

Abstract
Cancer dormancy is a poorly understood stage of cancer progression. However, the ability to control this step of the disease offers novel therapeutic opportunities. Here we summarize recent findings that implicate the extracellular matrix and adhesion receptor signaling in the escape or induction of tumor dormancy. We further review evidence suggesting that imbalances in the activity ratio of ERK to p38 signaling may determine the fate (i.e., tumorigenicity vs. dormancy) of different carcinoma cells. Special attention is placed on the mechanisms that p38 signaling regulates during the induction of dormancy and how modulation of these pathways may offer a therapeutic opportunity. We also review evidence for a novel drug-resistance mechanism in dormant tumor cells that when blocked may enable killing of dormant tumor cells. Finally, we explore the notion that dormancy of tumor cells may be the result of a selective adaptive response that allows disseminated tumor cells to pause their growth and cope with stress signaling imposed by dissemination and/or treatment until growth can be restored.
Keywords: metastasis, quiescence, ERK, p38, unfolded protein response, uPAR, tumor dormancy, microenvironment
INTRODUCTION
Tumorigenesis is a multi-step process involving genetic and epigenetic changes. These provide the cells with a growth advantage promoting rapid and autonomous cell proliferation and survival that ultimately results in a primary tumor. During this process single or small groups of tumor cells may be shed from the primary tumor and spread to distant organs to eventually develop metastatic lesions. Metastatic disease, rather than the primary tumor itself, is the major cause of morbidity and mortality among cancer patients as in this stage the disease is highly refractory to current therapies.1 The development of metastasis follows a sequence of events that a tumor cell must complete successfully. These include local invasion, intravasation, dissemination through the blood and/or lymphatic systems, extravasation and, if the surrounding milieu is propitious, growth at distant secondary sites (excellent reviews on the metastatic cascade can be found elsewhere).2-4 However, there are still questions as to what determines the fate of disseminated cells and how is this linked to prognosis. For example, what differences determine the proliferation or dormancy of disseminated tumor cells? Furthermore, are these differences linked to the outcome of patients where relapse occurs within months vs. those that are disease-free for more than five years (Fig. 1A)? Answers to these questions have been hard to obtain mostly due to the lack of appropriate models to understand how tumor cells behave after dissemination to target organs.
Figure 1.
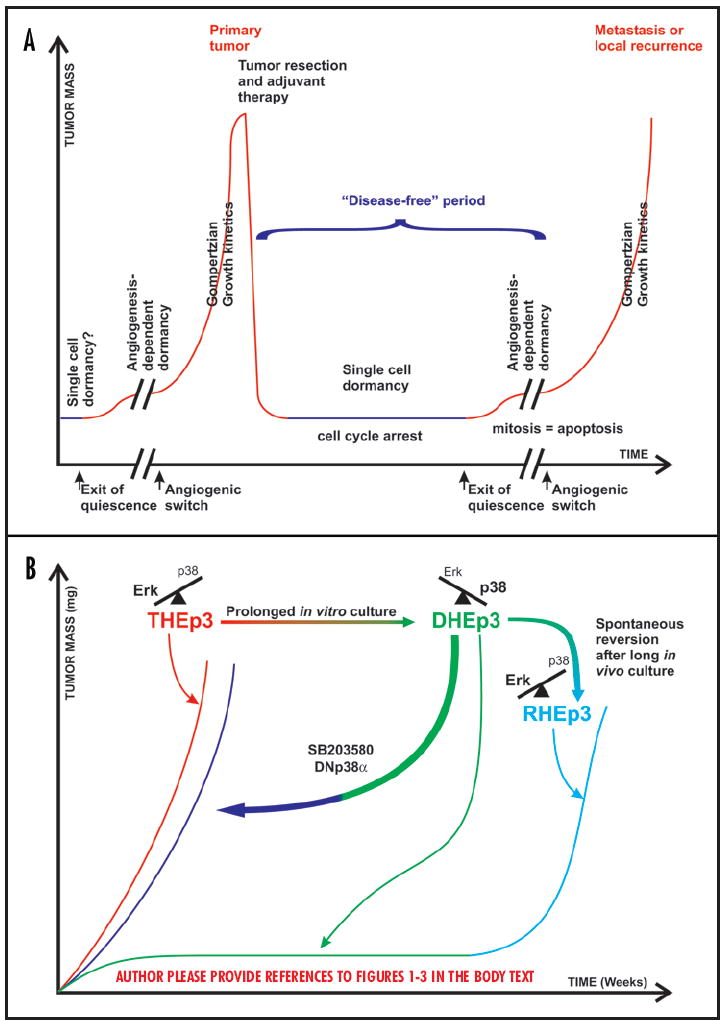
(A) A graphical representation of the progression of a tumor mass in patients. Over long periods (years) the accumulation of genetic and epigenetic changes that confer growth advantages lead to cell transformation and tumor development. Upon diagnosis, treatment involves primary tumor resection and or adjuvant chemotherapy. Nevertheless, in situ or disseminated single growth arrested cells or small groups of cells lacking vascularization may persist undetected for months to decades. At any given moment, tumor cells may emerge from quiescence and/ or acquire angiogenic potential and resume uncontrolled proliferation in situ or in distant organs. (B) Schematic depiction of the HEp3 tumor dormancy model. Tumorigenic cells with high ERK and low p38 signaling (T-HEp3) can proliferate efficiently when inoculated in vivo. In contrast cells with high p38 and low ERK activity (dormant, D-HEp3) are unable to grow in vivo and hence form small nodules that contain tumor cells in a G0/G1 arrest and are dormant for months. However these cells can revert to a tumorigenic phenotype either spontaneously after prolonged passaging in vivo (R-HEp3) or following downregulation of p38 activity with a dominant negative p38α or with a p38 inhibitor (SB203850), all of which tip the balance towards ERK signaling.
It is known that most of the intravasated cells die in circulation explaining why metastasis is a highly inefficient process (less than 0.01% of the cells that are released into circulation go on to form clinically detectable metastases).2 Those cells that survive and are equipped with all the proper genetic and epigenetic alterations to grow in the secondary site may give rise to metastasis in patients that show rapid recurrence. Alternatively, tumor cells may survive as solitary nonproliferating dormant cells that are clinically undetectable. These dormant cancer cells pose a major threat for cancer patients because although they may be in a quiescent state, years or decades after the removal of primary tumor they might be capable of resuming uncontrolled proliferation and give origin to untreatable overt metastases. Late recurrence in situ and as metastasis accounts for more than 50% of mortality among cancer patients. Thus, it is possible that the inherent ability of dormant tumor cells to resume a proliferative program increases the long-term risk of tumor recurrence.
