

Abstract
G-proteins transduce signals along diverse pathways, but the factors involved in pathway selection are largely unknown. Here, we have studied the ability of Gαq to select between two effectors—mammalian inositide-specific phospholipase Cβ (PLCβ) and phosphoinositide-3-kinase (PI3K)—in human embryonic kidney 293 cells. These studies were carried out by measuring interactions between eCFP- and eYFP-tagged proteins using Forster resonance energy transfer in the basal state and during stimulation. Instead of association of Gαq with effectors through diffusion and exchange, we found separate and stable pools of Gαq-PLCβ and Gαq-PI3K complexes existing throughout the stimulation cycle. These separate complexes existed despite the ability of Gαq to simultaneously bind both effectors as determined by in vitro measurements using purified proteins. Preformed G-protein/effector complexes will limit the number of pathways that a given signal will take, which may simplify predictive models.
INTRODUCTION
G-protein-coupled receptors (GPCRs) are the largest family of mammalian transmembrane receptors and are activated by agonists ranging from light to hormones to neurotransmitters (1). Binding of an agonist to its specific GPCR enables the receptor to activate heterotrimeric (GαGβγ) G-proteins by catalyzing the exchange of GTP for GDP on the α-subunit. In principle, G-proteins can receive signals from multiple GPCRs and have the potential to activate multiple pathways. However, most signals only result in activation of a single pathway, and our understanding of the factor(s) causing this selection is limited.
Here, we have studied the ability of the Gαq family heterotrimeric G-proteins to discriminate between two effector pathways: mammalian inositide-specific phospholipase Cβ (PLCβ) and phosphoinositide 3-kinase (PI3K). Gαq-subunits are coupled to receptors that bind agonists such as catecholamines, bradykinin, endothelin-1, prostaglandin F2, and angiotensin II. Their main effector is PLCβ, which catalyzes the hydrolysis of the minor lipid phosphatidylinositol 4,5 bisphosphate (PIP2) to produce second messengers that lead to activation of protein kinase C and an increase in intracellular calcium (2,3). These events in turn result in proliferative and mitogenic changes in the cell. Several types of PLCβ are found in all mammalian cell lines, and all are activated by Gαq. Activation of PLCβs by Gαq involves a large increase in affinity between the two proteins and changes in the nature of their association (4). There are four known PLCβ enzymes (PLCβ1–2) that differ in their tissue distribution, and all are strongly activated by Gαq.
It was recently shown that Gαq has another effector: PI3K. Class I PI3K enzymes phosphorylate PI(4,5)P2 to produce PI(3,4,5)P3, which plays a key role in intracellular vesicle trafficking including transport of glucose transporters to the plasma membrane surface needed for glucose uptake (5–7). Class I PI3K enzymes are heterodimers composed of a regulatory subunit, p85, and a catalytic subunit, p110 (8). There are several subtypes of p85 and p110 but only the p85α/p110α subtype is a Gαq effector. The high correlation between human cancers and mutations in p110α (9) has attracted keen interest in this protein. All PI3K subtypes are ubiquitously expressed and activated by receptor tyrosine kinases (RTKs) in response to stimulation by growth factors (e.g., insulin, epidermal growth factor, insulin growth factor (IGF), etc.). Activation is thought to occur by recruitment of the p85 subunit through binding of its two SH2 domains to the phosphorylated tyrosine residue of the activated RTK. This recruitment brings the entire PI3K in close proximity to the membrane surface and its PI(4,5)P2 substrate. Membrane-bound PI3K then phosphorylates PI(4,5)P2 to produce PI(3,4,5)P3, which then activates a number of downstream pathways that control cell growth and survival. Unlike PLCβ, where GTP-bound Gαq increases its activity several-fold, binding of PI3K to Gαq results in inhibition (10–12).
In a previous study, we characterized the cellular localization and association of Gαq and PLCβ1 in two different cell lines: the rat pheochromocytoma (PC12) and human embryonic kidney 293 (HEK293) cell lines (13). We found that Gαq is localized almost entirely on the plasma membrane, whereas PLCβ1 had a significant cytosolic population and a large plasma membrane population. We also found that the amount of Gαq expressed in these cell lines exceeded PLCβ1. Using Forster resonance energy transfer (FRET), we found that Gαq and PLCβ1 were associated on the plasma membrane in the basal state of both cell types. Activation of Gαq did not increase the observed FRET and did not alter the amount of PLCβ1 in the cytosol, suggesting that the generation of activated Gαq does not drive movement of the cytosolic PLCβ1 population to the plasma membrane. It is therefore possible that this excess Gαq is free to interact with its effector (PI3K) when PI3K is driven from the cytosol to the plasma membrane upon RTK activation.
