

Abstract
Lantibiotics are ribosomally synthesized and post-translationally modified peptide antibiotics. The modifications involve dehydration of Ser and Thr residues to generate dehydroalanines and dehydrobutyrines, followed by intramolecular attack of cysteines onto the newly formed dehydro amino acids to produce cyclic thioethers. LctM performs both processes during the biosynthesis of lacticin 481. Mutation of the zinc ligands Cys781 and Cys836 to alanine did not affect the dehydration activity of LctM. On the other hand, these mutations compromised cyclization activity when investigated with full length or truncated peptide substrates. Mutation of His725, another residue that is fully conserved in lantibiotic cyclases, to Asn resulted in a protein that still catalyzed dehydration of the substrate peptide and also retained cyclization activity, but at a decreased level compared to wild type enzyme. Collectively, these results show that the C-terminal domain of LctM is responsible for cyclization, that the zinc ligands are critical for cyclization, and that dehydration takes place independently from the cyclization activity. Furthermore, these mutant proteins are excellent dehydratases and provide useful tools to investigate the dehydration activity as well as to generate dehydrated peptides for study of the cyclization reaction by wild type LctM.
Keywords: Lantibiotics, thioether, cyclase, dehydratase, zinc protein
The lantibiotics are a class of post-translationally modified peptides that have attracted much attention for their potential applications (1, 2) exemplified by nisin’s commercial use as a food preservative in the dairy industry for over thirty years (3). Understanding the biosynthetic pathways that generate these natural products offers information to improve their production as well as the opportunity to generate novel molecules with promising biological properties. The lantibiotics are produced by Gram-positive bacteria as prepeptides consisting of a leader sequence followed by a propeptide region. This peptide is acted upon by a set of enzymes that successively dehydrate serine and threonine residues in the propeptide to the corresponding dehydroalanine (Dha) and dehydrobutyrine (Dhb) residues, and catalyze the addition of cysteines onto the dehydro amino acids resulting in thioether formation (Figure 1A). These sulfide crosslinks are called lanthionines (from Ser) or methyllanthionines (from Thr). The class I lantibiotics, like nisin and subtilin, require two distinct enzymes in their biosynthetic pathway to catalyze the dehydration (LanB) and cyclization (LanC) reactions, whereas class II lantibiotics like lacticin 481 have a single enzyme (LanM) that performs both activities (Figure 1B) (4). The mechanisms by which the LanM enzymes carry out the dehydration and cyclization reactions are just beginning to be understood (Figure 1A). Studies conducted in vitro on the bifunctional lacticin 481 and haloduracin synthetases provided the first clues into the mechanism of dehydration (5–8) and the recent crystal structure of the nisin cyclase (NisC) has provided insight into thioether formation (9).
Figure 1.

A. Dehydration of serine and threonine residues by the LanB dehydratase results in the formation of dehydroalanine (Dha) or dehydrobutyrine (Dhb), respectively. The LanC cyclase subsequently catalyzes lanthionine formation through cysteine addition onto the unsaturated amino acids. Alternatively, the bifunctional LanM protein can catalyze both reactions. B. The biosynthesis of lacticin 481. Following ribosomal synthesis, lacticin 481 synthetase LctM dehydrates serine and threonine residues in the propeptide region of the substrate peptide LctA and catalyzes cyclization of cysteines onto the dehydro amino acids. This leads to the formation of two lanthionines and one methyllanthionine. Subsequent proteolytic cleavage of the leader sequence from lacticin 481 by LctT and transport of the final product across the cell membrane leads to production of the mature lantibiotic in Lactococcus lactis (14). The truncated substrate (but with Ser 35 mutated to Ala) used in this study is boxed.
