

Abstract
Highly fluorescent nanoparticles of the conjugated polymer poly(9,9-dioctylfluorene) (PFO) with distinct phases were prepared, and their photophysical properties were studied by steady state and time-resolved fluorescence spectroscopy. An aqueous suspension of PFO nanoparticles prepared by a reprecipitation method was observed to exhibit spectroscopic characteristics consistent with the glassy phase of the polymer. We demonstrate that controlled addition of organic solvent leads to partial transformation of the disordered polymer chains into the planarized conformation (β-phase), with the fractions of each component phase dependent on the amount of solvent added. Fluorescence spectroscopy of the PFO nanoparticles containing β-phase indicates efficient energy transfer from the glassy-phase regions of the nanoparticles to the β-phase regions. Salient features of the nanoparticles containing β-phase include narrow, red-shifted fluorescence and increased fluorescence quantum yield as compared to the glassy-phase nanoparticles. Fluorescence lifetime measurements indicate that the increased quantum yield of the β-phase PFO originates from a decrease in the nonradiative decay rate, with little change in the radiative rate. This decrease is likely due to exciton trapping by the β-phase, which leads to a reduction in the energy transfer efficiency to quencher species present within the nanoparticle.
1. Introduction
Conjugated polymers have attracted great interest due to their potential application in optoelectronic devices such as light-emitting displays and photovoltaic cells.1–3 These semiconducting macromolecules offer the advantage of combining the useful electrical and optical properties of semiconductors with the outstanding processability and mechanical characteristics of plastics. The device properties such as emission color and quantum efficiency depend sensitively on the molecular conformation and polymer chain packing, as confirmed by a number of studies indicating that the properties of solution-processed conjugated polymer thin films and thin film devices are highly dependent on various factors and processing steps such as choice of solvent, polymer concentration, and thermal annealing.4–8 Recently, we reported a facile reprecipitation method for preparation of a variety of conjugated polymer nanoparticles.9,10 The preparation method involves the rapid addition of a dilute solution of hydrophobic conjugated polymer dissolved in a water-miscible organic solvent to water. The rapid mixing with water leads to a sudden decrease in solvent quality, resulting in the formation of a stable suspension of hydrophobic conjugated polymer nanoparticles. Possible target applications of these nanoparticles include biological imaging and sensing due to the high fluorescence brightness of the nanoparticles under both one-photon and two-photon excitation.9,11
As a promising class of blue-emitting conjugated polymers, polyfluorenes display complex structure–property relationships.4 In particular, the dependence of photophysical properties on polymer morphology for poly(9,9-dioctylfluorene) (PFO) has garnered much attention.12–15 In PFO, distinct phases have been identified: principally a disordered glassy phase and a crystalline β-phase containing planar polymer chains.16,17 The presence of β-phase in thin films can be influenced by thermal and vapor treatment of as-cast films or by varying the solvent from which the film is spin-cast.12,14,18 Significantly, the fraction of β-phase in the films was found to affect the generation of polarons and triplet excitons13,15,19 as well as the photoluminescence quantum efficiency.20 These results indicate that control over conformation is important for optimizing performance. Here, we report on the swelling-induced conformational transition of PFO nanoparticles. PFO nanoparticles prepared by the reprecipitation method were observed to exhibit the spectroscopic characteristics of the glassy polymer phase. Addition of organic solvent to the aqueous suspension resulted in the formation of β-phase, presumably by solvent-induced swelling, which facilitates formation of the thermodynamically favored β-phase. The β-phase persists after the removal of the organic solvent. The fraction of the polymer in the β-phase was observed to depend on the concentration of the organic solvent. The resulting mixed-phase PFO nanoparticles exhibit efficient energy transfer from the glassy phase to the β-phase, resulting in narrower, red-shifted fluorescence and increased quantum yield, as compared to the all glassy PFO nanoparticles. The mixed-phase nanoparticles also exhibit reduced energy transfer to dye dopants incorporated in the nanoparticles, consistent with competitive energy transfer to polymer chains in the β-phase. The results indicate that the energy transfer and fluorescence properties of PFO nanoparticles can be tailored to a specific application by a combination of doping and control of polymer conformation.
2. Experimental Section
The polyfluorene derivative poly(9,9-dioctylfluorenyl-2,7-diyl) (PFO, MW 147 000, polydispersity 3.0) was purchased from ADS dyes (Quebec, Canada). The fluorescent dye tetraphenylporphyrin (TPP) and the solvents toluene (HPLC grade, 99.7%) and tetrahydrofuran (THF, anhydrous, 99.9%) were purchased from Sigma-Aldrich (Milwaukee, WI). All chemicals were used without further purification.