The presence of two Gαq effectors provides an opportunity to determine how Gαq may discriminate between different effectors under different conditions in living cells. In this study, we used FRET to monitor the localization and Gαq association of PI3K in living cells under basal conditions and stimulation of RTK, GPCR, or both. We found that, like PLCβ1, there is a stable plasma membrane population of preassociated Gαq-PI3K that is distinct from Gαq-PLCβ complexes. Thus, direct competition between the two effectors does not occur; instead, there are different G-protein populations that are dedicated to the two effectors. Interestingly, this population of Gαq-PI3K complexes does not significantly change upon stimulation of either RTKs or GPCRs. We propose that Gαq-PI3K complexes function to preserve the amount of PIP2 substrate available for PLCβ and keep PI3K localized to the plasma membrane and inhibited until displacement of Gαq by activated RTKs.
MATERIALS AND METHODS
Recombinant proteins
Purification of the p85α/p110α complex from baculovirus-infected Sf9 cells was described previously (12). PLCβ2 and Gαq were also purified from an Sf9 expression system (4). eCFP-Gαq and the constitutively active eCFP-Gαq(R183C) were a generous gift from Dr. Catherine Berlot (Geisinger Clinic, Danville, Pennsylvania). The constructs were derived from Gαq-GFP, as described previously (14). The eCFP construct was originally obtained from Clontech (Mountain View, CA). The mouse p110α subunit of PI3 kinase (a generous gift from Dr. Richard Lin, Stony Brook University, Stony Brook, New York) was amplified from the p3XFLAG-CMV-10 vector using polymerase chain reaction and the following primers: forward: CCG GGT ACC ATG CCT CCA CGA CCA; reverse: CGC GGA TCC TCA GTT CAA AGC ATG CTG. It was then inserted into the eYFP-C1 vector between the Kpn1 and BamH1 sites. To create eCFP-p110a we inserted p110a obtained from the previous construct into the eCFP-C1 vector.
Cell culture and transfection
HEK293 and A10 cells were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 50 U/mL of penicillin, and 50 μg/mL streptomycin sulfate at 37°C in a 5% CO2 incubator. C6 cells were cultured in Roswell Park Memorial Institute-1640 medium supplemented with 7.5% FBS, 50 U/mL of penicillin, and 50 μg/mL streptomycin sulfate at 37°C in a 5% CO2 incubator.
C6, A10, and HEK293 cells were transfected using Lipofectamine reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Briefly, cells were grown on 60-mm dishes for 24–48 h to achieve 80–90% confluence. At 1 h prior to transfection, the growth medium was replaced with Opti-MEM (Invitrogen, Eugene, OR) I reduced serum medium. Then, 5–10 μg of DNA diluted with 300 μL Opti-MEM I was mixed with 20 μL of Lipofectamine diluted with 300 μL Opti-MEM I and incubated at room temperature for 30 min to form complexes. The DNA-Lipofectamine complexes were added to the cells, and the cells were returned to the incubator and kept at 37°C for 8–14 h. The Opti-MEM I medium was changed to full-growth medium, and the cells were allowed to recover for 8–14 h. Next, the cells were divided and placed into separate 35-mm glass-bottom culture dishes (MatTek, Ashland, MA) and imaged 48–72 h later. HEK293 cells were also transfected using the calcium phosphate coprecipitation method in which 5–10 μg of plasmid was mixed with 120 mM CaCl2 and HBS buffer (21 mM HEPES, 123 mM NaCl, 5 mM KCl, and 0.9 mM Na2HPO4, pH 7.1), incubated on ice for 10 min, and added to cells maintained in 60-mm dishes. Subsequent steps were identical with the Lipofectamine transfection.