CANCER CELL DORMANCY AND MINIMAL RESIDUAL DISEASE (MRD)
Mathematical modeling of tumor growth based on the analysis of the rates of breast cancer recurrence at local and distant sites predicted tumor growth kinetics in which a dormancy period preceded the Gompertzian growth of the relapse (Fig. 1A).5,6 The latency period that precedes the relapse presupposes that cells that were able to grow as a primary now become quiescent or that micrometastatic lesions do not achieve a net increase in mass to become clinically detectable. Whether this applies to all tumor types is unknown but examples of dormant tumor masses are the existence of metastasis with occult primaries or the patients that develop cancer in transplanted organs (these could be primary or secondary tumors) (see also Klein and Hölzel this issue). Furthermore, while the presence of dormant tumor cells was mostly inferred from the clinical retrospective data,7-10 recently, using more sensitive techniques, several groups showed the existence of such residual “dormant” populations in patients with breast and prostate cancer among other types.11 These cells were detected as nonproliferating tumor cells in the bone marrow of those patients.11,12 It is estimated that almost 20–40% of patients with breast, colorectal, small cell lung and probably other cancers have disseminated tumor cells in their bone marrow11 (See Laufs et al., and Klein and Hölzel this issue). The detection of these cells is predictive for metastases to the bone or other organs such as liver or lung and is associated with poor prognosis.13-15 Thus, it is possible that the bone marrow may either be a final destination for metastasis or a reservoir where dormant tumor cells resume growth prior to their final dissemination to their target organs.
While the clinical evidence for residual dormant disease continues to grow, the underlying mechanisms that are involved in the induction, maintenance and escape from dormancy are poorly characterized (Fig. 1A). Modeling cancer dormancy is important for three main reasons: first it will help identify prognostic markers for better staging of cancer (see Laufs et al. and Klein and Hölzel in this issue), second it will help designing novel therapeutics to induce and/or maintain dormancy of tumor cells, and third it may lead to the understanding of the survival mechanisms that render dormant tumor cells resistant to chemotherapy, which could subsequently lead to therapies to fully eradicate disseminated dormant tumor cells.
DORMANT SINGLE CELLS VS. DORMANT MICROMETASTASES
Tumor dormancy has been proposed to exist either at the single cell level, where tumor cells enter a growth arrest or as small masses of tumor cells that are unable to grow beyond a certain size.2 Studies pioneered by the Folkman laboratory (reviewed by Naumov et al., and Indraccolo et al., this issue) indicate that dormancy of micro-metastases ensues because of a balance between the proliferation and apoptotic rates of the tumor cells, resulting from the inability of those small tumor masses to recruit new blood vessels.16,17 Thus, while in these tumor masses there are proliferating cells because of their avascular environment, there is no net increase in the tumor mass. However, such a stage can be reverted if the tumor cell population (or a subset) acquires the ability to recruit new blood vessels either by downregulating angiogenesis inhibitors or by upregulating pro-angiogenic factors (Fig. 1A).16,17
The dormancy of single tumor cells, where cells that arrive at secondary sites enter a growth arrest represents a different scenario. Here single cells or small cohorts of cells enter growth arrest and dormancy. The fact that staining of single disseminated cells is usually negative for markers of proliferation such as PCNA, suggests that these cells are in a G0/G1 arrest.18 Such a shift from proliferation to dormancy might occur if the tumor cells that survive dissemination are unable to cope with the cellular stress induced by this process and/or the new microenvironment. Recent work (see Townson et al., this issue) demonstrated that many tumor cells that arrive at the secondary site and survive fail to proliferate.19-21 While angiogenesis-dependent dormancy is a direct consequence of suppression of neovascularization and occurs at the population level, in the second case dormancy independently of angiogenesis and occurs at the level of individual cells. This suggests that mechanisms other than those that control angiogenesis can regulate the induction and maintenance of growth arrest and dormancy of single cells. Although intuitively nonoverlapping, the two mechanisms of dormancy may not be mutually exclusive and it is possible that dormancy of single cells is followed by dormancy of a tumor mass due to hypo-vascularization (Fig 1A). Work from our lab suggests that an interplay between surface receptors (e.g., uPAR, integrins), mitogenic (e.g., ERK) and stress (e.g., p38) signaling pathways may determine the shift from proliferation to G0/G1 arrest and dormancy of single tumor cells.22 In this review we will discuss the progress made on these and other mechanisms that might explain the growth arrest and dormancy of tumor cells (Fig. 1B) (see also White and Muller and Townson et al., this issue).
SIGNALING BY p38SAPK CAN SUPPRESS CELL TRANSFORMATION AND TUMOR METASTASIS
The role of ERK1/2 in the promotion of mitogenesis is well known.23,24 However, the function of SAPKs is less understood. p38 was initially cloned as a cytokine regulated kinase inhibited by the anti-inflammatory drug SB203580.25 Its sequence homology to ERKs generated the notion that p38 may also be involved in cell proliferation and malignancy.26,27 However, a growing body of evidence suggested that p38 isoforms are involved in the induction of cell cycle arrest28 and apoptosis29 and in the negative regulation of transformation and tumorigenesis.28,30,31 For example, forced activation of p38 in vitro antagonizes Ras-induced transformation in both fibroblasts and epithelial cells32,33 and Ras-induced inhibition of p38 is critical for transformation.34 Moreover, hyper-activation of p38 signaling inhibits tumorigenicity in vivo35,36 and inhibition of p38 activity by inactivation of both MKK3 and MKK6 in mice led to increased tumorigenesis.37 Evidence supporting that p38 activation may be important to suppress malignancy is derived from studies showing that inactivation of Wip1, a phosphatase for p3838,39 leads to inhibition of Ha-Ras-induced tumorigenesis of mouse embryonic fibroblasts.36 Also, when Wip1 null mice were crossed with MMTV-regulated ErbB2, Ha-Ras or Wnt1 transgenic mice Wip1 deficiency inhibited mammary tumorigenesis induced by ErbB2 or Ha-Ras, due to restored p38 signaling, but not Wnt induced tumorigenesis.40 One interesting effect of p38 that might explain its anti-tumorigenic effect is its activation during cellular senescence,41,42 which has been proposed to protect against cancer.43,44
Recent evidence supports a role for p38 in mediating the inhibition of metastasis formation as well. For example MKK4 was postulated to function as a metastasis suppressor gene45 in prostate46 and ovarian carcinoma.47 By expressing MKK4, 6 and 7 constructs in SKOV3ip.1 ovarian carcinoma cells, Hickson et al.48 showed that p38 mediates MKK4- and also MKK6-dependent inhibition of metastasis. It will be of interest to know whether the inhibition of tumor and/or metastasis growth by p38 observed by these authors is due to an increase in apoptosis or a growth arrest in vivo. In addition, recent publications by Ossowski et al, and from our lab indicate that p38 may induce dormancy of tumor cells through a mechanism that requires inhibition of metastasis promoting genes such as the urokinase receptor (uPAR) (see below).22,49 These published reports allow us to propose that while p38 activation could result in suppression of primary tumor formation, it could also serve as a tool to suppress growth of metastatic cells that retain an intact p38 signaling pathway by inducing either apoptosis or dormancy. Both outcomes may be life saving or life prolonging for patients with refractory disseminated disease.
A LINK BETWEEN UROKINASE RECEPTOR EXPRESSION AND TUMOR DORMANCY
As proposed earlier, after dissemination tumor cells that are equipped with the proper genetic and epigenetic mechanisms will resume growth in the metastatic site. Thus, insight into the mechanisms by which cell might evade dormancy could be found in the signaling properties of genes that are known to promote metastases. Urokinase receptor (uPAR) has been functionally associated both in experimental systems and in the clinic with the ability of tumors to metastasize. Interestingly, uPAR blockade can induce in some cases apoptosis, such as in brain tumors, but in other cases it can induce a state of dormancy (Fig. 2).50 For an in depth analysis of the proteolytic and nonproteolytic functions of uPAR and its link to MRD see the accompanying review by Laufs et al. In this review we will focus on the mechanisms by which uPAR expression and signaling through α5β1-integrin and the EGF receptor (EGFR) controls the decision between proliferation and dormancy of human carcinoma cells (Fig. 2).51
Figure 2.