Previous investigations of the lantibiotic cyclization reactions focused on the nisin and subtilin (SpaC) cyclases and revealed that these proteins are metalloenzymes that contain zinc (10). The crystallographic study of NisC showed that the enzyme is homologous in structure to the β-subunit of mammalian farnesyl transferase (9, 11). Hence, the lantibiotic cyclases appear to be members of a family of proteins that utilize zinc for activation of a thiol of their substrates (12, 13). Two cysteines and one histidine serve as the zinc ligands in NisC with water occupying a fourth coordination site. The LanM proteins display low but detectable sequence homology in their C-terminal regions with the LanC enzymes including conservation of the three metal ligands suggesting that this segment in each protein is responsible for catalyzing cyclization (Figure 2) (1, 15). A working model has been proposed for the cyclization reaction (10) in which the zinc activates the Cys for nucleophilic attack, and an active site base and acid are involved in deprotonation of the thiol and protonation of the enolate intermediate, respectively (Scheme 1). To date, no direct experimental evidence has been reported to support the hypothesized role of zinc in the LanM proteins. The in vitro reconstitution of lacticin 481 biosynthesis (Figure 1B) (5) offers a complete system to probe the residues important for catalysis. Here, we describe initial mutagenesis studies aimed at elucidating the role of zinc in the cyclization reaction carried out by lacticin 481 synthetase (LctM).
Figure 2.

Partial sequence alignment of the C-terminal domains of select LanM proteins with LanC enzymes. The conserved metal ligands are in red font and the conserved active site His in green. The active site Arg that is conserved in the LanC proteins but not the LanM proteins is shown in blue font. LctM (accession number AAC72258) (16), MrsM (CAB60261) (17), CinM (CAD60521) (18), SpaC (AAA22777) (19), EpiC (CAA44254) (20), NisC (CAA48383) (21), and PepC (CAA90026) (22).
Scheme 1.
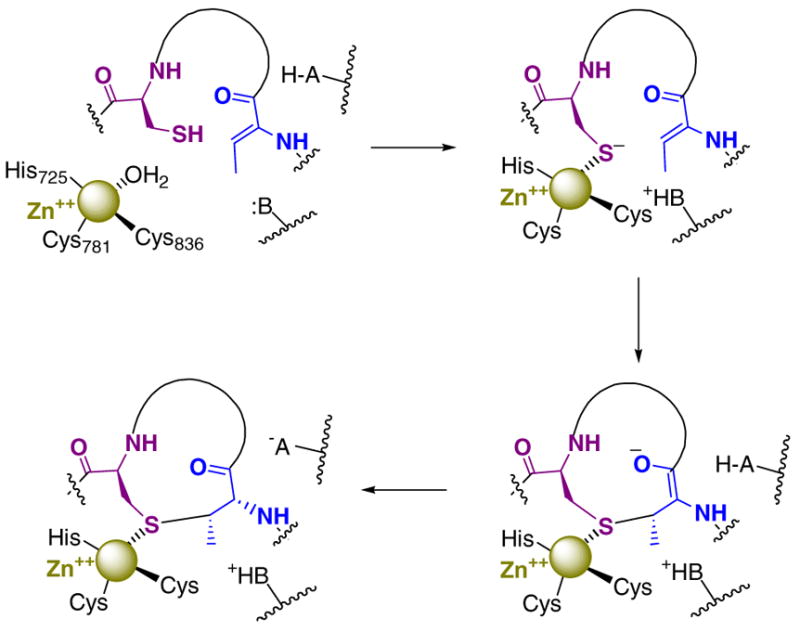
Proposed mechanism for the LctM-catalyzed formation of a methyllanthionine ring.
Materials and Methods
Construction of Expression Systems for LctM Mutants (23–25)
The mutants C781A, C836A and C781A/C836A were generated by overlap extension PCR with typical concentrations of template DNA (LctM wild type plasmid ( 5)) of ca. 0.1–0.2 ng/mL. Initially, two traditional PCRs were carried out for each mutant, one using the T7 forward primer paired with the internal mutant reverse primer and another reaction with the T7 reverse primer paired with the internal mutant forward primer (see Supporting Information for primer sequences). These PCR products were used for nested PCRs. First, all components except the primers were combined and subjected to 5 traditional PCR cycles, and subsequently the outside primers were added to the mixture and amplification over 25 additional cycles resulted in a piece of DNA that contained the correct mutation. Site directed mutagenesis using the Quikchange™ kit was performed using the protocol supplied by Stratagene for construction of the H725N mutant.