Fluorescence measurements and data analysis
Purified Gαq was stored in a solution containing GDP. To activate Gαq, the protein was incubated for 1 h at 30°C in buffer (50 mM HEPES (pH 7.2), 100 mM (NH4)2SO4, 150 mM MgSO4 and 1 mM EDTA) containing 100 μM GTPγS, followed by dialysis against the same buffer plus 20 mM 2-mercaptoethanol (15). To activate Ras with GTPγS, the protein was incubated with 20 mM HEPES (pH 7.2), 200 μM (NH4)2SO4, 5 mM EDTA, and 5 μM GTPγS for 1 h on ice. The reaction was stopped by adding 20 mM MgSO4 and dialyzed against 20 mM HEPES, 160 mM KCl, 6 mg GDP, and 1 mM 2-mercaptoethanol for 30 min.
Prior to labeling with coumarin (7-(dimethylamino)coumarin-4-acetic acid succinimidyl ester; Molecular Probes, Eugene, OR), Gαq was dialyzed against 20 mM HEPES and 160 mM KCl at pH 7.2 and then at the pH raised to 8.0 with the addition of a small amount of concentrated phosphate buffer before labeling with a fourfold molar excess of coumarin. The reaction mixture was incubated on ice for 1 h; the unreacted probe was then removed by dialysis against buffer at 4°C.
Fluorescence measurements were performed on a spectrofluorometer (ISS, Urbana, IL) using 3-mm pathlength cuvettes. Buffer controls used dialysis buffer (20 mM HEPES, 160 mM KCL, 1 mM DTT, pH 7.2). Samples contained 80 μM of LUV (POPC/POPS/POPE at a 1:1:1 molar ratio) to inhibit protein aggregation. Coumarin-labeled proteins were excited at 340 nM and scanned from 380 to 580 nM. Protein association was determined by the increase in coumarin fluorescence caused by the addition of the unlabeled PI3K at three different initial concentrations of Gαq(GTPγS). Signals were corrected for dilution and compared to the change in fluorescence caused by addition of buffer alone. The titration curves were analyzed as a bimolecular association to obtain the apparent dissociation constant (Kd). The values of Kd for the three initial Gαq concentrations (5, 25, and 50 nM) were within error of each other verify protein-protein interaction.
Immunofluorescence
For colocalization experiments, we first transfected HEK293 cells with p85α and eYFP-p110α. Transfected cells were grown in poly-D-lysine-coated, glass-bottom culture dishes (MatTek) for 48 h. The cells were washed with warm phosphate-buffered saline (PBS) and fixed with 1 mL of 3.7% formaldehyde at room temperature for 15 min. The fixing solution was removed, and the cells were washed three times with PBS. The cells were permeabilized 5 min with 1 mL of 0.2% Nonidet P-40 (Roche, Mannheim, Germany) in PBS. After permeabilization, the cells were blocked in Tris-buffered saline (TBS: 10 mM Tris, 150 mM NaCl, pH 7.2) containing 4% goat serum for 1 h. Cells were incubated with the primary antibody (rabbit anti-PLCβ1; Santa Cruz Biochemicals, Santa Cruz, CA) at 1:500 dilution in TBS containing 1% goat serum at room temperature for 1 h and then washed three times with TBS for 3 min each. Cells were incubated with Alexa647-conjugated antirabbit secondary antibody (Invitrogen, Eugene, OR) (1:500 dilution) in TBS containing 1% goat serum at room temperature for 1 h, and then washed three times with TBS for 3 min each.
In vivo single cell FRET measurements
In vivo FRET experiments were performed using a confocal laser scanning microscope (LSM 510 Meta/Confocor 2 system; Zeiss, Jena, Germany). Filter settings were as follows: 1), eCFP was excited by the 458 nm line of an argon-ion laser, and emission was collected using a 475- to 525-nm bandpass filter; 2), eYFP was excited by the 514 nm line of an argon-ion laser, and emission was collected using a 560- to 615-nm bandpass filter; and 3), for FRET experiments, the sample was excited by the 458 nm line of an argon-ion laser, and emission was collected using a 560- to 615-nm bandpass filter. Bleed-through from eCFP fluorescence into the FRET channel and direct excitation of eYFP by the 458 nm laser line values were estimated from cells transfected with 5 μg of free eCFP or free eYFP plasmids and from cells transfected with eCFP-Gαq and eYFP p110α alone and imaged under the appropriate filter sets. The maximum FRET value was determined from control cells transfected with a construct composed of eCFP and eYFP sandwiched between a 12-aa peptide (13,16).
