

Abstract
In the recent X-ray crystallographic structural models of photosystem II, Asp342 of the D1 polypeptide is assigned as a ligand of the oxygen-evolving Mn4 cluster. To determine if D1-Asp342 ligates a Mn ion that undergoes oxidation during one or more of the S0 → S1, S1 → S2, and S2 → S3 transitions, the FTIR difference spectra of the individual S state transitions in D1-D342N mutant PSII particles from the cyanobacterium Synechocystis sp. PCC 6803 were compared with those in wild-type PSII particles. Remarkably, the data show that the mid-frequency (1800 – 1200 cm−1) FTIR difference spectra of wild-type and D1-D342N PSII particles are essentially identical. Importantly, the mutation alters none of the carboxylate vibrational modes that are present in the wild-type spectra. The absence of significant mutation-induced spectral alterations in D1-D342N PSII particles shows that the oxidation of the Mn4 cluster does not alter the frequencies of the carboxylate stretching modes of D1-Asp342 during the S0 → S1, S1 → S2, or S2 → S3 transitions. One explanation of these data is that D1-Asp342 ligates a Mn ion that does not increase its charge or oxidation state during any of these S state transitions. However, because the same conclusion was reached previously for D1-Asp170, and because the recent X-ray crystallographic structural models assign D1-Asp170 and D1-Asp342 as ligating different Mn ions, this explanation requires that (1) the extra positive charge that develops on the Mn4 cluster during the S1 → S2 transition be localized on the Mn ion that is ligated by the α-COO− group of D1-Ala344 and (2) any increase in positive charge that develops on the Mn4 cluster during the S0 → S1 and S2 → S3 transitions be localized on the one Mn ion that is not ligated by D1-Asp170, D1-Asp342, or D1-Ala344. In separate experiments that were conducted with L-[1-13 C]alanine, we found no evidence that D1-Asp342 ligates the same Mn ion that is ligated by the α-COO− group of D1-Ala344.
The catalytic site of water oxidation in photosystem II (PSII)1 contains a cluster of four Mn ions and one Ca ion. The Mn4 cluster accumulates oxidizing equivalents in response to light-induced electron transfer reactions within PSII, thereby serving as the interface between one-electron photochemistry and the four-electron process of water oxidation (for reviews, see refs 1–4). During each catalytic cycle, the Mn4 cluster cycles through five oxidation states termed Sn, where “n” denotes the number of oxidizing equivalents that have been stored (n = 0 – 4). The S1 state predominates in dark-adapted samples. Most interpretations of Mn-XANES data have concluded that the S1 state consists of two Mn(III) and two Mn(IV) ions and that the S2 state consists of one Mn(III) and three Mn(IV) ions (for review, see refs 5 and 6). Whether the additional oxidizing equivalent of the S3 state is localized on a Mn ion (7, 8) or on a Mn ligand (9, 10) remains in dispute. The S4 state is a transient intermediate that reverts to the S0 state with the concomitant release of O2.
The amino acid residues that ligate the Mn4 cluster are provided mainly by the D1 polypeptide, one of the core subunits of PSII. One of these residues is D1-Asp342. This residue was originally proposed as a possible ligand of the Mn4Ca cluster on the basis of early mutagenesis studies (11, 12) and is assigned as a ligand to Mn in the recent X-ray crystallographic structural models (13, 14). In the ~ 3.5 Å structural model (13), this residue is a unidentate ligand of a single Mn ion. In the ~ 3.0 Å structural model (14), this residue bridges two Mn ions, including the Mn ion that is ligated (in this model) by the α-COO− group of D1-Ala344. The differences between the two structural models are probably caused by differences in data quality, extent of radiation damage, and approach to interpreting the electron density. Recent XANES and EXAFS studies of PSII single crystals (15) and PSII membrane multilayers (16) have provided compelling evidence that the X-ray doses that were used in the crystallographic studies to irradiate the PSII crystals would have rapidly reduced the Mn4Ca cluster’s oxidized Mn(III/IV) ions to their fully reduced Mn(II) states and that this radiation-induced reduction would have significantly perturbed the structure of the Mn4Ca cluster, disrupting μ-oxo bridges and altering Mn-ligand interactions (15, 16). The extent that the ~ 3.5 and ~ 3.0 structural models of the Mn4Ca cluster and its ligation environment are distorted from the native structure remains to be determined.
To obtain independent evidence that D1-Asp342 ligates the Mn4 cluster, and to determine if D1-Asp342 ligates a Mn ion that undergoes oxidation during one or more of the individual S state transitions, we have compared the FTIR difference spectra of the S0 → S1, S1 → S2, S2 →S3, and S3 → S0 transitions in D1-D342N mutant PSII particles from the cyanobacterium Synechocystis sp. PCC 6803 with those in wild-type PSII particles. Both wild-type and mutant PSII preparations exhibit an extensive array of vibrational modes that undergo shifts in frequency during the individual S state transitions. Many of these shifts produce strikingly large features in specific Sn+1-minus-Sn FTIR difference spectra, and many are clearly attributable to frequency shifts of carboxylate residues (17–29). The identification of these modes will complement the X-ray crystallographic studies by providing information about the dynamic structural changes that accompany water oxidation. Few of these modes have yet to be identified (25, 27, 30, 31). We recently employed isotopic labeling to identify the symmetric carboxylate stretching [νsym (COO−)] mode of the α-COO− group of D1-Ala344 in the S2-minus-S1 FTIR difference spectrum of wild-type PSII particles containing Ca or Sr (30, 31) [also see refs 25 and 29]. Because the substitution of Sr for Ca significantly altered several νsym (COO−) modes, including some that may correspond to one or more metal ligands, but importantly did not alter the νsym (COO−) mode of the α-COO− group of D1-Ala344, we concluded that D1-Ala344 ligates Mn rather than Ca (31) 2. The frequency of the νsym (COO−) mode of D1-Ala344 in the S1 state and its ~ 17 cm−1 or ~ 36 cm−1 downshift in the S2 state imply that this group is a unidentate ligand of a Mn ion whose charge increases during the S1 → S2 transition (25, 30, 31). A similar identification of one of the carboxylate stretching modes of D1-Asp342 in any of the Sn → Sn+1 transitions could provide unequivocal spectroscopic evidence for the ligation of the Mn4 cluster by D1-Asp342 and could provide information about the type of carboxylate ligation and the environment of the carboxylate group. If D1-Asp342 is ligand of a Mn ion whose charge increases during a specific Sn → Sn+1 transition, the increased charge should weaken the ligating Mn-O bond(s), thereby decreasing the frequency of the D1-Asp342 symmetric carboxylate stretching mode and possibly shifting the frequency of the D1-Asp342 asymmetric carboxylate stretching mode. The shifted mode(s) should appear in the corresponding Sn+1-minus-Sn FTIR difference spectrum of wild-type PSII particles but not in the spectra of D1-D342N PSII particles because the mutation has eliminated the carboxylate group. The absence of the mode in the mutant would permit its identification in the wild-type control.
Our data show that the mid- frequency (1800 – 1200 cm−1 ) S1-minus-S0, S2-minus-S1, S3-minus-S2, and S0-minus-S3 FTIR difference spectra of D1-D342N PSII particles are remarkably similar to those of wild-type PSII particles. Importantly, the D1-D342N mutation eliminates no carboxylate modes from any of the difference spectra. The simplest explanation for these data is that the Mn ion that is ligated by D1-Asp342 does not change its charge or oxidation state during the S0 → S1, S1 → S2, or S2 → S3 transitions. In separate experiments that were conducted with L-[1-13 C]alanine, we found no evidence that D1-Asp342 ligates the same Mn ion that is ligated by the α-COO− group of D1-Ala344.