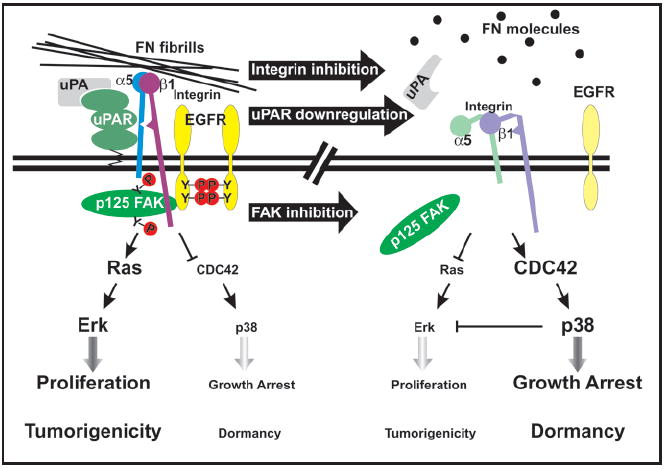
Cell surface regulation of the ERK/p38 ratio as a determinant of tumor dormancy. (Left) In uPAR rich HEp3 cells, the uPA bound receptor, through a region requiring Ser-245, interacts frequently with the FN receptor α5β1-integrin leading to efficient assembly of FN fibrils. Further, this interaction results in the formation of a functional complex involving FAK and EGFR, leading to a strong activation of the Ras-ERK mitogenic signaling and inhibition of p38 signaling via inactivation of Cdc42. This results in a high ERK/p38 ratio that favors proliferation and promotes tumorigenicity. (Right) Down regulation of uPAR expression, blocking of integrin function or inhibition of FAK results in the disassembly of the complex and reduced ERK activation. Consequently this leads to the activation of Cdc42 and subsequent activation of p38 resulting in a low ERK/p38 ratio, which favors growth arrest and forces the tumor cells into dormancy.
While initially the poor prognosis associated with overexpression of the uPA-uPAR system was linked with its proteolytic function,52-55 evidence from MRD (see review by Laufs et al.) and experimental studies49 suggested that it may require the ability of uPAR to propagate mitogenic signals. Clinical evidence for the growth-promoting role of uPAR was obtained by Heiss and colleagues56 where detection of uPAR in cytokeratin positive cells disseminated to bone marrow predicted for a short disease free period and recurrence (reviewed by Laufs et al., this issue). However, the mechanisms that allowed uPAR to promote proliferation of tumor cells were unknown.
Mechanistic insight into the role of uPAR in regulating tumor growth in vivo came from examining its role in the malignant behavior of HEp3 squamous carcinoma cells (Fig. 2). Using an antisense mRNA to block the expression of uPAR, it was demonstrated that the downregulation of uPAR in HEp3 cells while not affecting the proliferation rate in culture, caused a significant reduction in invasiveness in vivo. Surprisingly, uPAR antisense also resulted in a complete loss of tumorigenicity in vivo.57,58 Low uPAR-expressing cells formed very small nodules that remained dormant for several months and the loss of tumorigenicity was not due to a decrease in proteolytic activity, enhanced apoptosis or the lack of vasculature but due to an overall decrease in proliferation.58 Based on these and other published results, it was proposed that the uPA-uPAR interactions has a dual role in tumorigenesis—a well characterized one dependent on proteolysis that promotes local invasion, intravasation and metastasis and another as an initiator of a signaling cascade that regulates tumor proliferation in vivo.
This intriguing new function of uPAR led to test and demonstrate that in uPAR rich tumorigenic HEp3 cells, this receptor was physically associated with the fibronectin (FN) receptor α5β1 integrin.50 uPAR interaction with α5β1 integrin caused an increase in the activation/avidity of the integrin, strong adhesion to FN50 and enhanced FN fibrillogenesis.22 Strikingly, this interaction resulted in a strong and persistent FN-dependent activation of the mitogenic Ras-ERK pathway thereby promoting rapid cell proliferation in vivo. In contrast, in cells where uPAR expression was downregulated by an antisense,58 the frequency of interaction between uPAR and α5β1-integrin was greatly reduced. Detailed analysis, revealed that in the tumorigenic HEp3 cells that have a high level of ERK activation, downregulation of uPAR and disruption of the complex led to the deactivation of the ERK pathway and subsequent arrest of the cells in G0/G1 phase of cell cycle, triggering tumor dormancy in vivo (Fig. 2).22
INHIBITION OF uPAR SIGNALING INDUCES TUMOR DORMANCY BY ESTABLISHING AN IMBALANCE THAT FAVORS p38SAPK OVER ERKMAPK SIGNALING
Analysis of the uPAR-α5β1-integrin interaction revealed that a complex comprising uPAR, α5β1, FAK and EGFR is the minimal functional complex required to transduce uPA- and FN-dependent signals to the ERK pathway.22,51,59 Interventions that blocked the expression or function of uPAR, α5β1, FAK or the EGFR all resulted in decreased ERK activation and dormancy in vivo. Most importantly, Chaurasia et al., have recently identified a site on uPAR where α5β1 integrin binds specifically.60 Identification of this site and its mapping onto the 3D structure of uPAR is promising as it may aid in identifying inhibitors that disrupt the interaction, reducing ERK activation and inducing dormancy.
However, while ERK inhibition may be sufficient to induce dormancy, modulation of other pathways may be needed to force single disseminated tumor cells or tumor masses into dormancy. Signaling by uPAR was found to induce tumorigenicity not only by hyper-activating ERK signaling but also by inhibiting the p38SAPK pathway that is necessary to maintain the G0/G1 arrest.22,51 It was found that in cells with high uPAR levels the uPAR-α5β1 complex promoted the assembly of the FN fibrils. FN fibrils, through a pathway that required inactivation of Cdc42 were found to maintain the growth suppressive p38SAPK pathway in an low activation state. In contrast, when the uPAR-integrin complex or FN fibrils were disrupted the p38 pathway was activated favoring growth arrest in vivo. Such a disruption might occur in vivo since in patients soluble uPAR was found cleaved between D1 and D2 and we have shown that uPAR devoid of D1 is unable to interact with α5β1-integrins and activate EGFR->ERK signaling.51 These results allowed us to conclude that the level of uPAR expression and signaling influences the decision between tumorigenicity and dormancy in vivo by regulating the balance between ERK and p38SAPK pathways.49 This, ratio or balance was found to be predictive of the in vivo behavior of various tumor types including prostate, breast and fibrosarcoma indicating that it is not circumscribed to the HEp3 model (Figs. 1B and 2).49
Ossowski and Reich originated a confirmation of the generality of this mechanism, in experiments in the early 1980’s. While studying the malignant properties of HEp3 squamous carcinoma cells they found that when these cells are passaged in vivo on CAMs they remain highly tumorigenic and metastatic. However, they observed that the same cells passaged in culture progressively lost their in vivo tumorigenic and metastatic potential and formed small dormant tumor nodules in vivo.61,62 To further examine the cause for this phenotypic shift, the tumor nodules were harvested and subject to in vitro clonal analysis. To their surprise all of the examined clones (~ 57 clones) became spontaneously dormant after ~40 passages in culture, suggesting that the acquisition of the dormant phenotype occurred in a nonclonal manner.62 Further, dormancy was reversible as they found that tumorigenicity reappeared after prolonged passaging of the nontumorigenic cells on CAMs or nude mice in vivo.