FRET values were determined as follows:
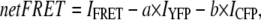 |
where a is the percentage of bleed-through of CFP through FRET filter set and b is the percentage of direct excitation of YFP by 458 nm light. To compare FRET values among cells with varying protein expression levels, we normalized the net FRET values (normalized FRET or NFRET) according to Xia et al. (17) as follows:
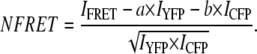 |
Colocalization
Cells were imaged using the multitrack mode of the Zeiss confocal laser scanning microscope system. EYFP was excited with a 514-nm laser line, and emission was measured using the LP530 filter. Alexa 647 was excited with a 633-nm line of an HeNe laser, and the emission spectrum was measured using the LP 650 filter. Filters were obtained from Zeiss; images were analyzed using software from Zeiss.
RESULTS
Localization of Gαq and its effectors in the basal and stimulated states in living cells
We have previously characterized the localization of PLCβ1 and Gαq in PC12 and HEK293 cells (13). In both cell lines, we found that Gαq is almost exclusively on the plasma membrane in the basal and stimulated states, whereas PLCβ1 has a significant cytosolic population and a plasma membrane population. The amount of PLCβ1 in these two cellular compartments did not change upon Gαq activation, showing no net movement to or from the plasma membrane.
PI3K has been found to be localized mainly in the cytosol, although a small plasma membrane fraction and a focal adhesion population has been reported (18). In cells, PI3K has been shown to exist as a tightly bound heterodimer consisting of p110 and p85 subunits (8). To determine the localization of PI3K in different cell lines, we overexpressed eYFP-p110α concomitantly with p85α and monitored the localization in the basal and stimulated states in HEK293, A10, and C6 cells. Because expression of the untagged p85α subunit cannot be visualized, we verified its expression by Western blot analysis. We find that, under our conditions, it is expressed at a level approximately twofold higher than endogenous.
In accordance with previous studies, we found that the overexpressed p85α/eYFP-p110α, which we will refer to as eYFP-PI3K, is mainly cytosolic but also has a small population localized on the plasma membrane (Fig. 1 A). Interestingly, the distribution of the enzyme is very punctuate and similar in appearance to images of some of its protein partners (5), which suggests that PI3K is contained in large domains or protein aggregates. An analysis of the intensity distributions of the eYFPp110α fluorescence coexpressed with p85α along the z axis of the cell shows that the intensity distribution is close to the plasma membranes in HEK293 cells (Fig. 1 C), whereas the intensity distribution is seen internally in C6 glial cells (Fig. 1 D). These varying cell distributions are thought to reflect the dynamic nature of PI3K localization and sharply contrast the more uniform localization and even distributions seen for PLCβ, Gαq, and GPCRs (13,14,16).
FIGURE 1.

Image of a representative HEK293 cell expressing p85α/eYFP-p110α showing its cellular distribution after serum starvation for 24 h (A) and stimulation with 100 ng/mL of IGF-1 for 15 min (B). (C and D) Distribution of the eYFP-p110α intensity along a 3 × 3 pixel point along the z axis in a HEK293 cell and a C6 glial cell where the error is the standard deviation derived from the average of the nine pixels in the 3 × 3 sampling at each point (see Materials and Methods). The integration time is 6.4 μs/pixel.
Cell fractionation studies have suggested that PI3K moves from the cytosol to the plasma membrane by activated RTK to access its PIP2 substrate (19). Additionally, live cell imaging studies that followed movement of GFP-p85α in NIH3T3, A431, and MCG-7 cells have shown redistribution from the cytosol to the plasma membrane upon epidermal growth factor stimulation (18). We monitored p85α/eYFP-p110α expressed in HEK293 and C6 cells upon stimulation with 100 ng/mL IGF-1 (Fig. 1 B). We observed translocation with stimulation, showing that the overexpressed p85α and eYFPp110α are complexed and allow for interactions with activated RTK. We note that the punctuate distribution of PI3K makes it difficult to quantify the overall amount of translocation in the various cell types by image analysis.