PFO nanoparticles were prepared using a method similar to the reprecipitation method described previously.9 The PFO polymer was dissolved in THF by heating up to ∼50 °C for 20 min under inert atmosphere. The solution was stirred overnight at room temperature, and then diluted to make a 400 ppm solution. A 200 μL quantity of the solution mixture was quickly injected into 8 mL of deionized water under sonication in an ultrasonic bath followed by 10 additional seconds of sonication. The resulting suspension was filtered through a 0.2 μm membrane filter. The THF was removed by partial vacuum evaporation, followed by filtration through a 0.2 μm filter. The resulting aqueous dispersions contain PFO nanoparticles which exhibit the spectroscopic characteristics of the glassy phase of the polymer. Two swelling methods were observed to cause partial phase transformation of the nanoparticles, resulting in mixed-phase particles. For one method, the water-miscible solvent THF was added to a suspension of polymer nanoparticles, while for the other method, toluene was used to swell the nanoparticles. For the method using THF, varying amounts of THF (0.5, 1, 2, and 4 mL) were mixed with 10 mL of aqueous nanoparticle suspensions, respectively. The mixtures were left for 2 h, and then the THF was removed by partial vacuum evaporation. For the other method, 1 mL of toluene was mixed with 10 mL of aqueous nanoparticle suspension. The mixture was sonicated for 2 min, and left for 12 h at room temperature. Due to the immiscibility of the two liquids, the mixture partitioned into a top layer (toluene) and bottom layer (water with nanoparticles). The top layer was removed by a pipet, and the residual toluene was removed by nitrogen stripping.
The shape and size distribution of the PFO nanoparticles were characterized by atomic force microscopy (AFM). For the AFM measurements, one drop of the nanoparticle dispersion was placed on a freshly cleaved mica substrate. After evaporation of the water, the surface was scanned with an Ambios Q250 multimode AFM in tapping mode. The UV–vis absorption spectra were recorded with a Shimadzu UV-2101PC scanning spectrophotometer using 1 cm quartz cuvettes. The as-prepared nanoparticle suspensions show a peak absorbance of ∼1. Each sample was diluted to yield an absorbance of 0.1 for fluorescence measurements. The fluorescence emission spectra were recorded using a commercial fluorometer (Quantamaster, PTI, Inc.). Fluorescence lifetimes were measured using the time-correlated single-photon counting technique (TCSPC). The sample was excited by the second harmonic (400 nm, ∼100 fs pulses) of a mode-locked femtosecond Ti:Sapphire laser (Coherent Mira 9000). The output of a fast PIN diode (Thorlabs, DET210) monitoring the laser pulse was used as the start pulse for a time-to-amplitude converter (TAC, Canberra model 2145). Fluorescence signal from the aqueous nanoparticle dispersion was collected in perpendicular to the excitation, passed through either a 420 or a 450 nm narrow (10 nm) bandpass filter, and detected by a single photon counting module (id Quantique, ID100–50). The output of the photon counting module was used as the stop pulse for the TAC. The laser was attenuated to maintain the count rate below 20 kHz. The signal from the TAC was digitized using a multichannel analyzer (FastComTec, MCA-3A). The instrument response function was measured before and after each fluorescence lifetime measurement using the scattered laser light from a dilute suspension of polystyrene beads. The combination of the detector and electronics results in an instrument response function with a width of ∼60 ps (fwhm). A custom algorithm employing an iterative deconvolution method was employed to determine the fluorescence lifetime, yielding an estimated uncertainty in lifetime of 20 ps or better.
3. Results and Discussion
Poly(9,9-dialkylfluorene)s are known to exhibit complex morphology. For instance, they can form a disordered glassy phase, a partially crystalline β-phase, or a liquid crystalline phase, depending on the side-chain structure and thermal treatment.4 Figure 1a shows the chemical structure of the PFO polymer and the β-phase chain segment that can be described as a “planar zigzag” or 21 helix conformation.12 The β-phase formation of PFO in solution can be observed in poor solvent by varying temperature21 or in solvent/nonsolvent mixtures by increasing the nonsolvent content.4 In a previous report, we developed a facile method for preparation of a variety of hydrophobic conjugated polymer nanoparticles, including the polyfluorene derivative poly(9,9-dihexylfluorene).9 Here we demonstrate the preparation of PFO nanoparticles with varying fractions of β-phase polymer and examine the photophysical properties of the mixed-phase nanoparticles. Rapid mixing of a dilute solution of PFO in THF (400 ppm, 200 μL) with water (8 mL) under sonication leads to the formation of PFO nanoparticles that exhibit the optical properties of the glassy phase. The THF was removed by partial vacuum evaporation. The resulting nanoparticle suspensions are clear (nonturbid), colorless, and stable for weeks, with no evidence of aggregation or decomposition. For AFM measurement, one drop of the nanoparticle dispersion was placed on a freshly cleaved mica substrate. As indicated in the AFM images of Figure 1b, the PFO nanoparticles exhibit an approximately spherical shape. The particle height histogram (Figure 1c) shows that most of the nanoparticles possess a particle size in the range of 30–60 nm, with small fraction of particles over 70 nm. The mean particle size of ∼50 nm corresponds to roughly 280 polymer molecules per nanoparticle, assuming dense packing of the polymer chains, from which a molar extinction coefficient of ∼5 × 109M−1cm−1 is estimated. Unlike inorganic semiconductor nanoparticles, which exhibit a pronounced quantum size effect, the particle size does not significantly affect the shape of the absorption and fluorescence spectra of nanoparticles, rather, an increase in particle size merely increases the optical cross-section.9,22 Particle size also affects intraparticle energy transfer in dye-doped conjugated polymer nanoparticles.23
Figure 1.