MATERIALS AND METHODS
Construction of Mutant and Propagation of Cultures
The D1-D342N mutation was constructed in the psbA-2 gene of Synechocystis sp. PCC 6803 (33) and transformed into a host strain of Synechocystis that lacks all three psbA genes and contains a hexahistidine-tag (His-tag) fused to the C-terminus of CP47 (34). Single colonies were selected for ability to grow on solid media containing 5 μg/mL kanamycin monosulfate. The control WT(D1) strain was constructed in an identical fashion except that the transforming plasmid carried no site-directed mutation. The designation “WT(D1)” differentiates this strain from the native wild-type strain that contains all three psbA genes, lacks a His-tag on the C-terminus of CP47, and is sensitive to antibiotics. Cells were propagated as described previously (30), but in the presence of 5 mM glucose and in three 7 L carboys instead of in multiple 1 L flasks. The light sensitivity of the D1-D342N strain that was noted previously for small scale cultures (12) was not a problem for cells propagated in carboys. For the isolation of isotopically-labeled PSII particles, the liquid media contained 0.5 mM L-[1-13 C]alanine (99% 13 C enrichment, Cambridge Isotope Laboratories, Andover, MA) (30, 31) (the presence of 5 mM glucose in the growth medium produced no noticeable difference in the FTIR difference spectra of isotopically labeled WT(D1) PSII particles). To verify the integrity of the mutant cultures that were harvested for the purification of PSII particles, an aliquot of each culture was set aside and the complete sequence of the psbA-2 gene was obtained after PCR amplification of genomic DNA (33). No trace of the wild-type codon was detected in any of the mutant cultures.
Purification of PSII particles
Isolated PSII particles were purified under dim green light at 4 °C with Ni-NTA superflow affinity resin (Qiagen, Valentia, CA) as described previously (31). For most samples, the purification buffer consisted of 1.2 M betaine, 10% (v/v) glycerol, 50 mM MES-NaOH (pH 6.0), 20 mM CaCl2, 5 mM MgCl2, 50 mM histidine, 1 mM EDTA, and 0.03% (w/v) n-dodecyl β-D -maltoside. For some samples, the MES-NaOH and CaCl2 concentrations were each lowered to 10 mM. This change produced no noticeable difference in the characteristics of the purified PSII particles. The purified PSII particles were concentrated to ~ 1.0 mg of Chl/mL by ultrafiltration, frozen in liquid N 2, and stored at −196 °C (vapor phase nitrogen). Mn-depleted PSII particles were prepared with hydroxylamine and EDTA, as described previously (30).
EPR Measurements
Continuous-wave EPR spectra were recorded with a Bruker ECS-106 X-band EPR spectrometer that was equipped with a Bruker ER-4116 dual mode cavity. Cryogenic temperatures were achieved with an Oxford ESR900 liquid helium cryostat. The temperature was controlled with an Oxford ITC503 temperature and gas flow controller that was equipped with a Cernox (Lake Shore Cryotronics, Westerville, OH) temperature sensor. Sample manipulations were conducted under dim green light at 4 °C. The samples were concentrated to approximately 7.5 mg of Chl/mL with Centricon-100 concentrators (Millipore Corp., Bedford, MA), deaerated by gently bubbling with a stream of Ar gas, transferred to standard quartz 4 mm OD EPR tubes (Wilmad LabGlass, Buena, NJ) under an atmosphere of Ar gas, dark-adapted for 2 h on ice, then frozen in liquid nitrogen. The S2 state was generated by illuminating samples for 5 min in a non-silvered Dewar at 198 K (dry ice/ethanol) with a focused, heat-filtered, 350-Watt Radiac light source. The samples were then immediately frozen in liquid nitrogen.
Preparation of FTIR Samples
All manipulations were conducted under dim green light at 4 °C. For experiments conducted at 250 K, samples were exchanged into FTIR analysis buffer [40 mM sucrose, 10 mM MES-NaOH (pH 6.0), 5 mM CaCl2, 5 mM NaCl, 0.06% (w/v) n-dodecyl β-D-maltoside (22, 26)] by repeated concentration/dilution cycles. For experiments conducted at 273 K, most samples were exchanged into FTIR analysis buffer by passage through a centrifugal gel filtration column (28), although one set of samples [WT(D1b) in Figure 4] was exchanged into FTIR buffer by repeated concentration/dilution cycles. Concentrated samples (approx. 10 μL in volume) were mixed with 1/10 volume of fresh 100 mM potassium ferricyanide (dissolved in water), spread to a diameter of about 10 mm on a 15 mm diameter BaF2 window, then dried lightly (until tacky) under a stream of dry nitrogen gas. For experiments conducted at 250 K, the lightly-dried samples were placed in a humidifier at 95% RH for 10 min. For experiments conducted at 273 K, 1 μL of 20% (v/v) glycerol (in water) was spotted onto the window, adjacent to the lightly-dried sample, but not touching it, to maintain the humidity of the sample in the FTIR cryostat at 99% RH (19). A second IR window with a Teflon spacer (0.5 mm thick) was placed over the first and sealed in place with silicon-free high-vacuum grease. The sample was immediately loaded into the FTIR cryostat and allowed to equilibrate in darkness to 250.0 K (for 2 – 4 h) or 273.0 K (for 2 h). Sample concentrations and thicknesses were adjusted so that the absolute absorbance of the amide I band at 1657 cm−1 was 0.7 – 1.1.
FIGURE 4.

Comparison of the mid-frequency FTIR difference spectra of WT(D1) and D1-D342N PSII particles in response to the first (A), second (B), third (C), and fourth (D) of six successive flash illuminations applied at 273 K [traces (a) in each panel]. The spectra (plotted from 1760 cm−1 to 1170 cm−1 ) correspond predominantly to S2-minus-S1, S3-minus-S2, S0-minus-S3, and S1-minus-S0 FTIR difference spectra, respectively. Spectra of two wild-type samples [WT(D1a) and WT(D1b)] are shown in each panel. The WT(D1a) and D1-D342N samples were exchanged into FTIR buffer by passage through a centrifugal gel filtration column, while the WT(D1b) sample was exchanged by repeated concentration/dilution cycles (see text for details). To facilitate comparisons, after normalization to the peak-to-peak amplitudes of the negative ferricyanide peak at 2115 cm−1 and the positive ferrocyanide peak at 2038 cm−1, the D1-D342N spectra in (A-D) were multiplied vertically by factors of ~ 1.3, ~ 1.5, ~ 1.5, and ~ 1.6, respectively, to maximize overlap with the WT(D1) spectra between 1500 and 1300 cm−1. In (A), the D1-D342N spectrum was corrected for the presence of a significant population of Mn-depleted mutant PSII reaction centers (see text). Double-difference spectra (b, c, d in each panel) were calculated by directly subtracting the normalized traces shown in (a). The horizontal dashed lines indicate the zero levels. In (E), dark-minus-dark control traces of the WT(D1a), WT(D1b), and D1-D342N PSII particles are included to show the noise levels. The color coding in (E) is the same as in (a) of panels A – D. The WT(D1a) and WT(D1b) spectra represent the averages from nine samples (10,800 scans). The D1-D342N spectra represent the averages from 18 samples (21,600 scans).