FRET studies show that PI3K and Gαq are associated in unstimulated cells
To determine whether the plasma membrane population of PI3K is complexed with Gαq in the basal state, we measured the amount of FRET from eCFP-Gαq to eYFP-PI3K. In accordance with the cellular distribution of the proteins, we found FRET in a punctuate distribution only on the plasma membrane. Fig. 2 shows the raw images through the CFP (A), YFP (B), and FRET (C) channels and the corresponding NFRET image (see Materials and Methods) of a representative cell. The results of several studies are compiled in Table 1. It is surprising that the average amount of FRET between the two proteins is relatively high (in HEK293 cells, the overall NFRET is 0.48 ± 0.07) and on the order of the amount seen for Gαq-PLCβ1 complexes (13). This value can be compared to those obtained for eCFP and eYFP attached by a small peptide linker (80%) and free eCFP and eYFP (10%). Interestingly, the distribution of FRET values between Gαq and PI3K and between Gαq and PLCβ1 differs greatly. In Fig. 3, we compare the distribution of FRET values where we have normalized the sum of the values to 1.0. A majority of the FRET values for PI3K are very low, whereas the remainder is spread over higher values. In contrast, the distribution for Gαq-PLCβ is narrower and suggests more well-defined complexes.
FIGURE 2.

eCFP-Gαq-p85α/eYFPp110α FRET in a HEK293 cell. Image of a representative HEK293 cell as viewed through the CFP filer to image eCFP-Gαq (A), the YFP filer to image eYFP-PI3K (B), and the FRET filter (C). The normalized FRET is shown in panel D (see Materials and Methods for details).
TABLE 1.
Summary of FRET results
| Cell Type | Proteins expressed | Cell state | FRET |
|---|---|---|---|
| HEK293 | eYFP-p110α, eCFP-Gαq | Basal | 0.43 ± 0.02, n = 4 |
| HEK293 | eYFP-p110α, p85α, eCFP-Gαq | Basal | 0.49 ± 0.01, n = 7 |
| HEK293 | eYFP-p110α, p85α, eCFP-GαqRC | Basal | 0.33 ± 0.06, n = 4 |
| HEK293 | eYFP-p110α, p85α, eCFP-Gαq | Basal and carbachol stimulated | 0.45 ± 0.01, n = 5 basal |
| 0.45 ± 0.01, n = 3 with carbachol | |||
| HEK293 | eYFP-p110α, p85α, eCFP-Gαq | Basal and IGF stimulated | 0.40 ± 0.04, n = 5 basal |
| 0.40 ± 0.01, n = 3 with IGF | |||
| C6 | eYFP-p110α, p85α, eCFP-Gαq | Basal | 0.44 ± 0.05, n = 3 |
FIGURE 3.

Distributions of FRET values for Gαq and its effectors. Comparison of the distribution of the magnitude of the FRET values for eCFP-Gαq-eYFP-PLCβ (open squares) and eYFP-Gαq and p85α/eCFP-p110α (solid circles) in HEK293 cells. The FRET values for each complex were summed and normalized to 1.0. Standard deviation is shown.
In previous studies using purified proteins, we found that PI3K binds to activated Gαq with an affinity at least 10-fold stronger than the deactivated form (20). Based on these studies, we expected an increased association between PI3K and activated Gαq. To determine whether the level of FRET changes with Gαq stimulation, we performed two types of studies. In the first series of experiments, we monitored the FRET signal before and after stimulation with the Gαq-coupled agonist carbachol. No significant changes in the magnitude and distribution of FRET were observed. In a second series of experiments, we repeated FRET studies using a constitutively active form of Gαq—Gαq R183C (21). This construct lacked GTPase activity and thus remained in the activated state. Again, no significant differences in FRET between this construct and the wild-type construct were observed, suggesting that Gαq-PI3K association in living cells is independent of the state of activation.
Stimulation of cells with RTK activators, such as IGF-1, results in an increase in the membrane population of PI3K and thus we expected an increase in the plasma membrane-bound PI3K-Gαq complexes. However, the extent of FRET between Gαq and PI3K remained constant upon stimulation with 100 ng/mL IGF-1. This result implies that the membrane binding sites of PI3K are distinct from those where Gαq is localized and that Gαq-PI3K complexes are stable through the activation cycle. To determine whether activation of Gαq together with RTK activation changes the amount of complexation, we simultaneously stimulated the cells with both IGF and carbachol. Again, the values of FRET remained constant. Taken together, these results strongly suggest that there is a constant pool of Gαq-PI3K that remains complexed through various types of cell stimulation.