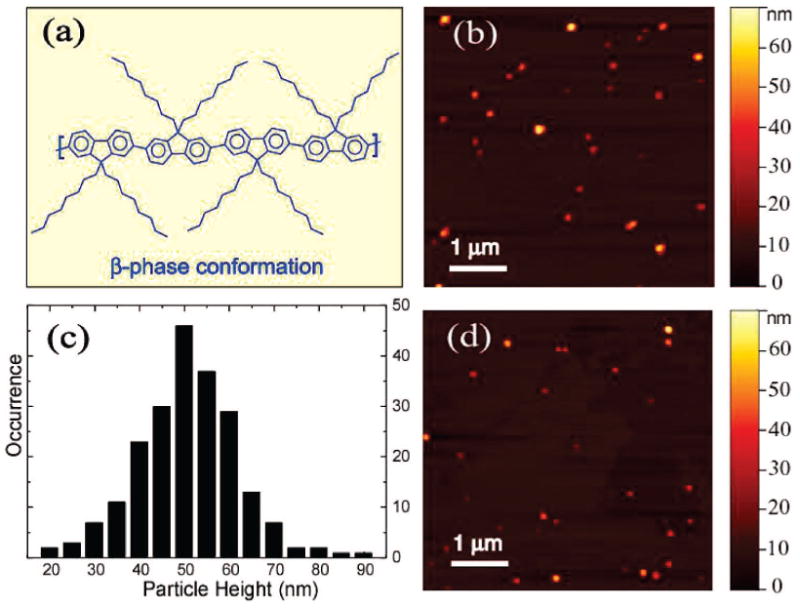
(a) Chemical structure and the β-phase conformation of the conjugated polymer PFO. (b) A representative AFM image of glassy PFO nanoparticles dispersed on mica substrate. (c) Histogram of particle height data taken from AFM images of glassy PFO nanoparticles. (d) A representative AFM image of mixed-phase PFO nanoparticles.
Conjugated polymers are characterized as rigid rod-like polymers due to the rigidity of the π-conjugated backbone. However, the presence of chemical defects, chain–chain interactions (including π-stacking between conjugated segments as well as interactions between side chains), and chain–solvent interactions result in complex morphology and phase behavior. The resulting structure and its effect on π-stacking and other interactions have a large impact on the optical and electronic properties of the polymer. At low concentration in a good solvent, PFO adopts an elongated rodlike conformation containing predominantly only a single polymer chain.24 Accordingly, PFO in dilute THF solution exhibits an unstructured absorption spectrum with maximum centered at ∼390 nm (Figure 2a, solid curve). The fluorescence spectrum exhibits a well-resolved vibronic structure with the 0–0 transition centered at ∼420 nm (Figure 2b, solid curve). During the nanoparticle preparation, the rapid mixing of a small volume of PFO in THF solution with water leads to a sudden decrease in solvent quality and the formation of nanoparticles due to aggregation of the polymer chains. As indicated in Figure 2a (dashed curve), the absorption spectrum of the as-prepared PFO nanoparticles is broadened and blue-shifted as compared to that of the polymer in THF solution. The blue-shifted absorption peak is consistent with an overall decrease in the conjugation length due to bending and kinking of the polymer backbone, and the red tail in the absorption spectrum is indicative of interchain interactions.9 The as-prepared PFO nanoparticles exhibit a slightly red-shifted fluorescence and a long red tail as compared to the polymer in THF solution. Since the nanoparticles possess a densely packed structure, the red-shift and the long red tail in fluorescence could be ascribed to increased interchain interactions, resulting in a small fraction of red-shifted aggregate species. The resulting energetic disorder, combined with multiple energy transfer, would result in a net red-shift in the fluorescence spectrum as compared to that of the polymer in solution. On the basis of these observations and comparison to thin film spectra,12,14 we conclude that the as-prepared nanoparticles consist of polymer molecules in a dense, disordered, “glassy” phase.
Figure 2.
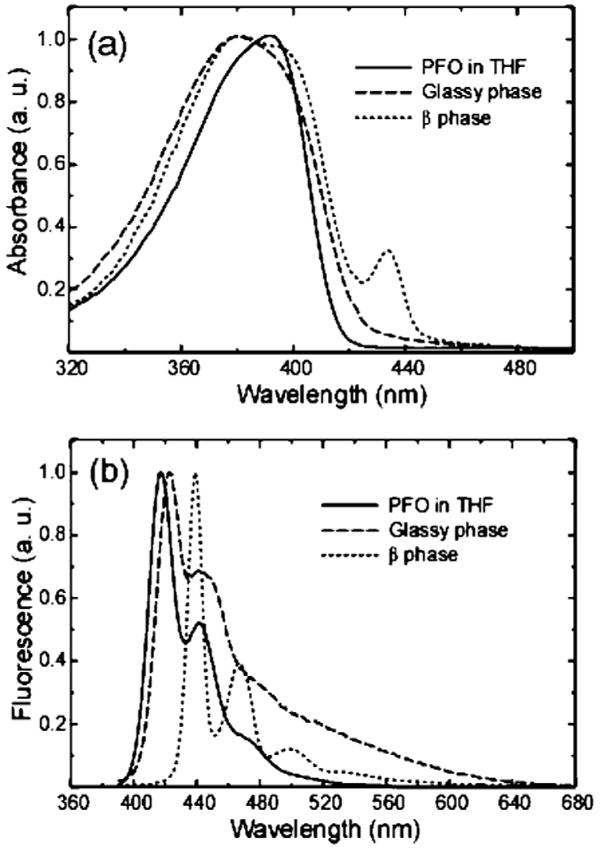
Absorption spectra (a) and fluorescence emission spectra (b) of PFO in dilute THF solution (solid), glassy PFO nanoparticles (dashed), and mixed-phase nanoparticles (dotted) obtained by toluene swelling.