Measurement of FTIR Spectra
Mid-frequency FTIR spectra were recorded with a Bruker Equinox 55 spectrometer (Bruker Optics, Billerica, MA) at a spectral resolution of 4 cm−1 as described previously (26, 28, 30, 31). The spectral resolution for all spectra was 4 cm−1. Flash-illumination (~ 20 mJ/flash, ~ 7 ns fwhm) was provided by a frequency-doubled Q-switched Nd:YAG laser [Surelite I (Continuum, Santa Clara, CA)]. For experiments conducted at 250 K (30, 31), each sample was flash-illuminated only once. The single beam spectrum that was recorded after the flash was divided by the single-beam spectrum that was recorded before the flash and the ratio was converted to units of absorption. Each single-beam spectrum consisted of 800 scans. For experiments conducted at 273 K (26, 28), after dark-adaptation, one pre-flash was applied followed by 5 min of additional dark-adaptation. This treatment was employed to oxidize YD and to maximize the proportion of centers in the S1 state. Six successive flashes then were applied with an interval of 12.2 sec between each. Two single beam spectra were recorded before the first flash and one single-beam spectrum was recorded starting 0.33 sec after the first and subsequent flashes (each single-beam spectrum consisted of 100 scans). The 0.33 sec delay was incorporated to allow for the oxidation of QA•− by the ferricyanide. To obtain difference spectra corresponding to successive S state transitions, the single-beam spectrum that was recorded after the nth flash was divided by the single-beam spectrum that was recorded immediately before the nth flash and the ratio was converted to units of absorption. To estimate the background noise level, the second pre-flash single-beam spectrum was divided by the first and the ratio was converted to units of absorption. The sample was dark-adapted for 30 min, then the entire cycle was repeated, including the pre-flash and the 5 min additional dark-adaptation period. The entire cycle was repeated 12 times for each sample and the difference spectra recorded with several samples were averaged.
Other Procedures
Chlorophyll concentrations and light-saturated rates of O2 evolution were measured as described previously (30).
RESULTS AND DISCUSSION
The O2 evolving activity of the purified D1 -D342N PSII particles was 1.4 – 1.8 mmol O2 (mg of Chl)−1 h−1 compared to 5.1 – 5.5 mmol O2 (mg of Chl)−1 h−1 for WT(D1) PSII particles. The lower O2 evolving activity of the D1-D342N PSII particles (25 – 35 % compared to wild-type) was expected because the O2 evolving activity of D1-D342N cells has been reported to be only ~ 33% compared to wild-type (12).
A parallel polarization, integer spin, multiline EPR signal was observed in dark adapted D1-D342N PSII particles (Figure 1A, lower trace). In terms of peak positions and spacings, this signal closely resembles the S1 state multiline EPR signal that is observed in WT(D1) PSII particles (Figure 1A, upper trace) and we assign it to the S1 state. A perpendicular polarization multiline EPR signal was observed in the same mutant PSII particles after illumination at 195 K (Figure 1B, lower trace). In terms of peak positions and spacings, this signal closely resembles the S2 state multiline EPR signal that is observed in WT(D1) PSII particles after illumination (Figure 1B, upper trace) and we assign it to the S2 state. Illumination produced no g ≈ 4.1 EPR signal nor any other recognizable Mn EPR signal in either sample (not shown). The similarity of the S1 and S2 state multiline EPR signals in the mutant compared to wild-type shows that the magnetic interactions that give rise to these signals (35, 36) are not appreciably altered by the D1-D342N mutation. In contrast, both the D1-H332E mutation (34, 37) and the substitution of Sr for Ca (e.g., see refs 31 and 38) perturb these interactions and substantially alter the appearance of the S2 state multiline EPR signal.
FIGURE 1.
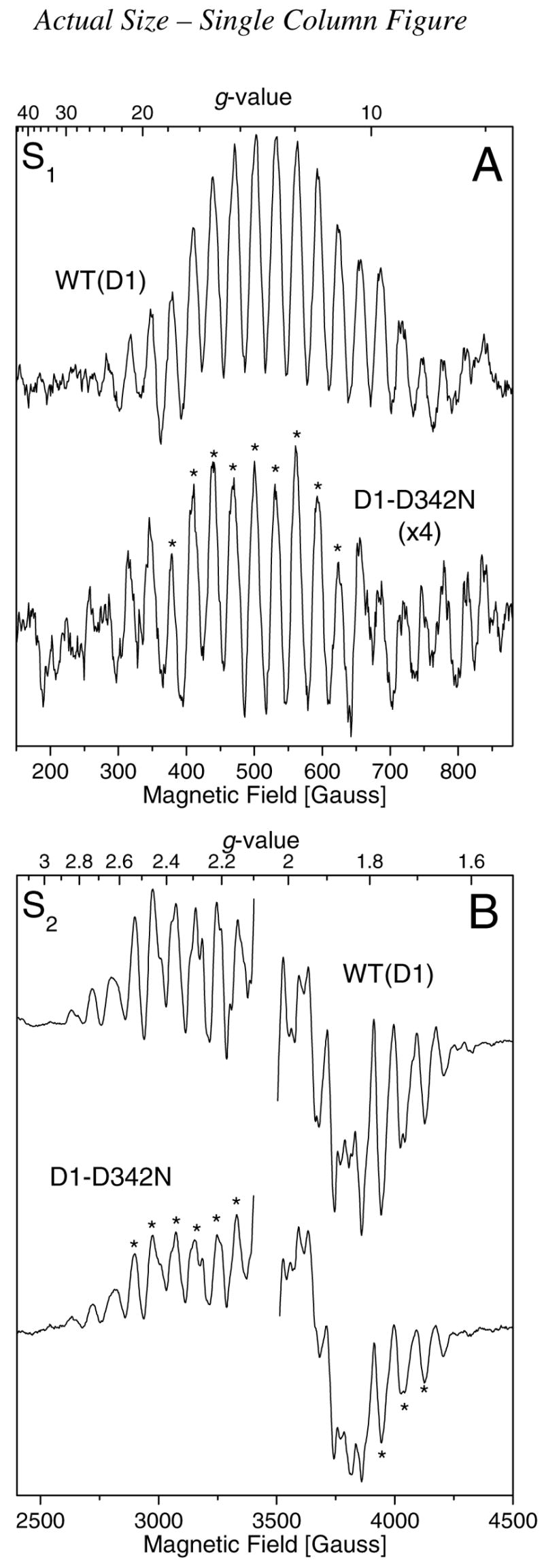
Comparison of the parallel polarization S1 state multiline EPR signals (A) and light-minus-dark perpendicular polarization S2 state multiline EPR signals (B) of Synechocystis WT(D1) (upper traces) and D1-D342N PSII particles (lower traces). The same samples were examined in both (A) and (B), with the data in (A) being recorded before that in (B). The WT(D1) and mutant samples each contained approx. 7.5 mg of Chl/mL in 1.2 M betaine, 10% (v/v) glycerol, 10 mM MES-NaOH (pH 6.0), 10 mM CaCl2, 5 mM MgCl2, 50 mM histidine, 1 mM EDTA, and 0.03% (w/v) n-dodecyl β-D-maltoside. Samples were deaerated prior to measurement to remove dissolved O 2. For panel (A), the experimental conditions were as follows: microwave frequency of 9.42 GHz, microwave power of 50 mW, modulation amplitude of 9 G, modulation frequency of 100 kHz, time constant of 81 ms, conversion time of 162 ms, and temperature of 3.5 ± 0.5 K. For panel (B), the experimental conditions were as follows: microwave frequency of 9.68 GHz, microwave power of 10 mW, modulation amplitude of 9 G, modulation frequency of 100 kHz, time constant of 40 ms, conversion time of 81 ms, and temperature of 7.0 ± 0.5 K. To generate the S2 state, the samples in (B) were illuminated for 5 min at 195 K before being flash-frozen in liquid nitrogen. Both spectra in (B) have had the large signal of YD• at g = 2 excised for clarity. Each spectrum in (A) and (B) represents the accumulation of 60 scans. To facilitate comparison, the S1 state multiline EPR signal of the D1-D342N PSII particles in (A) has been multiplied vertically by a factor of 4.
