in free energy that arises when streptavidin-enriched domains translocate from the Ld to the Lo phase. As a first approximation, there is an increase in energy due to the transfer of lipid, as calculated above, and a decrease in energy due to the increased entropy of the avidin, which can explore the area previously occupied by the streptavidin. For a streptavidin domain containing NS molecules, this results in the following: 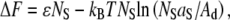 where aS is the area per streptavidin molecule, and Ad is the area of the disordered lipid phase. Because the energy cost ɛ is greater than kBT, this quantity is negative only when the total area of the streptavidin molecules exceeds that of the disordered lipid phase, but this requirement is not met for many of the lipid compositions used. However, we have neglected the effect of the ordering of the lipids bound to the streptavidin array in this calculation. We can argue qualitatively that, although lipid-lipid interactions are largely unchanged by protein crystallization, it is entropically favorable for lipids bound to a crystalline array of proteins to partition into the relatively ordered Lo phase. The entropy of the Ld phase is higher than that of the Lo phase, so that there should be a greater entropic penalty for lipids tethered to and immobilized by protein crystals to associate with the Ld phase. There are ∼40 lipids under each protein molecule, and two of them are tethered to each protein via biotin. This large difference in scale between the lattice spacing for the lipids and proteins should mean a low cost in energy for the two lattices to accommodate each other. A similar argument has been suggested to explain protein folding and assembly on membranes, where the entropy of the lipid phases and the proteins must be taken into account (35,36).
where aS is the area per streptavidin molecule, and Ad is the area of the disordered lipid phase. Because the energy cost ɛ is greater than kBT, this quantity is negative only when the total area of the streptavidin molecules exceeds that of the disordered lipid phase, but this requirement is not met for many of the lipid compositions used. However, we have neglected the effect of the ordering of the lipids bound to the streptavidin array in this calculation. We can argue qualitatively that, although lipid-lipid interactions are largely unchanged by protein crystallization, it is entropically favorable for lipids bound to a crystalline array of proteins to partition into the relatively ordered Lo phase. The entropy of the Ld phase is higher than that of the Lo phase, so that there should be a greater entropic penalty for lipids tethered to and immobilized by protein crystals to associate with the Ld phase. There are ∼40 lipids under each protein molecule, and two of them are tethered to each protein via biotin. This large difference in scale between the lattice spacing for the lipids and proteins should mean a low cost in energy for the two lattices to accommodate each other. A similar argument has been suggested to explain protein folding and assembly on membranes, where the entropy of the lipid phases and the proteins must be taken into account (35,36).
The dramatic change in the morphology of Lo domains shown in Fig. 3, G–I, which typically take on a rounded shape to minimize line energy (6), suggests another effect of the protein crystals on the lipid phases. Protein crystals pin bound lipids into an oriented lattice with long-range order, which may act as a template to promote ordering in the underlying lipid phases. Thus, the presence of such a coupled template may result in the transformation of the Lo phase into a phase with greater long-range order that no longer relaxes due to line tension. This hypothesis is confirmed by direct observation of the extremely slow dynamics of lipid phases in the presence of protein crystals that appear pinned in comparison with domains on bare membranes, which can be seen to diffuse readily (data not shown). The thin, elongated features connecting larger Lo domains as shown in Fig. 3 G further support the hypothesis that lipid phases are transformed by the presence of the crystal.
Effects of protein ordering on phase boundaries
We measured the lipid miscibility transitions for ternary lipid mixtures along two lines through phase space, approximating a tie line and a critical line for liquid-liquid phase coexistence, as shown in Fig. 5 A. We measured Tm for bare vesicles and for vesicles with a protein coat. This method enabled us to compare our measurements with published data on vesicles without biotinylated lipid (22) and to isolate the effect of the protein on phase behavior.
Phase boundaries for the bare vesicles are not measurably changed by the addition of the biotinylated lipid at the 5 mol% concentrations that we used. We also found that Tm was unchanged for vesicles coated with only noncrystallizing FITC-avidin. The presence of ordered protein domains also had little effect on lipid miscibility transition temperatures for compositions along the tie line, as shown in Fig. 5 B; however, they had a dramatic effect on the phase boundaries near critical points, as shown in Fig. 5 C. In particular, Lo phases with compositions approaching cholesterol/SM/DOPC in ratios of 1:1:1 become immiscible in the presence of protein crystals. One example of lipid domains that were stabilized by the presence of protein crystals is shown in Fig. 5 D. In this region of phase space, Tm was changed by up to 25°C in response to the presence of protein crystals. In contrast, lipid domains can also be destabilized, as in the case of gel phases at low cholesterol concentrations, which fail to appear in the presence of protein crystals. These results confirm our expectation that constraints on membranes should have the greatest impact near critical points, where the line energy between phases is at a minimum. This low line tension can lead to interesting effects such as fingering or stripe phases (23).