Addition of organic solvent to an aqueous suspension of hydrophobic polymer particles is known to induce swelling of the particles. The as-prepared (glassy) PFO nanoparticles exhibited clear changes in their spectroscopic properties upon exposure to either toluene or THF, and the changes persist after removal of the organic solvent. The observed shifts in the UV–vis absorption and fluorescence spectra are consistent with the formation of a β-phase.12 The spectroscopic properties of the β-phase PFO differ dramatically from those of the glassy phase, while the π−π* absorption of the glassy phase is relatively broad and featureless, the β-phase exhibits a narrow, red-shifted, and well-resolved absorption peak at 435 nm, with clear vibronic features superimposed on the main absorption band (Figure 2a, dotted curve). The fluorescence of the β-phase also displays a narrow, red-shifted emission peak at 439 nm and a well-resolved vibronic progression in the emission spectrum (Figure 2b, dotted curve). A clear photophysical picture has been derived from observing the spectral similarities between the β-phase of PFO and the planarized ladder-type poly(para-phenylene); the more extended π-conjugation in PFO β-phase accounts for the observed bathochromic shift and the sharp vibronic features observed in the spectra.12 It should be noted that β-phase formation was observed in PFO, but not in poly(9,9-dihexylfluorene) (PDHF) nanoparticles subjected to the same swelling protocol, although previous studies have indicated certain features related to β-phase formation in PDHF films.25 An alternative method for inducing the formation of β-phase is to anneal the glassy PFO nanoparticles at a temperature above the Tg of the polymer (∼75 °C).17 However, thermally annealed PFO nanoparticles (90 °C, for 1 h) exhibit only a small shoulder, rather than a well-resolved peak, at the red edge of the absorption spectrum (data not shown). A similar small shoulder is present in the absorption spectra of thermally annealed PFO films.12
We observed that addition of the water-miscible solvent THF can induce β-phase formation with reliable control of the relative fraction of β-phase. Aqueous dispersions of the glassy nanoparticles were mixed with varying amounts of THF to yield a THF/water ratio ranging from 0 to 40% by volume. The mixtures were left for 2 h, and then the THF was removed by partial vacuum evaporation prior to AFM and spectroscopic measurements. It should be noted that, over the range of 0–40% THF, the swelling step was not observed to induce nanoparticle aggregation or Ostwald ripening. Figure 1d shows the AFM results for the PFO nanoparticles swelled with the highest THF/water ratio (40%), which indicate no obvious changes in particle size distribution as compared to the glassy nanoparticles prior to swelling (Figure 1b). However, variation in the solvent/water ratio was observed to result in variations in the relative intensity of the spectroscopic features associated with the β-phase. Figure 3a shows the dependence of the absorption spectra of PFO nanoparticles on the THF fraction used in the swelling step. The β-phase absorption peak (435 nm) can be observed at the THF/water ratio of 5%, and the intensity increases upon further increase of the THF/water ratio. This change is also accompanied by a decrease in the 375 nm principal absorption feature and the appearance of the vibronic structure superimposed on the main absorption. At the highest THF/water ratio (40%) employed in the swelling experiments, the β-phase fraction was estimated to be ∼27% by comparing the intensity of β-phase 0–0 peak to the overall absorption from the planarized β-conformation.14 We found that the THF swelling must be done as a separate step after the glassy nanoparticle formation in order to achieve reliable control of the β-phase fraction. Mixed-phase PFO nanoparticles could be directly obtained by reducing the concentration of the polymer in THF (20 ppm) and increasing the amount of the polymer/THF solution (2 mL) relative to water (8 mL) in the reprecipitation; however, the particles prepared by this method exhibit smaller size (7–15 nm). Glassy-phase particles were not obtained using this preparation method, likely due to the larger amount of THF employed and its tendency to swell the particles.
Figure 3.
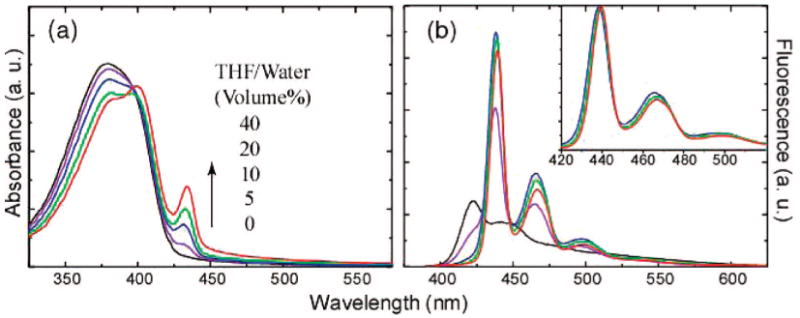
Absorption spectra (a) and fluorescence emission spectra (b) of PFO nanoparticles containing varying β-phase fractions. The absorption increase (435 nm) of the β-phase is indicated by an arrow, with increasing the THF/water ratio in the swelling. Each sample was diluted to yield an absorbance of 0.1 at 375 nm, and fluorescence spectra were obtained under the 375 nm excitation. The normalized emission spectra shown in the inset indicates a slight red-shift with increasing β-phase fractions.