For the S1 state multiline EPR signal, the integrated area of the nine peaks indicated in the mutant spectrum (asterisks in Figure 1A) was approximately 23% of the integrated area of the corresponding peaks in the WT(D1) spectrum. For the S2 state multiline EPR signal, the integrated area of the nine peaks indicated in the mutant spectrum (asterisks in Figure 1B) was approximately 54% of the integrated area of the corresponding peaks in the WT(D1) spectrum. Because the two EPR signals were recorded on the same sample, with the dark-adapted S1 state signal recorded before illumination to produce the S2 state, we conclude that about 54% of the D1-D342N PSII particles contained Mn4Ca clusters. The presence of a significant fraction of mutant PSII reaction centers without Mn4Ca clusters is expected because we previously estimated that a significant fraction (30 – 40%) of D1-D342N PSII reaction centers lack Mn4Ca clusters in vivo on the basis of Chl fluorescence studies of intact cells (12). That significant fractions of D1-D342N PSII reaction centers lack Mn4Ca clusters in cells and PSII particles implies that the Mn4Ca cluster is assembled less efficiently or is less stable in D1-D342N than in wild-type, as was concluded previously (12). That the steady- state rates of O2 evolution in D1-D342N cells and PSII particles (relative to wild-type) are less than the fractions of PSII reaction centers estimated to contain Mn4Ca clusters implies that the Mn4Ca clusters in D1-D342N produce O2 lessefficiently than those in wild -type. On the basis of the relative amplitudes of the S2 state multiline signals and the relative light-saturated rates of O2 evolution, we estimate that the Mn4Ca clusters in D1-D342N PSII particles evolve O2 at 50 – 65% the rate of the Mn4Ca clusters in wild-type PSII particles. The reasons for the different relative amplitudes of the S1 and S2 multiline EPR signals in the mutant and wild-type PSII particles are unknown. The discrepancy presumably reflects our limited understanding of the factors that influence the S1 state multiline EPR signal.
To determine if the D1-D342N mutation alters the νsym (COO− ) mode of the α-COO− group of D1-Ala344, the position of this mode was visualized by purifying PSII particles from cells that had been propagated in media containing L-[1-13 C]alanine. A comparison of the mid-frequency S2-minus-S1 FTIR difference spectra of WT(D1) and D1-D342N PSII particles containing either unlabeled (12 C) alanine (black traces) or L-[1-13 C]alanine (red traces) is shown in Figures 2A and 2B. The spectrum of D1-D342N PSII particles (Figure 2B) resembles that of WT(D1) (Figure 2A). The most prominent differences are (1) the differential peaks at (−)1705/(+)1696 cm−1 and (+)1552/(−)1543 cm−1 appear with significantly larger amplitudes in the mutant, (2) the single positive peak at ~ 1507 cm−1 in WT(D1) is split into a doublet at ~ 1511 and ~ 1504 cm−1 in the mutant, and (3) the positive peak at ~ 1365 cm−1 has a lower amplitude in the mutant. The larger amplitude differential peaks and the positive peak at ~ 1504 cm−1 are characteristic of the light-minus-dark FTIR difference spectrum of YD•-minus-YD that is observed in Mn-depleted PSII particles under the same experimental conditions (i.e., a single flash applied to dark-adapted samples at 250 K), as is the positive feature at ~ 1599 cm−1 that appears in L-[1-13 C]alanine-labeled Mn-depleted PSII particles (30). Previously, we showed that the incorporation of [1-13 C]alanine produces no changes between 1450 and 1200 cm−1 in the light-minus-dark FTIR difference spectrum of Mn-depleted PSII particles (see Figure 4 of ref 30). Consequently, the [1-13 C]alanine-induced changes that are apparent in the νsym (COO−) region of the S2-minus-S1 FTIR difference spectrum of D1-D342N PSII particles (Figure 2B) can be attributed to D1-D342N PSII particles that contain Mn4Ca clusters, and not to PSII particles that lack Mn4Ca clusters.
FIGURE 2.

Comparison of the mid-frequency (1800 – 1200 cm−1) S2-minus-S1 FTIR difference spectra of PSII particles purified from Synechocystis WT(D1) (A) or D1-D342N (B) cells that had been propagated in media containing either unlabeled (12 C) L-alanine (black traces) or L-[1-13 C]alanine (red traces). The sample temperature was 250 K. The spectra in (A) are reproduced from ref (31). The spectra of the unlabeled PSII particles have been normalized to the peak-to-peak amplitudes of the negative ferricyanide peak at 2115 cm−1 and the positive ferrocyanide peak at 2038 cm−1. The unlabeled and L-[1-13 C]alanine-labeled WT(D1) traces in (A) each represent the average of six samples (4,800 scans). The unlabeled and L-[1-13 C]alanine-labeled D1-D342N traces in (B) represent the averages of 12 samples (9,600 scans) and 16 samples (12,800 scans), respectively. Within each panel, the spectra have been normalized to maximize their overlap between 1500 and 1400 cm−1. Panel (C) shows the double difference spectra, 12 C-minus-13 C, of WT(D1) PSII particles (black trace) and D1-D342N PSII particles (red trace) that were obtained by subtracting the S2-minus-S1 FTIR difference spectra of L-[1-13 C]alanine-labeled PSII particles from the S2-minus-S1 FTIR difference spectra of unlabeled PSII particles (the spectra shown in Figures 2A and 2B were subtracted directly). Only the regions between 1380 and 1270 cm−1 are shown. To facilitate comparison, the 12 C-minus-13 C spectrum of D1-D342N PSII particles in (C) was multiplied vertically by a factor of 1.8.
To isolate the L-[1-13 C]alanine-shifted νsym(COO−) modes and to display them more clearly, the 12 C-minus-13 C double difference spectra of the region between 1380 and 1270 cm−1 in the WT(D1) and D1-D342N samples are presented in Figure 2C. The data show that the positions of the L-[1-13 C]alanine-shifted modes in the S1 and S2 states are nearly the same in D1-D342N mutant PSII particles as in WT(D1) PSII particles. In both samples, the data are consistent with a single S1 state mode at ~ 1356 cm−1 in the unlabeled samples shifting to ~ 1337 or ~ 1320 cm−1 after the incorporation of L-[1-13 C]alanine, and with a single S2 state mode at ~ 1320 or ~ 1337 cm−1 shifting to ~ 1302 cm−1. These L-[1-13 C]alanine-shifted modes were observed previously in wild-type PSII particles from Synechocystis sp. PCC 6803 and were shown to originate from the νsym(COO− ) mode of the α-COO− group of D1-Ala344 (25, 30, 31).3
The data of Figure 2C show that the D1-D342N mutation does not shift the νsym(COO−) mode of D1-Ala344 significantly in either the S1 or S2 states. If D1-Asp342 ligated the same Mn ion as D1-Ala344, changing the anionic carboxylate moiety of Asp to the neutral amide moiety of Asn should perturb the vibrational frequencies of the remaining ligands. This perturbation should be manifested as a shift of the symmetric carboxylate stretching mode of the α-COO− group of D1-Ala344 in both the S1 and S2 states. The data of Figure 2C show that this is not the case. Therefore, we conclude that D1-Asp342 does not ligate the same Mn ion as the α-COO− group of D1-Ala344.
To determine if D1-Asp342 ligates a Mn ion that undergoes oxidation during one or more of the S0 → S1, S1 → S2, and S2 → S3 transitions, we sought to compare the FTIR difference spectra of the individual S state transitions in WT(D1) and D1-D342N PSII particles. However, because our EPR and 250 K FTIR data (Figures 1 and 2) showed the absence of Mn4Ca clusters in significant fractions of purified D1-D342N PSII particles, we first compared the FTIR difference spectra of D1-D342N and Mn-depleted WT(D1) PSII particles to ascertain the possible contribution to the mutant spectra of PSII particles lacking the Mn4Ca cluster. In Figure 3, we compare the FTIR difference spectra of D1-D342N and Mn-depleted WT(D1) PSII particles that were induced by four successive flashes applied at 273 K under conditions that promote S-state turnover. The Mn-depleted WT(D1) PSII particles show substantial spectral features in response to the first flash, but show very small features in response to the second, third, and fourth flashes. Clearly, an oxidized electron donor is photoaccumulated with high quantum yield in Mn-depleted WT(D1) PSII particles under these conditions. The most prominent spectral features of this oxidized electron donor, apparent in the first flash spectrum, include differential peaks at (−)1706/(+)1698 cm−1, positive peaks at 1677, 1651, 1550, 1531, and 1512 cm−1, and negative peaks at 1624, 1453, and 1250 cm−1. These spectral features identify this FTIR difference spectrum as that of YZ•-minus-YZ (39). Evidently, in lightly dried Synechocystis PSII particles at 99% RH in the presence of excess potassium ferricyanide, YZ• is stable for tens of seconds at 273 K, persisting for sufficient time to contribute to the first flash spectrum and to remain present when the subsequent flashes are applied. After normalizing the first flash spectra to the peak-to-peak amplitudes of the negative ferricyanide band at 2115 cm−1 and the positive ferrocyanide band at 2038 cm−1, the amplitude of the (−)1707 cm−1 band in the D1-D342N PSII particles was found to be ~ 2.6-fold smaller than in the Mn-depleted WT(D1) PSII particles. If we assume that this band arises entirely from mutant PSII particles that lack Mn4Ca clusters, then we estimate that ~ 38% of the mutant D1-D342N PSII particles lack Mn4Ca clusters, an estimate that is in general agreement with our EPR data (described above) and with data that was presented earlier in ref 12. 4
FIGURE 3.