Interestingly, phase separation is not induced in uniform GUVs by protein crystals, unlike in the case of cross-linking GM1 gangliosides (30). This result suggests that, although these protein domains may stabilize lipid domains, they do not create lipid domains. These observations confirm that phase-separating membranes near critical points are highly sensitive to perturbation; this is an essential component of the raft hypothesis because most biologic membranes are composed of mixtures of saturated and unsaturated lipids and cholesterol that place them near critical points according to model membrane studies (3).
Implications
Our data demonstrate that streptavidin crystallization on the surface of lipid bilayers can alter the spatial distribution of the lipids in the bilayer. We attribute this to an entropic effect, whereby the overall free energy of the system is minimized when ordered protein domains are colocalized with ordered lipid domains. Entropic sorting in conjunction with lipid-lipid interactions may thus provide a mechanism for the cooperative sorting of protein coats and lipids, an important aspect of intracellular trafficking.
Most membrane-associated proteins form nanoscopic complexes; thus, it is difficult to study their effect on the local properties of the membrane. Here, we have taken advantage of large-scale complexes in the form of streptavidin crystals, which allowed us to directly visualize their influence on membrane phases. We show that protein ordering can have a dramatic effect on the membrane phase behavior, possibly creating more ordered lipid phases or inducing longer ranged order by providing a molecular template. Protein ordering can have the added effect of stabilizing raft phases, particularly in the vicinity of a critical point. Protein coats play an important role in the trafficking of lipids and proteins in the secretory pathway. Streptavidin- and avidin-coated GUVs provide a model system that may provide insight into the mechanisms for sorting by protein coats.
Acknowledgments
We thank Benny Davidovich, David R. Nelson, Jonathan Matchta, and Howard A. Stone for useful theoretical discussions, and Vernita D. Gordon, Paulina A. Achurra, and Jennifer McManus for helpful comments.
Alice P. Gast's present address is the Office of the President, Lehigh University, 27 Memorial Dr. West, Bethlehem, PA 18015.
Editor: David D. Thomas.
References
- 1.Bonifacino, J. S., and J. Lippincott-Schwartz. 2003. Coat proteins: shaping membrane transport. Nat. Rev. Mol. Cell Biol. 4:409–413. [DOI] [PubMed] [Google Scholar]
- 2.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 3.Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simons, K., and W. L. Vaz. 2004. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 33:269–295. [DOI] [PubMed] [Google Scholar]
- 5.Pralle, A., P. Keller, E. L. Florin, K. Simons, and J. Horber. 2000. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J. Cell Biol. 148:997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumgart, T., S. T. Hess, and W. W. Webb. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425:821–824. [DOI] [PubMed] [Google Scholar]
- 7.Keller, S. L. 2002. Coexisting lipid phases in lipid monolayers and bilayers. J. Phys. Condens. Matter. 14:4763–4766. [Google Scholar]
- 8.Bacia, K., P. Schwille, and T. V. Kurzchalia. 2005. Sterol structure determines the separation of phases and the curvature of the liquid-ordered phase in model membranes. Proc. Natl. Acad. Sci. USA. 102:3272–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silvius, J. R. 2005. Partitioning of membrane molecules between raft and non-raft domains: insights from model-membrane studies. Biochim. Biophys. Acta. 1746:193–202. [DOI] [PubMed] [Google Scholar]
- 10.Engelman, D. M. 2005. Membranes are more mosaic than fluid. Nature. 438:578–580. [DOI] [PubMed] [Google Scholar]
- 11.Bretscher, M. S., and S. Munro. 1993. Cholesterol and the Golgi apparatus. Science. 261:1280–1281. [DOI] [PubMed] [Google Scholar]
- 12.Casey, P. 1995. Protein lipidation in cell signalling. Science. 268:221–225. [DOI] [PubMed] [Google Scholar]
- 13.Peter, B. J., H. M. Kent, I. G. Mills, Y. Vallis, P. J. G. Butler, P. R. Evans, and H. T. McMahon. 2004. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 303:495–499. [DOI] [PubMed] [Google Scholar]