Each of the aqueous PFO nanoparticle dispersions was diluted to yield an absorbance of 0.1 at 375 nm, and fluorescence spectra were obtained using an excitation wavelength of 375 nm. As shown in Figure 3b, the fluorescence from the PFO glassy phase (∼423 nm) decreases dramatically and is eventually completely quenched as the THF/water ratio employed in the swelling increases. It should be noted that the observed changes in the fluorescence spectra are not likely due to residual THF, since little or no changes were observed in the spectra after additional vacuum evaporation steps to remove residual THF, indicating that any remaining THF has little effect on the particle properties. The fluorescence from the β-conformation (∼438 nm) increases and reaches a maximum for the PFO nanoparticles swelled at the THF/water ratio of 10%. Although further increase of the THF/water ratio increases the β-phase fraction (as determined from the absorbance), the nanoparticles exhibit a slight decrease in fluorescence intensity, together with a slight red-shift in the emission wavelength that is observed in the normalized emission spectra (Figure 3b, inset). This may be due to increased interactions between chains of the β-conformation or increases in the size of the β-phase domains resulting in an increase in conjugation length. It has previously been observed that increasing the conjugation length of conjugated polymers often results in both a red shift in the emission and a reduction in fluorescence quantum yield,26 and interchain aggregates are known to exhibit weak, red-shifted emission in some cases.7 Significantly, at some THF ratios the mixed-phase particles exhibit an increased fluorescence quantum yield relative to the glassy phase. Fluorescence quantum yields of ∼35% were determined for mixed-phase nanoparticles (swelled at the THF/water ratio of 10%) and ∼21% for the glassy-phase nanoparticles, respectively (coumarin 1 in ethanol was employed as a quantum yield standard). This is in sharp contrast to thin film results,20 where the glassy-phase and β-phase PFO were found to exhibit similar fluorescence quantum yields at room temperature. The spectroscopic characteristics of the mixed-phase nanoparticles, primarily the increased fluorescence quantum yield, narrower emission peak, and increased energy gap between excitation and emission as compared to the glassy-phase particles are also promising for fluorescence-based biosensing and imaging applications.
The glassy-phase emission is completely quenched and the β-phase emission is dominant in the PFO nanoparticles containing only a small fraction of β-phase, indicating significant energy transfer from the glassy-phase to β-phase domains. Efficient energy transfer from the glassy polymer to the β-phase has been observed in films as well.14 These results are not surprising, since the large spectral overlap between the glassy-phase emission and the β-phase absorption would allow efficient energy transfer to occur via the Förster mechanism. It should be noted that for appreciable energy transfer to occur, both phases must be present in the majority of the nanoparticles; significant particle-to-particle heterogeneity in the relative fraction of β-phase would significantly reduce energy transfer, since the interparticle distances in the dispersion are far too large for appreciable interparticle energy transfer to occur. Time-correlated single photon counting (TCSPC) was used to measure the fluorescence lifetimes of the mixed-phase nanoparticles swelled at the THF/water ratio of 10% (Figure 4). No rise time due to energy transfer was resolved in the β-phase emission, consistent with previous thin films results that indicate energy transfer occurs within 5 ps,14,27 below the time resolution of the TCSPC apparatus. However, the TCSPC measurements indicate a remarkable difference in fluorescence lifetimes of the glassy phase and β-phase. Fluorescence decay lifetimes were measured at the 0–0 peak for the two types of PFO nanoparticles, and lifetimes of 97 ps for the glassy-phase and 178 ps for mixed-phase nanoparticles were obtained. The increase in the lifetime for the mixed phase as compared to the glassy phase is also accompanied by an increase in the fluorescence quantum yield. The radiative rate constant kR and nonradiative decay rate constant kNR of the nanoparticles were determined by combining the quantum yield and fluorescence lifetime results. The obtained values are as follows: kR = 2.2 ns−1, kNR = 8.1 ns−1 for the glassy-phase nanoparticles and kR = 2.0 ns−1, kNR = 3.6 ns−1 for the mixed-phase nanoparticles. These results indicate that the increased quantum yield for the mixed-phase particles is due to their significantly lower nonradiative decay rate, while both types of particles exhibit similar radiative rates. There is currently not sufficient information to determine the origin of this difference in the nonradiative rate. One possibility is that differences in polymer morphology result in different rates of triplet or polaron generation/recombination between the two phases. The swelling process could also reduce disorder, which could reduce the number of the nonradiative channels. Additionally, the effect of disorder on the dipole–dipole coupling of excitons in many fluorescent aggregates28–30 including polyfluorene aggregates31 can lead to changes in radiative rate as well as in the exciton–phonon coupling, which could affect the nonradiative decay rate. Another possibility is that the minority β-phase domains act as exciton traps, reducing exciton mobility and therefore reducing the rate of energy transfer to quencher species such as hole polarons. This possibility is supported by our recent report on the effect of dopant dyes on the quantum yield and photostability of conjugated polymer nanoparticles.23
Figure 4.