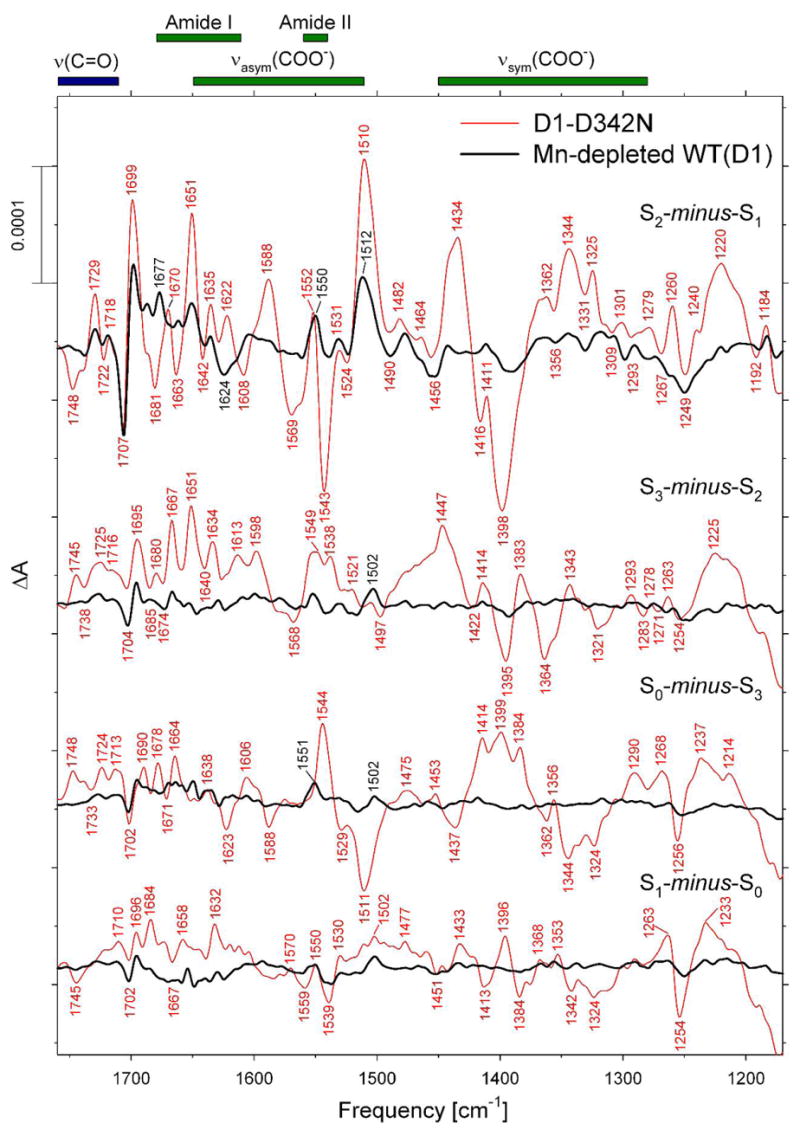
Comparison of the mid-frequency FTIR difference spectra of Mn-depleted WT(D1) and intact D1-D342N PSII particles in response to four successive flash illuminations applied at 273 K. The samples were exchanged into FTIR buffer by passage through a centrifugal gel filtration column. After normalization to the peak-to-peak amplitudes of the negative ferricyanide peak at 2115 cm−1 and the positive ferrocyanide peak at 2038 cm−1, the D1-D342N spectra were multiplied vertically by a factor of ~ 2.6 to maximize the overlap of the negative peak at ~ 1707 cm−1 in the first flash spectra. The Mn-depleted WT(D1) spectra represents the averages from nine samples (10,800 scans). The D1-D342N spectra represents the averages from 18 samples (21,600 scans).
On the basis of the data that is presented in Figure 3, the first flash spectrum of the D1-D342N PSII particles was corrected to eliminate the spectral contributions of mutant PSII particles lacking Mn4Ca clusters. This correction was accomplished by subtracting ~ 38% of the spectrum of Mn-depleted WT(D1) from the first flash spectrum, thereby eliminating nearly all of the (−)1707 cm−1 band. No correction was applied to the second, third, or fourth flash spectra.
The mid-frequency FTIR difference spectra of WT(D1) and D1-D342N PSII particles that were induced by four successive flashes applied at 273 K are compared in Figure 4 (panels A – D, traces a). (In Figure 4A, the spectrum of the D1-D342N PSII particles was corrected as described in the previous paragraph). The spectra that were induced by the first, second, third, and fourth flashes correspond predominantly to the S2-minus-S1, S3-minus-S2, S0-minus-S3, and S1-minus-S0 FTIR difference spectra, respectively. In a previous study (28), we emphasized the need for providing a control for indirect mutation-induced structural perturbations and showed that exchanging the same WT(D1) PSII particles into FTIR analysis buffer by different methods serves to mimic such structural perturbations. Consequently, in each panel of Figure 4, two WT(D1) spectra are shown. The spectra labeled “WT(D1a)” correspond to WT(D1) samples that were exchanged into FTIR analysis buffer by passage through a centrifugal gel filtration column (the same method that was employed for the D1-D342N samples). The spectra labeled “WT(D1b)” correspond to WT(D1) samples that were exchanged into FTIR analysis buffer by repeated concentration/dilution cycles, as in ref 31. Both WT(D1a) and WT(D1b) spectra resemble those that have been reported previously for wild-type Synechocystis PSII particles (22, 25-27). The small differences that are apparent between the WT(D1a) and WT(D1b) spectra can be attributed to minor structural perturbations in the environment of the Mn4Ca cluster that have little functional significance (28). Therefore, any spectral differences that are observed between D1-D342N and WT(D1) PSII particles would be significant only if they significantly exceed the spectral differences that are observed between the WT(D1a) and WT(D1b) PSII particles.