Expression and Purification of LctM Mutants
An overnight culture of E. coli BL21(DE3) cells carrying the pET28b plasmid containing the gene sequence for LctM-C781A, C836A, C781A/C836A, or H725N was grown in 100 mL of LB with 50 μg/mL kanamycin at 37 °C with shaking. The cells were divided into three aliquots of 30 mL and pelleted, followed by resuspension in 1 mL of fresh LB. Each 1 mL aliquot was added to 3 L of LB-kanamycin and grown at 37 °C with shaking until the OD600nm reached 0.8. Overexpression of the protein was induced by addition of IPTG to a final concentration of 0.5 mM. The cells were grown at 18 °C for 20–24 h and harvested by centrifugation at 12000 x g for 20 min at 4 °C. The cells (4.5 g/L) were resuspended in 1 M NaCl, 10% glycerol, 20 mM Tris, pH 8.3, frozen in liquid nitrogen, and stored at −80 °C.
Cells (4.5 g) were lysed by sonication, and cellular debris was pelleted by centrifugation at 29000 x g for 25 min at 4 °C. The resulting supernatant was passed through a 0.45 μm cellulose acetate filter, diluted 5-fold in 1 M NaCl, 10% glycerol, 20 mM Tris, pH 8.3, and loaded (2 mL/min) onto a 8 mL MC-20 POROS immobilized metal affinity chromatography (IMAC) column charged with 0.1 M NiSO4 and equilibrated in 1 M NaCl, 10% glycerol, 20 mM Tris, pH 8.3. The flow rate on the BioCAD FPLC was increased to 4 mL/min and the column was washed with 12 column volumes of 1 M NaCl, 10% glycerol, 20 mM Tris, pH 8.3. A second wash step consisted of 5 column volumes of 30 mM imidazole, 100 mM NaCl, 20 mM MOPS, pH 7.2. The protein was eluted using a 100 mL gradient of 30 to 500 mM imidazole in 100 mM NaCl, 20 mM MOPS, pH 7.2.
Fractions containing the protein of interest were combined and loaded slowly (0.5–1.0 mL/min) onto a 30 mL SP Sepharose™ FF cation exchange column equilibrated with 100 mM NaCl, 20 mM MOPS, pH 7.2. The column was washed extensively with this buffer until the UV absorbance at 280 nm was less than 0.02. The protein was eluted with a 100 mL gradient of 100 mM to 1 M NaCl in 20 mM MOPS, pH 7.2. Fractions containing protein as indicated by SDS-PAGE were combined and concentrated by Amicon using a YM-30 membrane (4.5 mg final yield from 1 L of culture). Glycerol was added to the concentrated protein to give a final concentration of 20%. The samples were frozen in liquid nitrogen and the stocks were stored at −80 oC.
Metal Analysis of LctM Mutants
After concentration utilizing a Amicon YM-30 ultrafiltration membrane, proteins were dialyzed against 2 x 2 L of chelex-treated buffer containing 20 mM MOPS, 300 mM NaCl, pH 7.1 for 18 h at 4 oC to remove weakly bound metals. The proteins were concentrated further on YM-30 centricons rinsed with chelex-treated deionized H2O (Millipore) to yield protein concentrations greater than 3 mg/mL. Protein concentrations were determined by averaging the values obtained from Bradford assay, BCA protein assay kit, and protein absorbance at 280 nm utilizing theoretically calculated extinction coefficients. Protein samples were submitted to the School of Chemical Sciences Microanalysis Laboratory at the University of Illinois at Urbana-Champaign for metal analysis by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Attempts to first remove the His-tag from the proteins prior to dialysis failed and resulted in precipitation during thrombin treatment and/or subsequent purification of the proteins.
Determination of the Solubility of LctA
Assessment of the solubility limits of His6-LctA were conducted on a 1 mL scale in buffer containing 50 mM Tris, pH 7.5, 10 mM MgCl2, 25 μg/mL bovine serum albumin, and 1 mM TCEP at 25 oC (standard assay conditions) for 4 h. A stock solution of His6-LctA was prepared at pH 2 containing a minimal amount of TFA (<0.1 %). The peptide has good solubility at this pH. A series of samples were then prepared with various peptide concentrations at pH 2. To these solutions was added the assay buffer described above, the pH was checked to be 7.5, and the samples were left for 4 h to mimic an enzymatic assay. No precipitation was visible when the final LctA concentration was less than 0.01 mg/mL whereas precipitated peptide was visible at higher concentrations. These conclusions were verified by centrifugation at 14000 rpm in an Eppendorf microcentrifuge for five minutes.