An initial analysis revealed that the phenotypic shift was not a genetic selection event but rather a conditional phenotype influenced by the growth environment.62 Experiments performed years later demonstrated that these spontaneous dormant cells (referred to as D-HEp3 cells) had low uPAR expression that depended on a low ERK/p38SAPK signaling ratio and was similar to that seen in the uPAR antisense cell line. Reexpression of uPAR, or constitutive activation of ERK in these DHEp3 cells was sufficient to restore the uPAR-α5β1-integrin complex, high ERK activity and interrupt dormancy of these cells in vivo (Figs. 1B and 2).
These studies established an important finding showing that in dormant cells p38 establishes a negative feedback loop on ERK activation. Both pharmacological (SB203580) and genetic inhibition (dominant negative p38α) of p38 activity in dormant cells resulted in the interruption of dormancy and restoration of in vivo growth. Inhibition of p38 also resulted in ERK dependent induction of uPAR transcription, assembly of the uPAR-α5β1-integrin complex and restored ERK activation. Hence inhibition of p38 restores the ERK pathway and the flow of mitogenic signaling resulting in enhanced proliferation in vivo.49,63
In conclusion these studies provided a novel insight into the opposing roles of both uPAR/α5β1/EGFR->ERK and p38 signaling in the induction and maintenance of dormancy in HEp3 cells. When uPAR-dependent activation of ERK predominates over p38 signaling, tumorigenicity is maintained. In contrast, when conditions are given (i.e., uPAR downregulation) that result in p38 activation predominating over and inhibiting ERK activity the result is tumor dormancy (Figs. 1B and 2).
MONITORING OF ERK AND p38 SIGNALING IN VIVO REVEALS PATHWAY ACTIVATION DYNAMICS THAT DEFINE DORMANT VS. PROLIFERATIVE TUMORS
The above-mentioned studies provided strong in vitro data that predicted accurately the behavior of tumorigenicity vs. dormancy in vivo. However, whether the intensity and thresholds of signals measured in vitro for ERK and p38 were recapitulated in vivo in primary tumors or metastasis remained unknown. To measure ERK or p38 pathway activation in vivo we designed a biosensor system that tracks ERK or p38 signaling by using a reporter construct where expression of a fusion protein between GAL4 DNA-binding domain and CHOP transactivation domain (p38 substrate) drives GFP expression from a 4xGAL4-UAS. In the ERK biosensor we replaced the CHOP transactivation domain with the transactivation domain of Elk-1 (ERK substrate). These reporters revealed that the high ERK activity measured in vitro persisted in vivo and both primary and lung metastatic lesions showed strong ERK-Elk activity. In contrast, using these reporters we discovered that while the low ERK activity detected in dormant DHEp3 cells in vitro decreases even further upon in vivo inoculation and is absolutely lost after 96 hrs in vivo, p38 activation increased over in vitro levels and persisted for at least two weeks. Thus, in order to achieve dormancy, an almost complete inhibition of ERK and activation of p38 must be achieved. ERK inhibition was dependent on p38 as pretreatment for only 48hrs with SB203580 increased ERK-Elk activity and these tumor cells interrupted dormancy in the chicken CAM22 and in nude mice (our unpublished results).
An unexpected finding was the behavior of the p38-CHOP in primary tumors and metastases of HT1080 fibrosarcoma cells that harbor an N-Ras mutation. These cells showed strong (as strong as dormant tumor cells) activation of p38-CHOP 24-48 hrs in vivo in primary sites. However, these cells were subsequently able to downregulate p38-CHOP activation and grow as a primary tumor that disseminated to form metastases in lungs and lymph nodes. Intriguingly, cells recovered from metastases that had the p38-CHOP reporter system were still susceptible to activation of p38 upon stress induction.63 Thus, these metastatic cells were able to silence p38 signaling through a mechanism that does not eliminate the ability of the pathway to become reactivated. This suggests that perhaps, cells harboring mutations such as N-Ras may be able to acquire mechanisms that help silence or ameliorate p38 signaling avoiding its deleterious effects.63 The p38 pathway was inhibited rapidly (48hrs) in vivo and was persistently inhibited in metastases. This suggests that in this model metastatic cells did not undergo a genetic selection to eliminate p38 signaling since they were still susceptible to p38-CHOP reporter activation.63 Thus, disseminated tumor cells that can maintain ERK signaling while suppressing p38 activation may go on to grow into secondary lesions. In contrast, tumor cells that are not be able to suppress or adapt to the stress signals and activate p38 may enter a growth arrest.63
p38 REGULATES DORMANCY AND SURVIVAL THROUGH AN ER-STRESS-LIKE RESPONSE
The previous findings supported a pivotal role for p38 in the induction and/or maintenance of dormancy. The fact that a transient inhibition of p38 with SB203580, which causes a burst in ERK activation,22,49 or siRNA (our unpublished results) interrupts dormancy suggested that p38 activates a dominant program that maintains the phenotype. However, the downstream mechanism that p38 engages to maintain dormancy was poorly understood. A proteomic search for p38 regulated genes comparing tumorigenic versus dormant D-HEp3 cells in which p38 signaling was high or inhibited with SB20358064 revealed that in D-HEp3 cells p38 upregulated four endoplasmic reticulum (ER)-resident chaperones (BiP, ER60, HSP47 and cyclophilin B). Expression of these proteins is normally induced during adaptation to stress signaling caused by unfolding of proteins in the ER (Fig. 3A), a response termed unfolded protein response (UPR).64
Figure 3.
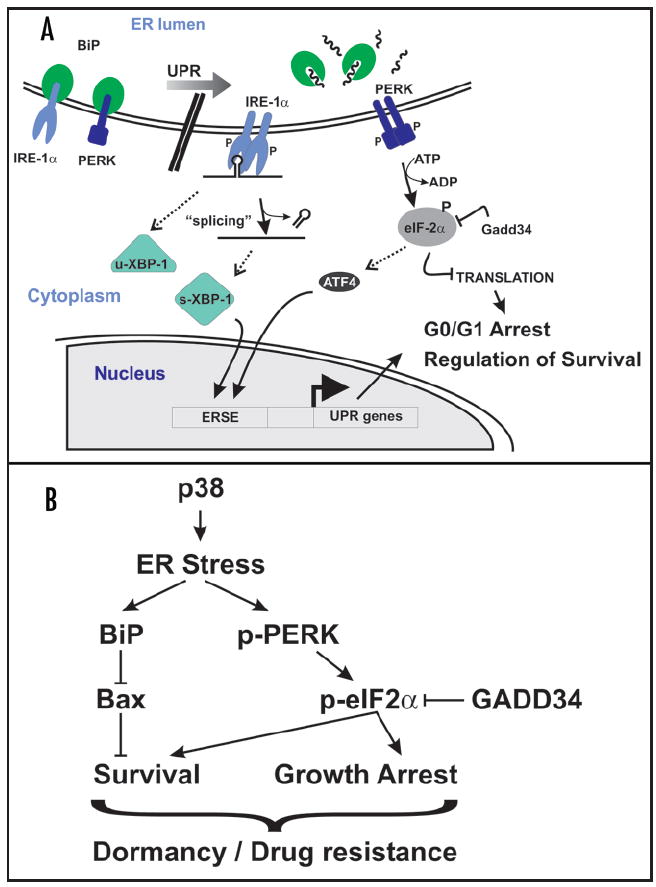
Regulation of ER-stress by p38 and tumor dormancy. (A) Simplified overview of pathways of the UPR relevant to this review. ER-stress induces dissociation of BiP from PERK and IRE1 and their subsequent activation via trans-phosphorylation. IRE1 activation leads to the noncanonical splicing of XBP-1 mRNA, causing a frame-shift in the ORF resulting in a potent transcription factor. Active PERK phosphorylates eIF2α on Ser 51, inhibiting translation and causing G0/G1 arrest. Preferential translation of ATF4 mRNA by phospho-eIF2α also induces (along with XBP1) the expression of UPR genes. P = phospho-group. ERSE = ER stress response element. Dephosphorylation of eIF2α by GADD34-PP1 restores translation. (B) Activation of p38 signaling in HEp3 cells regulates a program that favors growth arrest and survival through the activation of an ER stress response. While p38-dependent activation of PERK-eIF2α pathway coordinates both growth arrest and survival signaling, upregulation of BiP expression inhibits Bax activation and promotes survival and drug resistance in dormant cells without affecting the dormancy of the tumor cells.