The data of Figure 4 show that the Sn+1-minus-Sn FTIR difference spectra of D1-D342N PSII particles are remarkably similar to the corresponding spectra of WT(D1a) and WT(D1b) PSII particles, with the possible exception of some of the features in the νsym(COO−) and νasym(COO−) regions of the S2-minus-S1 FTIR difference spectrum (Figure 4A, trace a). Several features in the corresponding regions of the WT(D1a)-minus-D342N and WT(D1b)-minus-D342N double-difference spectra (Figure 4A, traces b) are somewhat larger than the features that are present in these regions of the WT(D1a)-minus-WT(D1b) double-difference spectra (Figure 4A, trace c), particularly the positive peaks near 1584, 1532, and 1502 cm−1, the negative peaks near 1560 and 1521 cm−1, and the differential peaks at (+)1365/(−)1352 cm−1 in the two WT-minus-D342N double difference spectra. If these small spectral differences correspond to a carboxylate group whose vibrational modes shift during the S1 to S2 transition, then these modes should be restored during the S3 to S0 and/or S0 to S1 transitions. Consequently, the differences between the mutant and wild-type S2-minus-S1 FTIR difference spectra should be mirrored in the S0-minus-S3, and/or S1-minus-S0 FTIR difference spectra (17–29). However, the D1-D342N S0-minus-S3, and S1-minus-S0 FTIR difference spectra look remarkably like the WT(D1a) and WT(D1b) spectra (Figures 1C and D, traces a). Notably, for all Sn+1-minus-Sn FTIR difference spectra with the exception of S2-minus-S1, the features that are present in the WT(D1a)-minus-D342N and WT(D1b)-minus-D342N double-difference spectra (Figures 4B – 4D, traces b) are similar in amplitude to the features that are present in the WT(D1a)-minus-WT(D1b) double-difference spectra (Figures 4B – 4D, traces c). Therefore, we conclude that the small differences between the S2-minus-S1 FTIR difference spectra of D1-D342N and WT(D1) PSII particles probably reflect imperfect subtraction of features arising from mutant PSII particles that lack Mn4Ca clusters.
On the basis of the comparisons shown in Figure 4, we conclude that our data provide no indication that the D1-D342N mutation eliminates a specific carboxylate vibrational mode from any of the Sn+1-minus-Sn FTIR difference spectra. If D1-Asp342 ligated a Mn ion whose charge or oxidation state increased during one or more of the S state transitions, then replacing Asp with Asn should eliminate 1 – 2 specific carboxylate modes from one or more of the Sn+1-minus-Sn FTIR difference spectra, replacing them with a combination of ν (C=O) and/or δ (NH2) modes. The elimination of the carboxylate modes should be as obvious as the [1-13 C]alanine-induced shift of the νsym (COO −) mode of the α-COO− group of D1-Ala344 that was observed in Figure 2 of this study and in earlier studies (25, 30, 31). Yet, no such elimination is observed (Figures 4A – 4D, traces b and d). Therefore, our data show that neither the symmetric nor the asymmetric carboxylate stretching mode of D1-Asp342 is altered significantly when the Mn4Ca cluster is oxidized during the S0 → S1, S1 → S2, or S2 → S3 transitions.
The inference from our results is that the carboxylate group of D1-Asp342 is not sensitive to the oxidation of the Mn4Ca cluster during the S0 → S1, S1 → S2, or S2 → S3 transitions. One explanation of these data is that D1-Asp342 ligates a Mn ion that does not increase its charge or formal oxidation state during any of the S0 → S1, S1 → S2, or S2 → S3 transitions. As previously discussed with respect to similar data obtained with mutants of D1-Glu189 (28), this explanation presents some challenges. First, in both of the recent X-ray crystallographic structural models, D1-Asp342 ligates one of the Mn ions that are located in the Mn3Ca portion of the Mn4Ca cluster (13, 14). If D1-Asp342 ligates one of these Mn ions, then its carboxylate group is insensitive to the Mn oxidations that must occur elsewhere in this portion of the Mn4Ca cluster during the S0 → S1, S1 → S2, or S2 → S3 transitions. Second, the same explanation was offered previously for D1-Asp170 to explain similar FTIR data that were obtained with D1-D170H mutant PSII particles (26). The recent X-ray crystallographic structural models assign D1-Asp170 and D1-Asp342 as ligands of different Mn ions (13, 14). Consequently, if neither of these Mn ions increases its charge or oxidation state during the S1 → S2 transition, then the extra positive charge that develops on the Mn4Ca cluster during this transition must necessarily be localized, probably to a single Mn ion. However, this conclusion conflicts with a resonant inelastic X-ray scattering (RIXS) study, whose authors concluded that this extra charge is strongly delocalized over the Mn4 cluster (42). Third, this explanation constrains the identity of the Mn ion(s) whose charge or formal oxidation state increase during the S0 → S1 and S2 → S3 transitions. Because there is evidence that the α-COO− group of D1-Ala344 is ligated to a Mn ion whose charge increases during the S1 → S2 transition (25, 30, 31), decreases during the S3 → S0 transition (25), and remains unchanged during the S0 → S1 and S2 → S3 transitions (25), any increase in positive charge that develops on the Mn4Ca cluster during the S0 → S1 and S2 → S3 transitions must necessarily be localized on the one Mn ion that is not ligated by D1-Asp170, D1-Asp342, or D1-Ala344 [Mn3 in the ~ 3.0 Å structural model (14)]. We are currently testing this possibility by analyzing mutations of the residue CP43-Glu354, identified as aMn ligand in both the ~ 3.0 Å (14) and ~ 3.5 Å (13) structural models.
An alternate explanation for our data is that D1-Asp342 does not ligate a Mn ion. We consider this explanation to be less likely because it would conflict with both recent X-ray crystallographic structural models (13, 14), and because D1-Asp342 was proposed to be a possible ligand to the Mn4Ca cluster on the basis of earlier mutagenesis studies (11, 12). These studies showed that O2 evolving activity is retained when D1-Asp342 is replaced with a possible metal ligand (e.g., Glu, Asn, or His) but not when D1-Asp342 is replaced by Ala or Val.
Recently, it has been suggested that the vibrational modes of the carboxylate ligands of the Mn ions in the Mn4Ca cluster may be insensitive to changes in the formal oxidation states of the Mn ions during any of the S state transitions (4, 32, 43). This suggestion could explain the observed insensitivity of D1-Asp170 (26, 40), D1-Glu189 (28), and D1-Asp342 (this work) to oxidations of the Mn4Ca cluster during the S0 → S1, S1 → S2, or S2 → S3 transitions. However, this suggestion does not account for the strikingly large number of features that are present in the individual Sn+1-minus-Sn FTIR difference spectra, all of which correspond to vibrational modes that change frequency in response to the oxidation of the Mn4Ca cluster, and many of which correspond to the symmetric and asymmetric modes of carboxylate groups. If these features do not correspond to metal-ligating carboxylate residues, then to what carboxylate residues do they correspond? They do not correspond to changes in the protonation states of free carboxylates because the intensities of the features that would correspond to such changes [those in the ν (C=O) regions of the Sn+1-minus-Sn FTIR difference spectra (44)] are very weak in comparison to the strong intensities of the features in the νsym(COO−) and νasym(COO−) regions (Figures 4A-4D, traces a).
CONCLUDING REMARKS
The mid-frequency Sn+1-minus-Sn FTIR difference spectra of D1-D342N PSII particles are remarkably similar to those of wild-type PSII particles. In particular, there is no indication that the D1-D342N mutation eliminates any carboxylate modes. Therefore, either D1-Asp342 ligates a Mn ion that does not increase its charge or oxidation state during any of the S0 → S1, S1 → S2, or S2 → S3 transitions or D1-Asp342 does not ligate the Mn4 cluster. If D1-Asp342 does ligate the Mn4 cluster (as is expected), then the FTIR data provides a constraint on the identity of the Mn ion(s) whose charge or oxidation state increases during the S0 → S1 and S2 → S3 transitions.
Acknowledgments
We are grateful to Anh P. Nguyen for maintaining the wild-type and mutant cultures of Synechocystis sp. PCC 6803 and for purifying the thylakoid membranes that were used for purifying the PSII particles.
Footnotes
Abbreviations: Chl, chlorophyll; EDTA, ethylenediaminetetraacetic acid; EPR, electron paramagnetic resonance; EXAFS, extended X-ray absorption fine structure; FTIR, Fourier transform infrared; MES, 2-(N-morpholino)-ethanesulfonic acid; NTA, nitrilotriacetic acid; P680, chlorophyll species that serves as the light-induced electron donor in PSII; PSII, photosystem II; QA, primary plastoquinone electron acceptor; RH, relative humidity; WT(D1), wild -type control strain of Synechocystis sp. PCC 6803 that was constructed in identical fashion as the D1-D342N mutant but that contains the wild-type psbA-2 gene; XANES, X-ray absorption near edge structure; YZ, tyrosine residue that mediates electron transfer between the Mn cluster and P 680 +•, YD, second tyrosine residue that can reduce P680 +• in PSII.