Assays of LctM Mutants with LctA and truncated substrates
Hexa-His tagged wild type LctA (His6-LctA) was expressed and purified as described previously (5, 6). Assays of His6-LctA (65 μM) were conducted on a 300 μL scale in the presence of 50 mM Tris, pH 7.5, 10 mM MgCl2, 25 μg/mL bovine serum albumin, 1 mM TCEP, and 0.75 μM enzyme at 25 oC for 4 hours. Assays were subsequently checked for loss of four waters by MALDI-TOF MS after acidification and Zip-tip (C-18, Millipore) treatment of the samples using the protocol provided by the supplier. The LctA substrate is only sparingly soluble under these conditions (approximately 0.01 mg/mL or 1.3 μM) but all substrate is eventually consumed and the product is soluble. Assays of truncated LctA (50 μM) were conducted on a 20 or 40 μL scale in the presence of 50 mM Tris, pH 7.5, 10 mM MgCl2, 25 μg/mL bovine serum albumin, 1 mM TCEP, and 2 μM enzyme at 25 oC for 1–4 h. Assays were also conducted under identical conditions but with the addition of 5 μM ZnCl2. To monitor the reactions, aliquots of 10 μL were removed, acidified with TFA, desalted and concentrated using a Zip-tip (C-18, Millipore), and monitored by MALDI-TOF MS confirming a single dehydration.
Expression and Purification of LctA(1–37)S35A thioester
BL21 (DE3) cells containing the pET15-LctA(1–37)S35A-intein-CBD plasmid (6) were grown in LB containing 100 μg/mL ampicillin at 37 oC and peptide expression was induced with 0.65 mM IPTG when the OD600nm reached 0.5 to 0.6. The cells were grown at 25 oC for an additional 6 h and then were harvested at 12,000 x g at 4 oC for 15 min. The cell paste was resuspended in 20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM EDTA and 0.1% Triton-X-100, frozen with liquid nitrogen, and stored at −80 oC until use. Cells were lysed by sonication on ice and then centrifuged at 23,700 x g for 30 min at 4 oC. The supernatant was filtered through a 0.45 μm cellulose acetate filter onto a chitin (NEB, 20 mL bed volume) column equilibrated with 100 mM HEPES, pH 7.2, 500 mM NaCl and 1 mM EDTA at 4 oC. The fusion protein was allowed to bind to the chitin column for 2 h with gentle shaking. The column was washed with 30 column volumes of 100 mM HEPES, pH 7.2, 500 mM NaCl, and 1 mM EDTA followed by 5 column volumes of 20 mM HEPES, pH 7.75, 200 mM NaCl, and 1 mM EDTA. To catalyze intein cleavage and generate the His6-LctA(1–37)S35A MES-thioester, the column was equilibrated with 75 mL of 20 mM HEPES, pH 7.75, 200 mM NaCl, 1 mM EDTA, and 50 mM MESNa at 4 oC for 14 h. The His6-LctA(1–37)S35A thioester was eluted from the column, the salt concentration was diluted 5-fold by addition of deionized water, and the peptide was concentrated on a YM1 amicon membrane to approximately 20 mL. The remaining solution was lyophilized and the peptide thioester was resuspended in 10 mL of H2O with 0.1% TFA for RP-HPLC. The LctA(1–37)S35A thioester was purified by HPLC on a preparative C4 column utilizing a gradient from 2% B and 98% A to 100% B over 45 min (A: H2O with 0.1 % TFA; B: 80 % acetonitrile with 0.086 % TFA).
Generation of His6-LctA(1–38)S35A by expressed protein ligation
His6-LctA(1-38)S35A was obtained by ligating the truncated LctA thioester with cysteine using expressed protein ligation (26, 27). L-Cysteine (2 mg) was dissolved in 300 μL of ligation buffer containing 100 mM HEPES, pH 7.75, 200 mM NaCl and 50 mM MESNa. The pH of the solution was adjusted to ~ 7.5, added to lyophilized His6-LctA(1–37)S35A thioester, and allowed to react at 4 oC for 14 h. Crude ligation mixtures were analyzed by MALDI-TOF to ensure a successful ligation and purified by RP-HPLC on a C4 analytical column utilizing a gradient from 30% to 100% B over 45 min.
p-Hydroxymercuribenzoic Acid (PHMB) Assays (28)
Assays of His6-LctA(1–38)S35A with either wild type or mutant LctM were carried out as described (5) on a 20 or 40 μL scale. Upon completion of the single dehydration of the substrate as determined by MALDI-TOF MS, a 10 μL aliquot of the assay mixture was placed in a new tube and evaporated to dryness. This crude sample was then incubated in 5 μL of 10 mM TCEP and 4 M guanidine hydrochloride at 25 oC for 10 min. Subsequently, 10 μL of 10 mM PHMB was added, incubated at 25 oC for 12 h and checked for thiol modification by MALDI-TOF MS. For MS data, see Supporting Information.