In normal conditions, secreted proteins are folded in the ER and the folding “capacity” of this organelle depends on molecular chaperones that coordinate this process in steady-state conditions. However, several pathophysiological conditions can compromise the folding capacity of the ER resulting in protein aggregation and induction of signals that activate the UPR. For reviews see refs. 65–67. The UPR serves as a stress-induced checkpoint by concomitantly (1) inducing the expression of ER chaperones to cope with the misfolded proteins and (2) decreasing rate of protein synthesis to relieve the protein load in the ER. While ER stress forces the cells into growth arrest (G0/G1) to pause and restore homeostasis, chronic ER stress can result in apoptosis (Fig. 3A). The ability of ER-stress to concomitantly induce survival and growth arrest prompted us to study its link to dormancy of HEp3 cells, because these are the two parameters that define the dormancy of these cells. A critical pathway in the UPR is the one regulated by protein kinase/RNase and RNA-dependent protein kinase-like ER kinase (PERK). PERK is maintained in an inactive state by the association of BiP with its lumenal domains. Upon protein unfolding, BiP dissociates from PERK and preferentially binds to the unfolded proteins resulting in PERK activation and phosphorylation of eukaryotic translation initiation factor 2α (eIF2α). This event leads to protein synthesis repression and induction of G0/G1 arrest (Fig. 3A). The UPR also induces GADD34, a regulatory subunit of the eIF2α phosphatase PP1 that relieves translation repression,68,69 allowing translation of the UPR response genes to favor proper protein folding and return to homeostasis.
Most of the published data (reviewed in refs. 25, 70-73) suggested that p38 mainly functions in promoting cell cycle arrest and apoptosis. Although this might be true, p38 may also signal to promote cell survival.64,74-77 In our model it appears that the level of p38 signaling that is achieved is coupled to survival and not apoptosis. Mechanistic analysis revealed that in dormant HEp3 cells p38-dependent upregulation of BiP correlated with constitutive activation PERK and phosphorylation of eIF2α.64 Activation of PERK signaling during ER stress was shown to induce G0/G1 arrest and cell survival,78 the two components of dormancy. BiP has also been shown to promote survival following ER stress79 as well as in response to DNA-damaging agents and other stresses.80,81 Thus, we further hypothesized that PERK and BiP may provide a survival advantage to D-HEp3 cells (Fig. 3B). We found64 that p38 activation of PERK and upregulation of BiP in D-HEp3 cells renders them highly resistant to ER stress-inducing agents such as tunicamycin but also to TOPOII inhibitors such as doxorubicin. Further, the BiP-mediated resistance of D-HEp3 cells to doxorubicin treatment was also evidenced in vivo, where the cells are not dividing (our unpublished results). In addition, inhibition of PERK using a dominant negative PERKΔC-term mutant or overexpression of GADD34, increased the cell death of DHEp3 cells after etoposide treatments in vitro. Our results have identified two pathways that control survival of dormant, but not proliferative tumor cells. We have also determined that eIF2α phosphorylation by PERK, but not BiP expression, might be involved in the induction of growth arrest and dormancy in vivo (Ref. 64 and our unpublished results). This may be linked to the finding that PERK mediated growth arrest results from reduced translation of cyclin D1 (Fig. 3B).82-84
The ability of cells to co-opt stress signaling pathways, such as the p38SAPK and/or PERK to cope with stress, may be a trait that allows tumor cells to proliferate in adverse environments or to enter a state of dormancy that protects them from deleterious conditions. Translational repression is a theme in tumor cell adaptation to stress,85 as pathways that are inactivated upon nutritional stress, such as the AKT->mTOR pathway, or classical pathways that sense genotoxic damage such as p5386 will reduce CAP-dependent translation through the modulation of 4E-BP and eIF4E.
These findings may help to unravel an unexplored link between p38, translational control and the regulation of cell cycle and survival and may serve as a starting point to the understanding of cell cycle-independent mechanisms that make dormant metastasis resistant to chemotherapy. Further, that BiP inhibition renders dormant cells susceptible to TOPOII inhibition without interrupting dormancy provides a novel conceptual framework to design strategies to induce killing and to eradicate the residual dormant tumor cells after surgery and/or radio- or chemotherapy without interrupting their growth arrest.
SUPPRESSION OF METASTASIS THROUGH THE INDUCTION OF DORMANCY
Some key questions regarding tumor progression in humans emerge from understanding dormancy. Can an adverse or foreign microenvironments or stress (i.e., chemotherapy, hormonal therapy) sensed by the cancer cell result in tumor dormancy. Why tumor cells that are equipped with a repertoire of genetic and epigenetic alterations that allowed them to form a primary tumor, are then unable to resume growth at secondary sites? A potential answer is that there is still plasticity in these phenotypes to reverse the malignant phenotype and that the hallmarks may be not at a single cell level but provided by the population in the primary tumor (Fig. 4).
Figure 4.
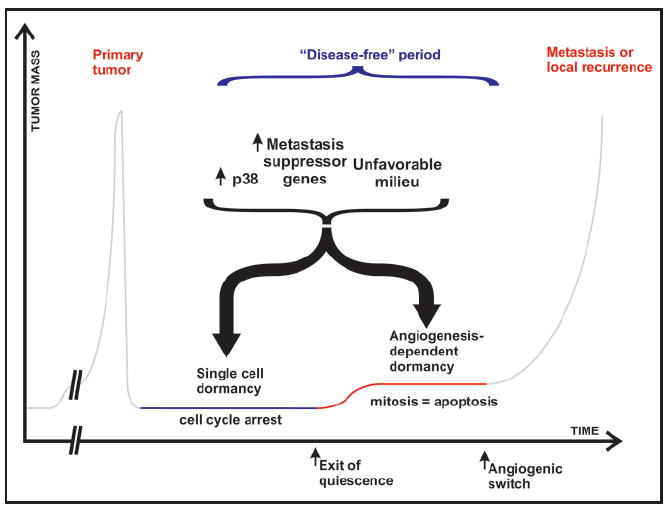
Potential mechanisms that may explain the dormancy of tumor cells. Disseminated cells are subject to strenuous conditions during the metastatic cascade. Tumor cells that are unable to cope with the stress induced by dissemination and/or an unfavorable milieu may pause growth but survive as isolated cells. Growth of these cells into a mass that remains avascular will result in dormancy as well. While single disseminated cells may enter dormancy through a growth arrest, dormancy of an avascular tumor mass results from the apoptotic rate balancing the proliferation rate. Although these mechanisms are different, they may share some regulatory pathways dependent on activation of p38 and/or metastasis suppressor genes that may become dominant in an unfavorable microenvironment. Spontaneous inhibition of these signals may interrupt dormancy and promote secondary growth.