- 14.Kundu, A., R. Avalos, C. Sanderson, and D. Nayak. 1996. Transmembrane domain of influenza virus neuraminidase, a type II protein, possesses an apical sorting signal in polarized MDCK cells. J. Virol. 70:6508–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahya, N., D. A. Brown, and P. Schwille. 2005. Raft partitioning and dynamic behavior of human placental alkaline phosphatase in giant unilamellar vesicles. Biochemistry. 44:7479–7489. [DOI] [PubMed] [Google Scholar]
- 16.Edidin, M. 2003. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32:257–283. [DOI] [PubMed] [Google Scholar]
- 17.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 2000. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shroeder, R., E. London, and D. A. Brown. 1994. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc. Natl. Acad. Sci. USA. 91:12130–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, A. P., and D. A. Fletcher. 2006. Actin polymerization serves as a membrane domain switch in model lipid bilayers. Biophys. J. 91:4067–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ratanabanangkoon, P., M. Gropper, R. Merkel, E. Sackmann, and A. P. Gast. 2002. Two-dimensional streptavidin crystals on giant lipid bilayer vesicles. Langmuir. 18:4270–4276. [Google Scholar]
- 21.Veatch, S. L., I. Polozov, K. Gawrisch, and S. L. Keller. 2004. Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophys. J. 86:2910–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veatch, S. L., and S. L. Keller. 2005. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys. Rev. Lett. 94:148101. [DOI] [PubMed] [Google Scholar]
- 23.Veatch, S. L., and S. L. Keller. 2003. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85:3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelova, M. I., F. Soléau, P. Méléard, J. F. Faucon, and P. Bothorel. 1992. Preparation of giant vesicles by external AC electric fields. Kinetics and application. Prog. Colloid Polym. Sci. 89:127–131. [Google Scholar]
- 25.Angelova, M. I., and D. S. Dimitrov. 1986. Liposome electroformation. Faraday Discuss Chem. Soc. 81:303–311. [Google Scholar]
- 26.Knobler, C. 1990. Seeing phenomena in flatland: studies of monolayers by fluorescence microscopy. Science. 249:870–874. [DOI] [PubMed] [Google Scholar]
- 27.McIntosh, T. J., A. Vidal, and S. A. Simon. 2003. Sorting of lipids and transmembrane peptides between detergent-soluble bilayers and detergent-resistant rafts. Biophys. J. 85:1656–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber, P. C., D. H. Ohlendorf, J. J. Wendoloski, and F. R. Salemme. 1989. Structural origins of high-affinity biotin binding to streptavidin. Science. 243:85–88. [DOI] [PubMed] [Google Scholar]
- 29.Parthasarathy, R., C.-h. Yu, and J. T. Groves. 2006. Curvature-modulated phase separation in lipid bilayer membranes. Langmuir. 22:5095–5099. [DOI] [PubMed] [Google Scholar]
- 30.Hammond, A., F. Heberle, T. Baumgart, D. Holowka, B. Baird, and G. W. Feigenson. 2005. Crosslinking a lipid raft component triggers liquid ordered-liquid disordered phase separation in modle plasma membranes. Proc. Natl. Acad. Sci. USA. 102:6320–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan, C., J. Furlong, P. Burgos, and L. J. Johnston. 2002. The size of lipid rafts: an atomic force microscopy study of ganglioside GM1 domains in sphingomyelin/DOPC/cholesterol membranes. Biophys. J. 82:2526–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietrich, C., Z. N. Volovyk, M. Levi, N. L. Thompson, and K. Jacobson. 2001. Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl. Acad. Sci. USA. 98:10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paladino, S., D. Sarnataro, S. Toivodar, and C. Zurzolo. 2007. Oligomerization is a specific requirement for apical sorting of glycosyl-phosphatidylinositol-anchored proteins but not for non-raft associated apical proteins. Traffic. 8:251–258. [DOI] [PubMed] [Google Scholar]
- 34.Radhakrishnan, A., and H. M. McConnell. 2005. Condensed complexes in vesicles containing cholesterol and phospholipids. Proc. Natl. Acad. Sci. USA. 102:12662–12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helms, V. 2002. Attraction within the membrane: forces behind transmembrane protein folding and supramolecular complex assembly. EMBO Rep. 3:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chow, W. S. 1999. Grana formation: entropy-assisted local order in chloroplasts? Aust. J. Plant Physiol. 26:641–647. [Google Scholar]