In vitro purified Gαq binds independently to either PLCβ2 or PI3K
The studies described above show that a population of Gαq is stably complexed with PI3K; also, in previous work (13), we showed that a population of Gαq is stably complexed with PLCβ1. It could be possible that Gαq is associated to both effectors in cells in higher-order complexes. We first tested this idea in vitro using purified proteins. These studies were carried out by labeling purified Gαq with the fluorescence probe coumarin, placing a small amount (1, 5, or 20 nM) of the activated (i.e., GTPγS-bound) protein in a cuvette and measuring the increase in affinity when PI3K (p110α/p85α) binds (for complete details see Ballou et al. (20)). This titration curve was then repeated in presence of PLCβ2 at a concentration ∼2 orders of magnitude higher than its dissociation constant for Gαq (80 nM). The results show that the presence of PLCβ2 has no measurable effect on the binding of PI3K to Gαq (Fig. 4), which suggest that Gαq can bind both effectors simultaneously.
FIGURE 4.

Binding of PI3K to 5 nM Gαq(GTPγS) in the absence and presence of PLCβ. In vitro fluorescence binding assay showing the change in the normalized fluorescence intensity of 5 nM activated CM-Gαq as purified PI3K is added, where the total increase in intensity was ∼18%. Experiments were performed in triplicate; the results are mean ± SE.
Separate pools of Gαq associate with each effector
The observation that Gαq may be capable of binding both effectors in vitro leads to the possibility that ternary complexes may form in cells. However, based on the high amount of FRET between Gαq and each effector that remains unchanged in the basal and stimulated states, we propose that cells contain separate pools of Gαq-PLCβ and Gαq-PI3K complexes. If this is the case, we predict that PI3K and PLCβ should exist in separate regions in the cell. We first tested this idea by measuring the amount of colocalization between PI3K and PLCβ by viewing expressed eYFP-PI3K fluorescence in HEK293 cells and viewing endogenous PLCβ by immunostaining. Colocalization between the two effectors was only seen in very sparse points at adhesion sites (Fig. 5 A), suggesting that colocalization may be due to crowding rather than ternary PI3K-Gαq-PLCβ complexes. We then directly tested for ternary complexes by measuring the ability of eCFP-PI3K to FRET with eYFP-PLCβ in HEK293 cells. The normalized FRET value (0.16 ± 0.02; n =26) was significantly lower than the value obtained for eYFP-Gαq and eCFP-p110α (0.48 ± 0.07; n =115) and close to the value measured for non-interacting proteins (0.10, see Methods). Interestingly, we found FRET from a few pixels in the cell images (Fig. 5 B), suggesting that the two enzymes are not in close proximity; instead, their FRET is due to stochastic diffusion on the plasma member or they interact by virtue of being part of larger membrane protein aggregates.
FIGURE 5.

Localization studies of PLCβ and PI3K in HEK293 cells. (A) Images of HEK293 immunostained for PLCβ (left) and overexpressing p85α/eYFP-p110α (center), showing only negligible colocalization (right). (B) Example of a normalized FRET image from eCFP-PI3K to eYFP-PLCβ expressed in an HEK293 cell.
DISCUSSION
Cells receive signals from their environment; these signals have the potential to activate multiple pathways. Although some pathways are parallel, others converge onto modules that may allow for signals to be redirected depending on the circumstances. Here, we have investigated the ability of a signal transducer to select two complementary pathways: 1), activation of PLCβ to increase intracellular Ca2+ signals through PIP2 hydrolysis, and 2), inhibition of vesicle trafficking events through inhibition of phosphorylation of PIP2 by PI3K. We found that, instead of Gαq selecting a specific effector during a stimulation event, separate pools of Gαq-effector complexes exist, thus making the signaling process less dynamic than expected. These separate pools keep signals along a particular pathway, reducing the likelihood of cross talk. As argued below, we propose that, although Gαq stimulation of PLCβ represents a forward motion pathway to stimulate cellular events, Gαq inhibition of PI3K serves as a backward motion to suppress cellular events.