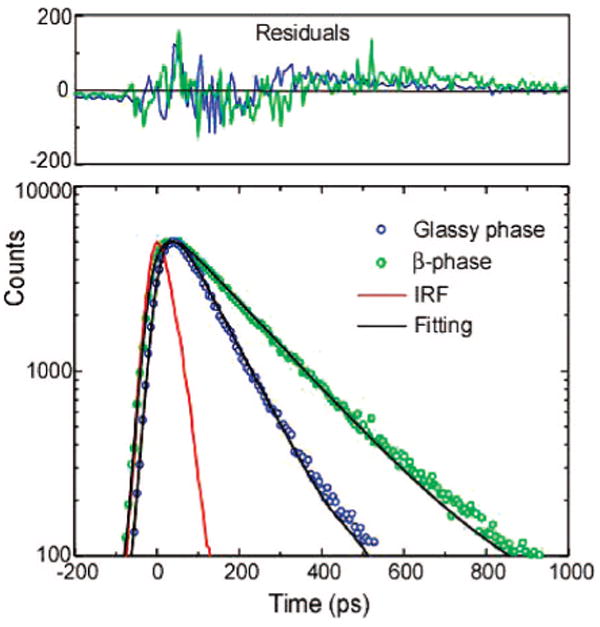
Semi-log plot of fluorescence decays of the glassy and β-phase PFO nanoparticles measured by a TCSPC setup. The red curve shows the instrumental response function (IRF). The scattered symbols represent experimental data, and the solid lines are fitting curves obtained by employing an iterative deconvolution method. Residuals are shown above the fits.
In order to test the hypothesis that the β-phase domains act as exciton traps that reduce the efficiency of energy transfer to quenchers or other energy acceptors, we measured the effect of polymer morphology on the efficiency of energy transfer in dye-doped PFO nanoparticles. We previously demonstrated that dye-doped and polymer-doped polyfluorene nanoparticles (containing PDHF in the glassy phase) can exhibit efficient energy transfer to the dopant.22,23 The efficiency of energy transfer from the polymer to the dopant depends strongly on the spectral overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor. Since the absorption and emission spectra of glassy-phase and β-phase PFO are significantly different, polymer morphology is likely to have a strong affect on energy transfer efficiency. TPP-doped glassy nanoparticles were prepared by the reprecipitation method described above, but with 1% TPP relative to polymer in the precursor solution. The β-phase was formed in TPP-doped nanoparticles by THF swelling with a THF/water ratio of 10%. On the basis of our previous experiments,23 the actual concentration of TPP dye in the nanoparticles is consistent with the composition of the precursor mixture, and a small amount of THF in the swelling step does not lead to appreciable dye leakage. Figure 5a shows the absorption spectra of the TPP-doped glassy and β-phase nanoparticles. No obvious absorption peak from TPP is observed due to the low doping concentration, while the β-phase feature appears in the absorption band of PFO. Figure 5b presents the fluorescence spectra of the TPP-doped glassy and β-phase nanoparticles (diluted to yield an absorbance of 0.1 at the 375 nm excitation wavelength). The TPP-doped glassy nanoparticles exhibit a strong emission from the TPP acceptors, indicating efficient energy transfer from the PFO to the TPP. This is consistent with the Förster transfer mechanism: there is sufficient spectral overlap between the glassy PFO emission and the TPP absorption (inset of Figure 5b), and the Förster radius for this donor–acceptor pair is determined to be 3.6–3.8 nm.32,33 However, the mixed-phase particles show a clear decrease of the TPP fluorescence combined with the appearance of β-phase emission and a complete quenching of the emission of the glassy phase. The Förster radius for energy transfer from the glassy phase to the β-phase is relatively large as compared to that of TPP,14 and the effective concentration of the β-phase is relatively high, both of which should result in a significantly higher energy transfer rate from the glassy phase to the β-phase as compared to the energy transfer rate from the glassy phase to the TPP dye. Furthermore, there is a small spectral overlap between the β-phase emission and the TPP absorption and a relatively large average distance between β-phase polymer chains and the nearest TPP molecule, which should result in very low energy transfer efficiency from the β-phase to the TPP dye. To quantify the competitive energy transfer to the energy acceptors (TPP and β-phase PFO), the energy transfer efficiency (Q) can be calculated from
Figure 5.