During primary tumor formation, a strong adaptive pressure on the tumor cell population favors the acquisition of traits that promote proliferation and block apoptosis despite inhibitory signals present in that environment. An exciting corollary to that hypothesis is that in some circumstances disseminated tumor cells that lack the mechanisms to immediately grow as a secondary lesion may have acquired the ability activate stress-signaling pathways that enable the cells to adapt and stop proliferation until the conditions are propitious. The finding that several metastasis suppressor genes specifically inhibit metastasis formation and not primary tumor growth (reviewed in refs. 45, 87, 88) strongly supports the above hypotheses that the activation of a growth-inhibitory response in the metastatic environment is functionally different from the responses to the primary tumor milieu (Fig. 4). To date, 11 metastasis suppressor genes have been identified. Of interest are those that specifically inhibit growth in the environment of the target organ, as most patients display disseminated disease at the time of diagnosis and targeting mechanisms that inhibit for example intravasation may be of limited utility in the current clinical setting. Disseminated cells in a stressful milieu might be able to maintain a high level of p38 signaling that either induces apoptosis or dormancy. Indeed, two metastasis suppressor genes, RKIP and MKK4, are capable of inhibiting MEK/ERK and promoting JNK and p38 signaling, respectively (reviewed in refs. 25, 89), although it is still unclear if they induce apoptosis or dormancy in the target organ. Hence, a tumor cell that has reached the secondary organ and is able to bypass the negative signaling from the host micro-environment will start to grow (Fig. 4). Most likely, successful metastatic cells are those that can shape their microenvironment creating a niche for mitogenesis and tumor growth (i.e., FN matrix deposition and ERK activation).22 Thus, as proposed by Steven Paget more than a century ago, a proper niche (i.e., soil) may determine the ability of secondary tumors to grow.90
Although long periods may elapse before a tumor cell will start growing in the new microenvironment, a sine qua non is its ability to survive throughout this quiescence period. Is it possible that the ability to adapt to the stress imposed by dissemination and other exogenous factors is coupled to the acquisition of resistance to chemotherapy? It is assumed that chemotherapeutic drugs are inefficient at killing dormant cells because they do not divide. While this may be true, a formal proof of that phenomenon is still lacking, and our findings reviewed above suggest that other mechanisms may be operational.
CONCLUSIONS AND FUTURE DIRECTIONS
Our findings using a model of squamous carcinoma dormancy allow us to propose that the proliferative state of malignant cells appears to be dominant over the dormant phenotype. This conclusion is supported by the fact that inhibition of ERK signaling alone does not seem to reprogram the cells into a dormant phenotype (i.e., transient inhibition of ERK does not reprogram the cells into dormancy) and only a distinct qualitative signal generated by the combined inhibition of ERK and activation of p38 is able to engage a state of quiescence.49 Furthermore, our studies and those reviewed by Indraccolo et al., in this issue suggest that while dormancy can persist for long periods in animal models it is a fragile state that can be readily interrupted. In the HEp3 model, a simple perturbation of the signaling network caused by inhibition of p38 kinase activity for 48 hrs22,49 or transient transfection with uPAR cDNA22,49 or an shRNA that targets MKK6 (our unpublished results) is sufficient to reprogram these cells and interrupt dormancy. Indraccolo et al., recently showed91 that a similar transient burst in angiogenic activity can interrupt the dormancy of proliferation arrested Kaposi’s sarcoma cells. Thus, although the initiating stimuli may be different, these findings suggest that the dormant phenotype may be robust and persistent while cells are in a tissue in homeostasis. However, if key pathways maintaining dormancy are perturbed cells can switch back to tumorigenicity more readily than anticipated.
What is the short-term benefit of developing models to understand dormancy? The system we have developed is generating a wealth of information regarding mechanisms and potential markers of dormancy. It has been demonstrated that circulating tumor cells can be recovered from the bloodstream or bone marrow of patients considered “cured” of a previous cancer1,12 or in a disease free period. It would be interesting to determine whether markers such as the state of ERK, p38 and eIF2α phosphorylation in cytokeratin positive cells might be informative as to the outcome of those patients. In addition, it would be interesting to determine if the genetic changes detected in disseminated cells by comparative genomic hybridization (CGH) (see Klein and Hölzel this issue) have any correlation with the pathways that are being implicated in induction of dormancy in experimental models. An exciting long-term possibility is combining the inhibitors of ERK signaling (i.e., Mek, EGFR, ErbB2 inhibitors) with activators of p38 to induce dormancy of otherwise growing micro-metastasis.
The presence of tumor cells even after decades after primary tumor eradication suggests that the goal of minimal residual disease in cancer is achievable. Thus, models of dormancy may allow us to develop therapeutic strategies to induce and/or maintain dormancy. A drawback with this clinical alternative is the inherent risk of metastasis due to the presence of quiescent but still living cancer cells. However, considering that recurrence can occur >10 years after diagnosis, maintaining cells dormant may be a practical strategy. Moreover, as we summarized knowledge about the survival pathways that protect tumor cells while dormant may offer an additional therapeutic opportunity to fully eradicate the dormant cells.
Finally, it will be important to develop mouse models that faithfully recapitulate the mass of primary tumors and the pace at which cancer progresses in a human patient to be able to model MRD and metastasis. The use of inducible oncogene systems92,93 may help model this step of cancer progression. These models and the ones developed using human cancer cell lines described in this review may be useful to explore and understand the mechanisms of cancer dormancy.
Acknowledgments
This work is supported by a grant from the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program and the NIH/National Cancer Institute grant CA109182 (to J.A. Aguirre-Ghiso), and Ruth L. Kirschstein National Research Service Award (NIH/National Cancer Institute) Fellowship (to A.C. Ranganathan).