We first characterized the cellular localization of PI3K using a fluorescent-tagged chimera. We found that, in direct contrast to the plasma membrane localization of Gαq, PI3K was widely distributed throughout the cytoplasm and plasma membrane (Fig. 1). This distribution correlates well with the function of PI3K and with previous imaging studies (18). It is interesting to note that, unlike the even distributions of Gαq and PLCβ in cells, PI3K is punctuated in appearance. This punctuate distribution of PI3K most likely reflects high concentrations of the enzyme on internal vesicles correlating to its role in endocytic trafficking. The punctuate distribution of PI3K on the plasma membrane is also seen for RTKs (5), suggesting colocalization of these proteins.
We found a significant amount of FRET between Gαq and PI3K. The distance at which half of the donor fluorescence is lost to transfer (i.e., Ro) for the eCFP and eYFP pair is 30 Å (22). Control studies using noninteracting donor/acceptors on the instrumentation used in this study showed the stochastic FRET to be 10%. Although most of the values for Gαq-PI3K are lower than this value, there is a broad range of high FRET values that contrast sharply with the more narrow distribution seen for Gαq-PLCβ complexes (Fig. 3). We speculate that this broad range of FRET values reflects the punctuate distribution of PI3K aggregates on the plasma membrane that contain varying amounts of Gαq, possibly resulting from inefficient dissolution of internal vesicles containing PI3K as seen in Fig. 2 D.
The key finding of this study is that separate pools of Gαq exist for both effectors. PLCβ has long been established as the main effector of Gαq, but it was puzzling that the cellular amount of PLCβ was far less than Gαq (13), suggesting that Gαq may interact with other cellular proteins. Recently, Lin and colleagues have discovered another Gαq effector (PI3K) that linked two distinct cell signaling pathways—GPCRs and RTKs (10–12). Although the affinity of activated Gαq is approximately threefold stronger for PLCβ than for PI3K (20), the affinities for deactivated Gαq are comparable, suggesting that Gαq can be bound to either depending on their local concentrations and presence of competing proteins. Thus, Gαq will bind to whichever effector is available, thereby directing the signal in either direction. We found that, instead of Gαq transducing a signal through diffusion and binding with effectors, there were at least two populations of Gαq preassociated with each effector in the basal and stimulated states. Whereas the advantage of preformed Gαq-PLCβ can be understood in terms of rapid signal transduction, the role of preassociated Gαq-PI3K complexes is not as clear. We speculate that the function of the Gαq-PI3K association is to prevent PIP2 phosphorylation by the plasma membrane pool of PI3K during GPCR activation, thus ensuring that the PLCβ substrate does not become depleted. In this way, Gαq could serve as an indirect local regulator of the PIP2 level on the plasma membrane.
Our in vitro studies suggest the possibility that Gαq can bind both effectors simultaneously. We tested this idea by determining the amount of colocalization of the two effectors and the amount of FRET between their fluorescent constructs in cells. We found that PLCβ and PI3K are only associated in a few points in the cell. This result suggests that the two signaling pathways are isolated, although it is possible that a few complexes containing these proteins exist, thereby allowing for cross talk between the pathways.
Although the delineation of signaling pathways is difficult, the finding that cells may have preselected pathways for signals to follow may help to simplify predictive models. It would be very interesting to determine whether other pathways are also preselected.
Acknowledgments
The authors thank Richard Lin (Department of. Hematology, Stony Brook University) for assistance with the PI3K constructs and Stuart McLaughlin (Department of Physiology and Biophysics, Stony Brook University) for use of his microscope.
This work was supported by a National Institutes of Health grant (GM053132 to S.S.) and a National Research Service Award from the Diabetes and Metabolic Diseases Research Center (T32 to U.G.).
Editor: Enrico Gratton.
References
- 1.Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 1994. Molecular Biology of the Cell, 3rd ed. Garland Publishing, New York.