The authors of ref 29 have recently arrived at the same conclusion. However, it should be noted that some authors continue to argue that the α-COO− group of D1-Ala344 ligates Ca rather than Mn (4, 32).
Specific L-[1-13 C]alanine-labeling of PSII shifts the νsym (COO− ) mode of the α-COO− group of D1-Ala344 by either ~ 19 or ~ 36 cm−1 (25, 30, 31). It is not yet possible to distinguish between these two possibilities.
It is striking that no evidence for the flash-induced formation of YZ• was found in D1-D170H PSII particles (26, 40), despite evidence that ~ 50% of these mutant PSII particles lack Mn4Ca clusters (26, 41). This difference between D1-D170H and D1-D342N PSII particles may be related to the substantial mutation-induced increase of the YZ•/YZ midpoint potential that was reported in D1-D342N cells (12), although it is unclear why the increased YZ•/YZ midpoint potential would increase the lifetime of YZ• in the FTIR samples at 273 K.
Support for this work was provided by the National Institutes of Health (Grants GM 066136 and GM 076232 to R. J. D. and Grant GM 048242 to R. D. B.). Additional support to W. H. was provided by the Human Frontiers Science Program (Grant Number RGP0029/2002).
References
- 1.Goussias C, Boussac A, Rutherford AW. Photosystem II and Photosynthetic Oxidation of Water: An Overview. Phil Trans R Soc London, Ser B. 2002;357:1369–1381. doi: 10.1098/rstb.2002.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sauer K, Yachandra VK. The Water-Oxidation Complex in Photosynthesis. Biochim Biophys Acta. 2004;1655:140–148. doi: 10.1016/j.bbabio.2003.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hillier W, Messinger J. Mechanism of Photosynthetic Oxygen Production. In: Wydrzynski T, Satoh Ki, editors. Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase. Springer; Dordrecht, The Netherlands: 2005. pp. 567–608. [Google Scholar]
- 4.McEvoy JP, Brudvig GW. Water-Splitting Chemistry of Photosystem II. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 5.Sauer K, Yano J, Yachandra VK. X-ray Spectroscopy of the Mn4Ca Cluster in the Water-Oxidation Complex of Photosystem II. Photosyn Res. 2005;85:73–86. doi: 10.1007/s11120-005-0638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yachandra VK. The Catalytic Manganese Cluster: Organization of the Metal Ions. In: Wydrzynski T, Satoh Ki, editors. Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase. Springer; Dordrecht, The Netherlands: 2005. pp. 235–260. [Google Scholar]
- 7.Haumann M, Müller C, Liebisch P, Iuzzolino L, Dittmer J, Grabolle M, Neisius T, Meyer-Klaucke W, Dau H. Structural and Oxidation State Changes of the Photosystem II Manganese Complex in Four Transitions of the Water Oxidation Cycle (S0 → S1, S1 → S2, S2 → S3, and S3,4 → S0 ) Characterized by X-ray Absorption Spectroscopy at 20 K and Room Temperature. Biochemistry. 2005;44:1894–1908. doi: 10.1021/bi048697e. [DOI] [PubMed] [Google Scholar]
- 8.Haumann M, Liebisch P, Müller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 Formation Tracked by Time-Resolved X-ray Experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- 9.Roelofs TA, Liang W, Latimer MJ, Cinco RM, Rompel A, Andrews JC, Sauer K, Yachandra VK, Klein MP. Oxidation States of the Manganese Cluster During the Flash-Induced S-state Cycle of the Photosynthetic Oxygen-Evolving Complex. Proc Natl Acad Sci USA. 1996;93:3335–3340. doi: 10.1073/pnas.93.8.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Messinger J, Robblee JH, Bergmann U, Fernandez C, Glatzel P, Visser H, Cinco RM, McFarlane KL, Bellacchio E, Pizarro SA, Cramer SP, Sauer K, Klein MP, Yachandra VK. Absence of Mn-Centered Oxidation in the S2 → S3 Transition: Implications for the Mechanism of Photosynthetic Water Oxidation. J Am Chem Soc. 2001;123:7804–7820. doi: 10.1021/ja004307+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nixon PJ, Diner BA. Analysis of Water-Oxidation Mutants Constructed in the Cyanobacterium Synechocystis sp. PCC 6803. Biochem Soc Trans. 1994;22:338–343. doi: 10.1042/bst0220338. [DOI] [PubMed] [Google Scholar]
- 12.Chu HA, Nguyen AP, Debus RJ. Amino Acid Residues that Influence the Binding of Manganese or Calcium to Photosystem II. 2 The Carboxy-terminal Domain of the D1 Polypeptide. Biochemistry. 1995;34:5859–5882. doi: 10.1021/bi00017a017. [DOI] [PubMed] [Google Scholar]
- 13.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the Photosynthetic Oxygen-Evolving Center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 14.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards Complete Cofactor Arrangement in the 3.0 Å Resolution Structure of Photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 15.Yano J, Kern J, Irrgang KD, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. X-ray Damage to the Mn4Ca Complex in Single Crystals of Photosystem II: A Case Study for Metalloprotein Crystallography. Proc Natl Acad Sci USA. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabolle M, Haumann M, Müller C, Liebisch P, Dau H. Rapid Loss of Structural Motifs in the Manganese Complex of Oxygenic Photosynthesis by X-ray Irradiation at 10-300 K. J Biol Chem. 2006;281:4580–4588. doi: 10.1074/jbc.M509724200. [DOI] [PubMed] [Google Scholar]
- 17.Noguchi T, Sugiura M. Flash-Induced Fourier Transform Infrared Detection of the Structural Changes during the S-State Cycle of the Oxygen-Evolving Complex in Photosystem II. Biochemistry. 2001;40:1497–1502. doi: 10.1021/bi0023807. [DOI] [PubMed] [Google Scholar]
- 18.Hillier W, Babcock GT. S-State Dependent FTIR Difference Spectra for the Photosystem II Oxygen Evolving Complex. Biochemistry. 2001;40:1503–1509. doi: 10.1021/bi002436x. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi T, Sugiura M. Flash-Induced FTIR Difference Spectra of the Water Oxidizing Complex in Moderately Hydrated Photosystem II Core Films: Effect of Hydration Extent on S-State Transitions. Biochemistry. 2002;41:2322–2330. doi: 10.1021/bi011954k. [DOI] [PubMed] [Google Scholar]
- 20.Noguchi T, Sugiura M. FTIR Detection of Water Reactions During the Flash-Induced S-State Cycle of the Photosynthetic Water-Oxidizing Complex. Biochemistry. 2002;41:15706–15712. doi: 10.1021/bi020603i. [DOI] [PubMed] [Google Scholar]
- 21.Noguchi T, Sugiura M. Analysis of Flash-Induced FTIR Difference Spectra of the S-State Cycle in the Photosynthetic Water-Oxidizing Complex by Uniform 15 N and 13C Isotope Labeling. Biochemistry. 2003;42:6035–6042. doi: 10.1021/bi0341612. [DOI] [PubMed] [Google Scholar]