General Procedure for Bioassays
RP -HPLC purified His6-LctA was dissolved in deionized water (18 MOhm Cm) to a final concentration of 2 mg/mL; this solution had a pH of 2 due to residual TFA from HPLC purification. An assay (total volume of 6 mL) was conducted with 0.6 mg of LctA in the presence of 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 25 μg/mL BSA, 1 mM TCEP, and 5 mM ATP. His6-LctM (60 μg of wild type or LctM mutant) was added twice over four hours at 25 oC to ensure reactions went to completion. The assay volume was reduced to 1 mL on a Labconco Centrivap Concentrator. Samples were acidified with 10 μL of TFA and then purified by C4 analytical RP-HPLC utilizing a gradient from 2% B and 98% A to 100% B over 45 min with monitoring at 220 nm. Fractions were lyophilized, resuspended in 30 μL of Millipore water, and samples containing fully dehydrated material were identified by MALDI-TOF MS. Fully dehydrated material obtained from incubating LctA with LctM-C781A, LctM-C836A, or LctM-C781A/C836A had a slightly shorter retention time (29.5 min) than assays with wild type LctM (30.1 min). Dehydrated material produced from incubation with LctM-H725N eluted at both 29.5 and 30.1 min, likely indicating cyclized material elutes at higher acetonitrile concentrations. Lys-C (Roche) was resuspended in 100 mM Tris, pH 8.3, to give a final concentration of 0.4 μg/μL. The purified assay product was resuspended in 16 μL of 100 mM Tris, pH 8.3, with 4 μL of endoproteinase Lys-C and incubated for 3 h at 37 oC. Confirmation of the removal of the leader peptide was obtained by MALDI-TOF MS and leaderless products were concentrated to dryness. The compounds were redissolved in 5 μL of sterile deionized water. GM17 agar (4 % M17, 0.5 % glucose, and 1.5 % agar) was autoclaved, transferred to a sterile flask (25 mL/plate), and cooled to 50 oC in a water bath. An aliquot of 750 μL of an overnight culture of L. lactis CNRZ 117 grown at 25 oC in GM17 broth (4 % M17 and 0.5 % glucose) was added to the melted agar and a plate was poured. After the plate solidified, a sterile pipet tip was utilized to create holes in the agar and the cleavage products were applied. Lacticin 481 from cell free broth of L. lactis CNRZ 481 was utilized as a positive control. The plate was grown overnight at 25 oC and monitored for clearance. L. lactis CRNZ 481 and the lacticin sensitive strain L. lactis CRNZ 117 were attained from the Centre National de Recherches Zootechniques (CNRZ) culture collection, Jouy-enJosas, France.
RESULTS
Overexpression and Purification of LctM Mutants
Based on sequence homology with LanC enzymes and the X-ray structure of NisC (9), Cys781 and Cys836 are likely ligands to the zinc in LctM (Figure 2). Furthermore, His725 is proposed to be either the general base for deprotonation of a cysteine of the substrate peptide or the general acid for protonation of the resultant enolate intermediate (Scheme 1). Therefore, the LctM mutants C781A, C836A, and C781A/C836A were chosen to investigate the importance of an intact zinc binding site for dehydration and cyclization and H725N was prepared to evaluate the importance of His725 in these processes. The C781A, C836A and C781A/C836A mutants were generated via overlap extension PCR (OE-PCR) (23–25), and the H725N mutant was generated using the Quickchange™ site directed mutagenesis kit. The four mutant proteins (109 kDa) were heterologously expressed in E. coli and purified by immobilized metal affinity chromatography, followed by cation exchange chromatography, providing proteins of > 95 % purity as judged by SDS-PAGE. The zinc content of these proteins was analyzed after extensive dialysis using ICP-MS, demonstrating 0.94 equivalents of Zn2+ in His6-LctM, 0.93 equivalents in His6-LctM-H725N, 0.8 equiv. in the His6-LctM-C836A mutant, 0.65 equiv. in His6-LctM-C781A, and 0.45 equiv. in the double mutant His6-LctM-C781A/C836A. Attempted removal of the His-tag of these proteins by thrombin prior to metal analysis was not successful. Thus, these values are upper limits for the Zn content in the active site. However, since essentially no Ni2+ or other divalent metal ions were detected in the ICP-MS analysis and since all proteins contain the same His-tag, the observed Zn-content is believed to be representative of the effect of the mutations on the zinc content. This assumption is supported by the very similar zinc content for the His725Asn mutant, which does not involve a zinc ligand, and the wild–type protein.