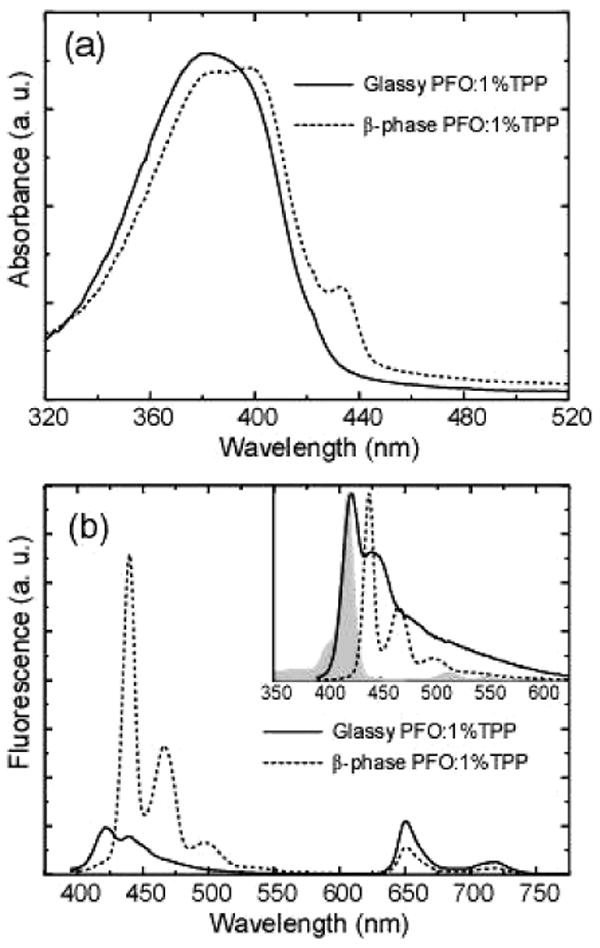
Absorption spectra (a) and fluorescence emission spectra (b) of TPP-doped glassy PFO (solid) and β-phase nanoparticles (dotted). The inset shows spectral overlap between the TPP absorption (gray) and fluorescence emission of glassy PFO (solid) and β-phase nanoparticles (dotted).
| (1) |
Here the resonance energy transfer rate (kET) was estimated by the expression32
| (2) |
where ID and IA are the emission intensities of the donor and acceptor, and φA is the quantum yield of the acceptor. The integrated emission intensities (ID and IA) of the donor and acceptor can be obtained according to the emission spectra in Figure 5b. The fluorescence quantum yield (φA) of TPP in the PFO host was determined to be 8%. In the TPP-doped glassy nanoparticles, the energy transfer efficiency to TPP was calculated to be 66%. In the β-phase nanoparticles, the overall energy transfer efficiency to both TPP and β-phase PFO is nearly 100%, since the donor's fluorescence is completely quenched. The relative transfer efficiencies to TPP and β-phase PFO were estimated to be 31% and 69%, respectively. On the basis of the above results, it is concluded that the β-phase effectively out competes the TPP as an energy acceptor (at small TPP loading ratios), and the β-phase does not subsequently transfer an appreciable amount of its energy to the TPP; in effect, the β-phase acts as an exciton trap, reducing the efficiency of energy transfer to the TPP. The observed reduction in polymer → dye energy transfer efficiency due to the exciton trapping by the β-phase supports the hypothesis that the higher quantum yield and reduced nonradiative decay rate of (undoped) mixed-phase nanoparticles arises from reduced energy transfer to quencher species due to exciton trapping by the β-phase.
4. Conclusions
Effective control of polymer phase in fluorescent PFO nanoparticles was demonstrated, and the photophysical properties of mixed-phase nanoparticles were studied by steady state and time-resolved fluorescence spectroscopy. PFO nanoparticles were kinetically trapped into glassy phase during their initial formation using a reprecipitation method. We demonstrate that addition of organic solvent leads to the formation of a fraction of β-phase within the particles, with the relative amount of β-phase and glassy phase controlled by the amount of solvent added. The resulting mixed-phase PFO nanoparticles exhibit narrow, red-shifted fluorescence spectra and increased quantum yield relative to the glassy phase, indicative of efficient energy transfer from the glassy polymer to the β-phase. Fluorescence lifetime measurements indicate that the increased quantum yield of β-phase PFO originates from a dramatic decrease in the nonradiative decay rate, rather than significant changes in the radiative rate. This decrease is likely due in part to exciton trapping by the β-phase, which leads to a reduction in the efficiency of energy transfer to quencher species present within the nanoparticle. Similarly, reduced energy transfer to dye dopants was observed in particles containing β-phase, which suggests that manipulating polymer morphology by swelling methods is a viable strategy for suppressing undesired energy transfer processes such as energy transfer to quencher species.
Acknowledgments
The authors gratefully acknowledge financial support from the NSF/EPSCoR under Grants No. 2001RII-EPS-0132573 and No. 2004RII-EPS-0447660, NSF CAREER Grant No. CHE-0547846, and NIH Grant No. 1R01GM081040.