- 2.Rebecchi, M., and S. Pentylana. 2000. Structure, function and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80:1291–1335. [DOI] [PubMed] [Google Scholar]
- 3.Rhee, S. G. 2001. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70:281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Runnels, L. W., and S. Scarlata. 1999. Determination of the affinities between heterotrimeric G protein subunits and their phospholipase C-b effectors. Biochemistry. 38:1488–1496. [DOI] [PubMed] [Google Scholar]
- 5.Haj, F. G., P. J. Verveer, A. Squire, B. G. Neel, and P. I. H. Bastiaens. 2002. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science. 295:1708–1711. [DOI] [PubMed] [Google Scholar]
- 6.Katso, R., K. Okkenhaug, K. Ahmadim, S. White, J. Timms, and M. D. Waterfield. 2001. Cellular function of phosphoinositide-3-kinases: implications for development, immunity, homeostasis and cancer. Annu. Rev. Cell Dev. Biol. 17:615–675. [DOI] [PubMed] [Google Scholar]
- 7.Shepherd, P. R., D. J. Withers, and K. Siddle. 1998. Phosphoinositide 3-kinase: the key switch mechanism in insulin signaling. Biochem. J. 333:471–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geering, B., P. R. Cutillas, G. Nock, S. I. Gharbi, and B. Vanhaesebroeck. 2007. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA. 104:7809–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuels, Y., Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. V. Willson, S. Markowitz, K. W. Kinzler, B. Vogelstein, and V. E. Velculescu. 2004. High frequency of mutations of the PIK3CA gene in human cancers. Science. 304:554. [DOI] [PubMed] [Google Scholar]
- 10.Ballou, L. M., M. Cross, S. Huang, E. M. McReynolds, B. X. Zhang, and R. Z. Lin. 2000. Differential regulation of phosphatidylinositol-3-kinase /Akt and p70 S6 kinase pathways by the alpha(1A)-adrengeric receptor in rat-1 fibroblasts. J. Biol. Chem. 275:4803–4809. [DOI] [PubMed] [Google Scholar]
- 11.Bommakanti, R. K., S. Vinayak, and W. Simonds. 2000. Dual regulation of Akt/protein kinase B by heterotrimeric G proteins. J. Biol. Chem. 275:38870–38876. [DOI] [PubMed] [Google Scholar]
- 12.Ballou, L. M., H. Y. Lin, G. Fan, Y. P. Jiang, and R. Z. Lin. 2003. Activated G α q inhibits p110 alpha phosphatidylinositol-3-kinase and Akt. J. Biol. Chem. 278:23472–23479. [DOI] [PubMed] [Google Scholar]
- 13.Dowal, L., P. Provitera, and S. Scarlata. 2006. Stable association between G α (q) and phospholipase C β 1 in living cells. J. Biol. Chem. 281:23999–24014. [DOI] [PubMed] [Google Scholar]
- 14.Hughes, T. E., H. Zhang, D. Logothetis, and C. H. Berlot. 2001. Visualization of a functional Gaq-green fluorescent protein fusion in living cells. J. Biol. Chem. 276:4227–4235. [DOI] [PubMed] [Google Scholar]
- 15.Chiadac, P., V. S. Mavkin, and E. M. Ross. 1999. Kinetic control of guanine nucleotide binding to soluble Gaq. Biochem. Pharmacol. 58:39–48. [DOI] [PubMed] [Google Scholar]
- 16.Philip, F., P. Sengupta, and S. Scarlata. 2007. Signaling through a G protein coupled receptor and its corresponding G protein follows a stoichometrically limited model. J. Biol. Chem. 282:19203–19216. [DOI] [PubMed] [Google Scholar]
- 17.Xia, Z., and Y. Liu. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 81:2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillham, H., M. C. H. M. Golding, R. Pepperkok, and W. J. Gullick. 1999. Intracellular movement of green fluorescent protein–tagged phosphatidylinositol 3-kinase in response to growth factor receptor signaling. J. Cell Biol. 146:869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Susa, M., M. Keeler, and L. Varticovski. 1992. Platelet-derived growth factor activates membrane-associated phosphatidylinositol 3-kinase and mediates its translocation from the cytosol. Detection of enzyme activity in detergent-solubilized cell extracts. J. Biol. Chem. 267:22951–22956. [PubMed] [Google Scholar]
- 20.Ballou, L. M., M. Chattopadhyay, Y. Li, S. Scarlata, and R. Z. Lin. 2006. Gaq binds to p110a/p85a phosphoinositide 3-kinase and displaces Ras. Biochem. J. 394:557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conklin, B. R., O. Chabre, Y. H. Wong, A. D. Federman, and H. R. Bourne. 1992. Recombinant Gqa: mutational activation and coupling to receptors and phospholipase C. J. Biol. Chem. 267:31–34. [PubMed] [Google Scholar]
- 22.Patterson, G. H., D. W. Piston, and B. G. Barisas. 2000. Forster distances between green fluorescent protein pairs. Anal. Biochem. 284:438–440. [DOI] [PubMed] [Google Scholar]