References
- 1.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, Herlyn D, Terstappen LW, Fehm T, Tucker TF, Lane N, Wang J, Uhr JW. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 2.Chambers AF, Naumov GN, Varghese HJ, Nadkarni KV, MacDonald IC, Groom AC. Critical steps in hematogenous metastasis: An overview. Surg Oncol Clin N Am. 2001;10:243–55. vii. [PubMed] [Google Scholar]
- 3.Fidler IJ. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 4.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 5.Demicheli R, Retsky MW, Swartzendruber DE, Bonadonna G. Proposal for a new model of breast cancer metastatic development. Ann Oncol. 1997;8:1075–80. doi: 10.1023/a:1008263116022. [DOI] [PubMed] [Google Scholar]
- 6.Demicheli R. Tumour dormancy: Findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001;11:297–306. doi: 10.1006/scbi.2001.0385. [DOI] [PubMed] [Google Scholar]
- 7.Fehm T, Sagalowsky A, Clifford E, Beitsch P, Saboorian H, Euhus D, Meng S, Morrison L, Tucker T, Lane N, Ghadimi BM, Heselmeyer-Haddad K, Ried T, Rao C, Uhr J. Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin Cancer Res. 2002;8:2073–84. [PubMed] [Google Scholar]
- 8.Racila E, Euhus D, Weiss AJ, Rao C, McConnell J, Terstappen LW, Uhr JW. Detection and characterization of carcinoma cells in the blood. Proc Natl Acad Sci USA. 1998;95:4589–94. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pantel K, von Knebel Doeberitz M. Detection and clinical relevance of micrometastatic cancer cells. Curr Opin Oncol. 2000;12:95–101. doi: 10.1097/00001622-200001000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Braun S, Pantel K. Clinical significance of occult metastatic cells in bone marrow of breast cancer patients. Oncologist. 2001;6:125–32. doi: 10.1634/theoncologist.6-2-125. [DOI] [PubMed] [Google Scholar]
- 11.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 12.Pantel K, Otte M. Occult micrometastasis: Enrichment, identification and characterization of single disseminated tumour cells. Semin Cancer Biol. 2001;11:327–37. doi: 10.1006/scbi.2001.0388. [DOI] [PubMed] [Google Scholar]
- 13.Tsavellas G, Patel H, Allen-Mersh TG. Detection and clinical significance of occult tumour cells in colorectal cancer. Br J Surg. 2001;88:1307–20. doi: 10.1046/j.0007-1323.2001.01863.x. [DOI] [PubMed] [Google Scholar]
- 14.Coello MC, Luketich JD, Litle VR, Godfrey TE. Prognostic significance of micrometastasis in nonsmall-cell lung cancer. Clin Lung Cancer. 2004;5:214–25. doi: 10.3816/CLC.2004.n.002. [DOI] [PubMed] [Google Scholar]
- 15.Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, Schlimok G, Diel IJ, Gerber B, Gebauer G, Pierga JY, Marth C, Oruzio D, Wiedswang G, Solomayer EF, Kundt G, Strobl B, Fehm T, Wong GY, Bliss J, Vincent-Salomon A, Pantel K. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005;353:793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- 16.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 17.Folkman J, Hanahan D. Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symp. 1991;22:339–47. [PubMed] [Google Scholar]
- 18.Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, Chambers AF. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 2002;62:2162–8. [PubMed] [Google Scholar]
- 19.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–73. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000;60:2541–6. [PubMed] [Google Scholar]
- 21.Naumov GN, MacDonald IC, Chambers AF, Groom AC. Solitary cancer sells as a possible source of tumor dormancy? Semin Cancer Biol. 2001;11:271–6. doi: 10.1006/scbi.2001.0382. [DOI] [PubMed] [Google Scholar]
- 22.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–79. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 24.Seger R, Krebs EG. The MAPK signaling cascade. Faseb J. 1995;9:726–35. [PubMed] [Google Scholar]
- 25.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 26.Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–61. [PubMed] [Google Scholar]
- 27.Esteva FJ, Sahin AA, Smith TL, Yang Y, Pusztai L, Nahta R, Buchholz TA, Buzdar AU, Hortobagyi GN, Bacus SS. Prognostic significance of phosphorylated P38 mitogen-activated protein kinase and HER-2 expression in lymph node-positive breast carcinoma. Cancer. 2004;100:499–506. doi: 10.1002/cncr.11940. [DOI] [PubMed] [Google Scholar]
- 28.Bulavin DV, Fornace AJ., Jr p38 MAP kinase’s emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. doi: 10.1016/S0065-230X(04)92005-2. [DOI] [PubMed] [Google Scholar]
- 29.Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:20490–4. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- 30.Engelberg D. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin Cancer Biol. 2004;14:271–82. doi: 10.1016/j.semcancer.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Rennefahrt U, Illert B, Greiner A, Rapp UR, Troppmair J. Tumor induction by activated JNK occurs through deregulation of cellular growth. Cancer Lett. 2004;215:113–24. doi: 10.1016/j.canlet.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 32.Ellinger-Ziegelbauer H, Kelly K, Siebenlist U. Cell cycle arrest and reversion of Ras-induced transformation by a conditionally activated form of mitogen-activated protein kinase kinase kinase 3. Mol Cell Biol. 1999;19:3857–68. doi: 10.1128/mcb.19.5.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback for Ras proliferative signaling. J Biol Chem. 2000;275:38973–80. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 34.Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J Biol Chem. 2002;277:31808–17. doi: 10.1074/jbc.M203964200. [DOI] [PubMed] [Google Scholar]
- 35.Timofeev O, Lee TY, Bulavin DV. A subtle change in p38 MAPK activity is sufficient to suppress in vivo tumorigenesis. Cell Cycle. 2005;4:118–20. doi: 10.4161/cc.4.1.1342. [DOI] [PubMed] [Google Scholar]
- 36.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, Appella E, Fornace AJ., Jr Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 37.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–78. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamaguchi H, Minopoli G, Demidov ON, Chatterjee DK, Anderson CW, Durell SR, Appella E. Substrate specificity of the human protein phosphatase 2Cdelta, Wip1. Biochemistry. 2005;44:5285–94. doi: 10.1021/bi0476634. [DOI] [PubMed] [Google Scholar]
- 39.Takekawa M, Adachi M, Nakah ata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. Embo J. 2000;19:6517–26. doi: 10.1093/emboj/19.23.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 41.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 42.Sherr CJ, DePinho RA. Cellular senescence: Mitotic clock or culture shock? Cell. 2000;102:407–10. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 43.Ishikawa F. Cellular senescence, an unpopular yet trustworthy tumor suppressor mechanism. Cancer Sci. 2003;94:944–7. doi: 10.1111/j.1349-7006.2003.tb01382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trost TM, Lausch EU, Fees SA, Schmitt S, Enklaar T, Reutzel D, Brixel LR, Schmidtke P, Maringer M, Schiffer IB, Heimerdinger CK, Hengstler JG, Fritz G, Bockamp EO, Prawitt D, Zabel BU, Spangenberg C. Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer Res. 2005;65:840–9. [PubMed] [Google Scholar]
- 45.Shevde LA, Welch DR. Metastasis suppressor pathways-an evolving paradigm. Cancer Lett. 2003;198:1–20. doi: 10.1016/s0304-3835(03)00304-5. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida BA, Dubauskas Z, Chekmareva MA, Christiano TR, Stadler WM, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 17. Cancer Res. 1999;59:5483–7. [PubMed] [Google Scholar]
- 47.Yamada SD, Hickson JA, Hrobowski Y, Vander Griend DJ, Benson D, Montag A, Karrison T, Huo D, Rutgers J, Adams S, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002;62:6717–23. [PubMed] [Google Scholar]