References
- 1.Burroughes JH, Bradley DDC, Brown AR, Marks RN, Mackay K, Friend RH, Burns PL, Holmes AB. Nature. 1990;347:539–541. [Google Scholar]
- 2.Halls JJM, Walsh CA, Greenham NC, Marseglia EA, Friend RH, Moratti SC, Holmes AB. Nature. 1995;376:498–500. [Google Scholar]
- 3.Yu G, Gao J, Hummelen JC, Wudl F, Heeger AJ. Science. 1995;270:1789–1791. [Google Scholar]
- 4.Scherf U, List EJW. Adv Mater. 2002;14:477–487. [Google Scholar]
- 5.Nguyen TQ, Doan V, Schwartz BJ. J Chem Phys. 1999;110:4068–4078. [Google Scholar]
- 6.Nguyen TQ, Martini IB, Liu J, Schwartz BJ. J Phys Chem B. 2000;104:237–255. [Google Scholar]
- 7.Schwartz BJ. Annu Rev Phys Chem. 2003;54:141–172. doi: 10.1146/annurev.physchem.54.011002.103811. [DOI] [PubMed] [Google Scholar]
- 8.Padmanaban G, Ramakrishnan S. J Phys Chem B. 2004;108:14933–14941. [Google Scholar]
- 9.Wu C, Szymanski C, McNeill J. Langmuir. 2006;22:2956–2960. doi: 10.1021/la060188l. [DOI] [PubMed] [Google Scholar]
- 10.Szymanski C, Wu C, Hooper J, Salazar MA, Perdomo A, Dukes A, McNeill JD. J Phys Chem B. 2005;109:8543–8546. doi: 10.1021/jp051062k. [DOI] [PubMed] [Google Scholar]
- 11.Wu C, Szymanski C, Cain Z, McNeill J. J Am Chem Soc. 2007;129:12904–12905. doi: 10.1021/ja074590d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grell M, Bradley DDC, Ungar G, Hill J, Whitehead KS. Macromolecules. 1999;32:5810–5817. [Google Scholar]
- 13.Cadby AJ, Lane PA, Mellor H, Martin SJ, Grell M, Giebeler C, Bradley DDC, Wohlgenannt M, An C, Vardeny ZV. Phys Rev B. 2000;62:15604–15609. [Google Scholar]
- 14.Khan ALT, Sreearunothai P, Herz LM, Banach MJ, Kohler A. Phys Rev B. 2004;69:085201. [Google Scholar]
- 15.Hayer A, Khan ALT, Friend RH, Kohler A. Phys Rev B. 2005;71:241302. [Google Scholar]
- 16.Grell M, Bradley DDC, Long X, Chamberlain T, Inbasekaran M, Woo EP, Soliman M. Acta Polym. 1998;49:439–444. [Google Scholar]
- 17.Grell M, Bradley DDC, Inbasekaran M, Woo EP. Adv Mater. 1997;9:798–802. [Google Scholar]
- 18.Caruso ME, Anni M. Phys Rev B. 2007;76:054207. [Google Scholar]
- 19.List EJW, Kim CH, Naik AK, Scherf U, Leising G, Graupner W, Shinar J. Phys Rev B. 2001;6415:155204. [Google Scholar]
- 20.Ariu M, Lidzey DG, Sims M, Cadby AJ, Lane PA, Bradley DDC. J Phys: Condens Matter. 2002;14:9975–9986. [Google Scholar]
- 21.Dias FB, Morgado J, Macanita AL, da Costa FP, Burrows HD, Monkman AP. Macromolecules. 2006;39:5854–5864. [Google Scholar]
- 22.Wu C, Peng H, Jiang Y, McNeill J. J Phys Chem B. 2006;110:14148–14154. doi: 10.1021/jp0618126. [DOI] [PubMed] [Google Scholar]
- 23.Wu C, Zheng Y, Szymanski C, McNeill J. J Phys Chem C. 2008;112:1772–1781. doi: 10.1021/jp074149+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knaapila M, Garamus VM, Dias FB, Almasy L, Galbrecht F, Charas A, Morgado J, Burrows HD, Scherf U, Monkman AP. Macromolecules. 2006;39:6505–6512. [Google Scholar]
- 25.Teetsov JA, Vanden Bout DA. J Am Chem Soc. 2001;123:3605–3606. doi: 10.1021/ja0155534. [DOI] [PubMed] [Google Scholar]
- 26.Padmanaban G, Ramakrishnan S. J Am Chem Soc. 2000;122:2244–2251. [Google Scholar]
- 27.Ariu M, Sims M, Rahn MD, Hill J, Fox AM, Lidzey DG, Oda M, Cabanillas-Gonzalez J, Bradley DDC. Phys Rev B. 2003;67:195333. [Google Scholar]
- 28.Manas ES, Spano FC. J Chem Phys. 1998;109:8087–8101. [Google Scholar]
- 29.Spano FC. J Chem Phys. 2005;122:234701. doi: 10.1063/1.1914768. [DOI] [PubMed] [Google Scholar]
- 30.Hernando J, van Dijk EMHP, Hoogenboom JP, Garcia-Lopez JJ, Reinhoudt DN, Crego-Calama M, Garcia-Parajo MF, van Hulst NF. Phys Rev Lett. 2006;97:216403. doi: 10.1103/PhysRevLett.97.216403. [DOI] [PubMed] [Google Scholar]
- 31.Long X, Malinowski A, Bradley DDC, Inbasekaran M, Woo EP. Chem Phys Lett. 1997;272:6–12. [Google Scholar]
- 32.Lyons BP, Wong KS, Monkman AP. J Chem Phys. 2003;118:4707–4711. [Google Scholar]
- 33.Cabanillas-Gonzalez J, Fox AM, Hill J, Bradley DDC. Chem Mater. 2004;16:4705–4710. [Google Scholar]