- 22.Yamanari T, Kimura Y, Mizusawa N, Ishii A, Ono T-A. Mid- to Low-Frequency Fourier Transform Infrared Spectra of S-State Cycle for Photosynthetic Water Oxidation in Synechocystis sp. PCC 6803. Biochemistry. 2004;43:7479–7490. doi: 10.1021/bi0362323. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki H, Sugiura M, Noguchi T. pH Dependence of the Flash-Induced S-State Transitions in the Oxygen-Evolving Center of Photosystem II from Thermosynechococcus elongatus as Revealed by Fourier Transform Infrared Spectroscopy. Biochemistry. 2005;44:1708–1718. doi: 10.1021/bi0483312. [DOI] [PubMed] [Google Scholar]
- 24.Kimura Y, Ishii A, Yamanari T, Ono T-A. Water-Sensitive Low-Frequency Vibrations of Reaction Intermediates during S-State Cycling in Photosynthetic Water Oxidation. Biochemistry. 2005;44:7613–7622. doi: 10.1021/bi048203d. [DOI] [PubMed] [Google Scholar]
- 25.Kimura Y, Mizusawa N, Yamanari T, Ishii A, Ono TA. Structural Changes of D1 C-terminal α-Carboxylate during S-state Cycling of Photosynthetic Oxygen Evolution. J Biol Chem. 2005;280:2078–2083. doi: 10.1074/jbc.M410627200. [DOI] [PubMed] [Google Scholar]
- 26.Debus RJ, Strickler MA, Walker LM, Hillier W. No Evidence from FTIR Difference Spectroscopy That Aspartate-170 of the D1 Polypeptide Ligates a Manganese Ion That Undergoes Oxidation during the S0 to S1, S1 to S2, or S2 to S3 Transitions in Photosystem II. Biochemistry. 2005;44:1367–1374. doi: 10.1021/bi047558u. [DOI] [PubMed] [Google Scholar]
- 27.Kimura Y, Mizusawa N, Ishii A, Ono T-A. FTIR Detection of Structural Changes in a Histidine Ligand during S-State Cycling of Photosynthetic Oxygen-Evolving Complex. Biochemistry. 2005;44:16072–16078. doi: 10.1021/bi051306r. [DOI] [PubMed] [Google Scholar]
- 28.Strickler MA, Hillier W, Debus RJ. No Evidence from FTIR Difference Spectroscopy that Glutamate-189 of the D1 Polypeptide Ligates a Mn Ion that Undergoes Oxidation During the S0 to S1, S1 to S2, or S2 to S3 Transitions in Photosystem II. Biochemistry. 2006;45:8801–8811. doi: 10.1021/bi060583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki H, Taguchi Y, Sugiura M, Boussac A, Noguchi T. Structural Perturbations of the Carboxylate Ligands to the Mn Cluster upon Ca2+/Sr2+ Exchange in the S-state Cycle of Photosynthetic Oxygen Evolution as Studied by Flash-Induced FTIR Difference Spectroscopy. Biochemistry. 2006;45:13454–13464. doi: 10.1021/bi061232z. [DOI] [PubMed] [Google Scholar]
- 30.Chu H-A, Hillier W, Debus RJ. Evidence that the C-Terminus of the D1 Polypeptide is Ligated to the Manganese Ion that Undergoes Oxidation During the S1 to S2 Transition: An Isotope-Edited FTIR Study. Biochemistry. 2004;43:3152–3166. doi: 10.1021/bi035915f. [DOI] [PubMed] [Google Scholar]
- 31.Strickler MA, Walker LM, Hillier W, Debus RJ. Evidence from Biosynthetically Incorporated Strontium and FTIR Difference Spectroscopy that the C-Terminus of the D1 Polypeptide of Photosystem II Does Not Ligate Calcium. Biochemistry. 2005;44:8571–8577. doi: 10.1021/bi050653y. [DOI] [PubMed] [Google Scholar]
- 32.Sproviero EM, Gascón JA, McEvoy JP, Brudvig GW, Batista VS. QM/MM Models of the O2 -Evolving Complex of Photosystem II. J Chem Theory Comput. 2006;2:1119–1134. doi: 10.1021/ct060018l. [DOI] [PubMed] [Google Scholar]
- 33.Chu HA, Nguyen AP, Debus RJ. Site-Directed Photosystem II Mutants with Perturbed Oxygen Evolving Properties: 1 Instability or Inefficient Assembly of the Manganese Cluster In Vivo. Biochemistry. 1994;33:6137–6149. doi: 10.1021/bi00186a013. [DOI] [PubMed] [Google Scholar]
- 34.Debus RJ, Campbell KA, Gregor W, Li ZL, Burnap RL, Britt RD. Does Histidine 332 of the D1 Polypeptide Ligate the Manganese Cluster in Photosystem II? An Electron Spin Echo Envelope Modulation Study. Biochemistry. 2001;40:3690–3699. doi: 10.1021/bi002394c. [DOI] [PubMed] [Google Scholar]
- 35.Peloquin JM, Campbell KA, Randall DW, Evanchik MA, Pecoraro VL, Armstrong WH, Britt RD. 55 Mn ENDOR of the S2 -state Multiline EPR Signal of Photosystem II: Implications on the Structure of the Tetranuclear Mn Cluster. J Am Chem Soc. 2000;122:10926–10942. [Google Scholar]
- 36.Kulik L, Epel B, Lubitz W, Messinger J. 55 Mn Pulse ENDOR at 34 GHz of the S0 and S2 States of the Oxygen-Evolving Complex in Photosystem II. J Am Chem Soc. 2005;127:2392–2393. doi: 10.1021/ja043012j. [DOI] [PubMed] [Google Scholar]
- 37.Debus RJ, Campbell KA, Peloquin JM, Pham DP, Britt RD. Histidine 332 of the D1 Polypeptide Modulates the Magnetic and Redox Properties of the Manganese Cluster and Tyrosine YZ in Photosystem II. Biochemistry. 2000;39:470–478. doi: 10.1021/bi9917737. [DOI] [PubMed] [Google Scholar]
- 38.Boussac A, Rappaport F, Carrier P, Verbavatz JM, Gobin R, Kirilovsky D, Rutherford AW, Sugiura M. Biosynthetic Ca2+/Sr2+ Exchange in the Photosystem II Oxygen-Evolving Enzyme of Thermosynechococcus elongatus. J Biol Chem. 2004;279:22809–22819. doi: 10.1074/jbc.M401677200. [DOI] [PubMed] [Google Scholar]
- 39.Berthomieu C, Hienerwadel R, Boussac A, Breton J, Diner BA. Hydrogen-Bonding of Redox-Active Tyrosine Z of Photosystem II Probed by FTIR Difference Spectroscopy. Biochemistry. 1998;37:10547–10554. doi: 10.1021/bi980788m. [DOI] [PubMed] [Google Scholar]
- 40.Chu H-A, Debus RJ, Babcock GT. D1-Asp170 is Structurally Coupled to the Oxygen Evolving Complex in Photosystem II as Revealed by Light-Induced Fourier Transform Infrared Difference Spectroscopy. Biochemistry. 2001;40:2312–2316. doi: 10.1021/bi0022994. [DOI] [PubMed] [Google Scholar]
- 41.Debus RJ, Aznar C, Campbell KA, Gregor W, Diner BA, Britt RD. Does Aspartate 170 of the D1 Polypeptide Ligate the Manganese Cluster in Photosystem II? An EPR and ESEEM Study. Biochemistry. 2003;42:10600–10608. doi: 10.1021/bi034859f. [DOI] [PubMed] [Google Scholar]
- 42.Glatzel P, Bergmann U, Yano J, Visser H, Robblee JH, Gu W, De Groot FMF, Christou G, Pecoraro VL, Cramer SP, Yachandra VK. The Electronic Structure of Mn in Oxides, Coordination Complexes, and the Oxygen-Evolving Complex of Photosystem II Studied by Resonant Inelastic X-ray Scattering. J Am Chem Soc. 2004;126:9946–9959. doi: 10.1021/ja038579z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McEvoy JP, Gascón JA, Batista VS, Brudvig GW. The Mechanism of Photosynthetic Water Splitting. Photobiochem Photobiophys Sci. 2005;4:940–949. doi: 10.1039/b506755c. [DOI] [PubMed] [Google Scholar]
- 44.Barth A. The Infrared Absorption of Amino Acid Side Chains. Prog Biophys Molec Biol. 2000;74:141–173. doi: 10.1016/s0079-6107(00)00021-3. [DOI] [PubMed] [Google Scholar]