- 48.Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006;66:2264–70. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- 49.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–95. [PubMed] [Google Scholar]
- 50.Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell. 2002;1:445–57. doi: 10.1016/s1535-6108(02)00072-7. [DOI] [PubMed] [Google Scholar]
- 52.Choong PF, Nadesapillai AP. Urokinase plasminogen activator system: A multifunctional role in tumor progression and metastasis. Clin Orthop Relat Res. 2003:S46–58. doi: 10.1097/01.blo.0000093845.72468.bd. [DOI] [PubMed] [Google Scholar]
- 53.Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duffy MJ, O’Grady P, Devaney D, O’Siorain L, Fennelly JJ, Lijnen HJ. Urokinase-plasminogen activator, a marker for aggressive breast carcinomas. Preliminary report. Cancer. 1988;62:531–3. doi: 10.1002/1097-0142(19880801)62:3<531::aid-cncr2820620315>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 55.Fisher JL, Field CL, Zhou H, Harris TL, Henderson MA, Choong PF. Urokinase plas-minogen activator system gene expression is increased in human breast carcinoma and its bone metastases-a comparison of normal breast tissue, noninvasive and invasive carcinoma and osseous metastases. Breast Cancer Res Treat. 2000;61:1–12. doi: 10.1007/s10549-004-6659-9. [DOI] [PubMed] [Google Scholar]
- 56.Heiss MM, Allgayer H, Gruetzner KU, Funke I, Babic R, Jauch KW, Schildberg FW. Individual development and uPA-receptor expression of disseminated tumour cells in bone marrow: A reference to early systemic disease in solid cancer. Nat Med. 1995;1:1035–9. doi: 10.1038/nm1095-1035. [DOI] [PubMed] [Google Scholar]
- 57.Kook YH, Adamski J, Zelent A, Ossowski L. The effect of antisense inhibition of urokinase receptor in human squamous cell carcinoma on malignancy. Embo J. 1994;13:3983–91. doi: 10.1002/j.1460-2075.1994.tb06714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu W, Kim J, Ossowski L. Reduction in surface urokinase receptor forces malignant cells into a protracted state of dormancy. J Cell Biol. 1997;137:767–77. doi: 10.1083/jcb.137.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene. 2002;21:2513–24. doi: 10.1038/sj.onc.1205342. [DOI] [PubMed] [Google Scholar]
- 60.Chaurasia P, Aguirre-Ghiso JA, Liang OD, Gardsvoll H, Ploug M, Ossowski L. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J Biol Chem. 2006;281:14852–63. doi: 10.1074/jbc.M512311200. [DOI] [PubMed] [Google Scholar]
- 61.Ossowski L, Reich E. Loss of malignancy during serial passage of human carcinoma in culture and discordance between malignancy and transformation parameters. Cancer Res. 1980;40:2310–5. [PubMed] [Google Scholar]
- 62.Ossowski L, Reich E. Changes in malignant phenotype of a human carcinoma conditioned by growth environment. Cell. 1983;33:323–33. doi: 10.1016/0092-8674(83)90414-2. [DOI] [PubMed] [Google Scholar]
- 63.Aguirre-Ghiso JA, Ossowski L, Rosenbaum SK. Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res. 2004;64:7336–45. doi: 10.1158/0008-5472.CAN-04-0113. [DOI] [PubMed] [Google Scholar]
- 64.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced upregulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–11. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–8. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–98. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rutkowski DT, Kaufman RJ. A trip to the ER: Coping with stress. Trends Cell Biol. 2004;14:20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 68.Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–73. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 69.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 71.Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–67. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- 72.Obata T, Brown GE, Yaffe MB. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit Care Med. 2000;28:N67–77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- 73.Olson JM, Hallahan AR. p38 MAP kinase: A convergence point in cancer therapy. Trends Mol Med. 2004;10:125–9. doi: 10.1016/j.molmed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 74.Zechner D, Craig R, Hanford DS, McDonough PM, Sabbadini RA, Glembotski CC. MKK6 activates myocardial cell NF-kappaB and inhibits apoptosis in a p38 mitogen-activated protein kinase-dependent manner. J Biol Chem. 1998;273:8232–9. doi: 10.1074/jbc.273.14.8232. [DOI] [PubMed] [Google Scholar]
- 75.Weldon CB, Parker AP, Patten D, Elliott S, Tang Y, Frigo DE, Dugan CM, Coakley EL, Butler NN, Clayton JL, Alam J, Curiel TJ, Beckman BS, Jaffe BM, Burow ME. Sensitization of apoptotically-resistant breast carcinoma cells to TNF and TRAIL by inhibition of p38 mitogen-activated protein kinase signaling. Int J Oncol. 2004;24:1473–80. [PubMed] [Google Scholar]
- 76.Vega MI, Huerta-Yepaz S, Garban H, Jazirehi A, Emmanouilides C, Bonavida B. Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: Pivotal role of p38 MAPK in drug resistance. Oncogene. 2004;23:3530–40. doi: 10.1038/sj.onc.1207336. [DOI] [PubMed] [Google Scholar]
- 77.Carvalho H, Evelson P, Sigaud S, Gonzalez-Flecha B. Mitogen-activated protein kinases modulate H(2)O(2)-induced apoptosis in primary rat alveolar epithelial cells. J Cell Biochem. 2004;92:502–13. doi: 10.1002/jcb.20070. [DOI] [PubMed] [Google Scholar]
- 78.Niwa M, Walter P. Pausing to decide. Proc Natl Acad Sci USA. 2000;97:12396–7. doi: 10.1073/pnas.250476097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997;272:4327–34. doi: 10.1074/jbc.272.7.4327. [DOI] [PubMed] [Google Scholar]
- 80.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 81.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: Friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 82.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell. 2005;16:5493–501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci USA. 2000;97:12625–30. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci USA. 1999;96:8505–10. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bjornsti MA, Houghton PJ. Lost in translation: Dysregulation of cap-dependent translation and cancer. Cancer Cell. 2004;5:519–23. doi: 10.1016/j.ccr.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 86.Clemens MJ. Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene. 2004;23:3180–8. doi: 10.1038/sj.onc.1207544. [DOI] [PubMed] [Google Scholar]
- 87.Kauffman EC, Robinson VL, Stadler WM, Sokoloff MH, Rinker-Schaeffer CW. Metastasis suppression: The evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site. J Urol. 2003;169:1122–33. doi: 10.1097/01.ju.0000051580.89109.4b. [DOI] [PubMed] [Google Scholar]
- 88.Berger JC, Vander Griend DJ, Robinson VL, Hickson JA, Rinker-Schaeffer CW. Metastasis suppressor genes: From gene identification to protein function and regulation. Cancer Biol Ther. 2005:4. doi: 10.4161/cbt.4.8.1865. [DOI] [PubMed] [Google Scholar]
- 89.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 90.Paget S. The distribution of secondary growths in cancer of the breast. The Lancet. 1889;1:571–3. [PubMed] [Google Scholar]
- 91.Indraccolo S, Stievano L, Minuzzo S, Tosello V, Esposito G, Piovan E, Zamarchi R, Chieco-Bianchi L, Amadori A. Interruption of tumor dormancy by a transient angiogenic burst within the tumor microenvironment. Proc Natl Acad Sci USA. 2006;103:4216–21. doi: 10.1073/pnas.0506200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shachaf CM, Felsher DW. Tumor dormancy and MYC inactivation: Pushing cancer to the brink of normalcy. Cancer Res. 2005;65:4471–4. doi: 10.1158/0008-5472.CAN-05-1172. [DOI] [PubMed] [Google Scholar]
- 93.Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, Innocent N, Cardiff RD, Schnall MD, Chodosh LA. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–61. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]