Dehydration Assays of LctM Mutants with LctA
Assays testing the dehydration activities of the LctM mutants were carried out under the conditions described previously (5) using full length His6-LctA as substrate. The reaction with wild-type LctM served as a positive control. The assays were analyzed by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) displaying a peak at 7,709 Da for the substrate peptide. Treatment with wild type LctM in the presence of MgCl2 and ATP resulted in the appearance of minor peaks corresponding to M – 1 H2O (m/z 7791), M – 2 H2O (m/z 7673), M – 3 H2O (m/z 7655) and a major peak corresponding to the loss of 4 molecules of water (m/z 7637), consistent with previous results (Figure 3A) (5). Next, His6-LctA was treated with the LctM mutants C781A, C836A, C781A/C836A and H725N and the assay mixtures were analyzed by MALDI-MS (Figure 3). In all cases, four dehydrations were observed as the major product, indicating that these mutants retain dehydration ability comparable to wild type LctM.
Figure 3.
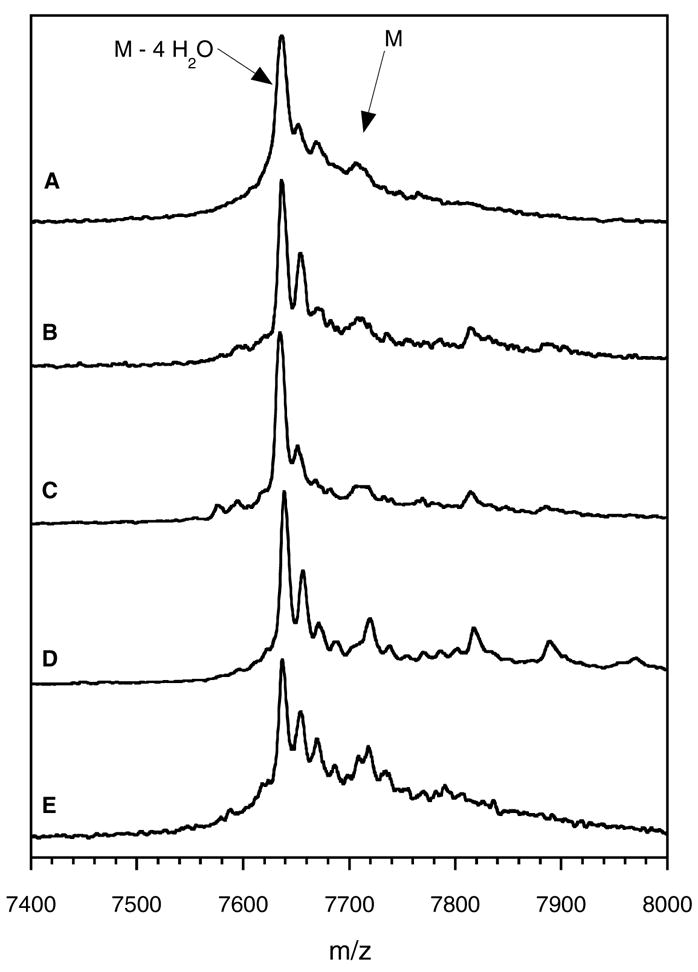
MALDI-TOF mass spectra of assays containing ATP, Mg2+, His6-LctA, and A) wild type LctM, B) LctM-H725N, C) LctM-C781A, D) LctM-C836A, and E) LctM-C781A/C836A.
Cyclization Assays with His6-LctA
Because the uncyclized and cyclized assay products have identical masses, they are indistinguishable by linear mode MALDI-TOF mass spectrometry. Previous studies have clearly shown that all three thioether rings are required for antimicrobial activity of lacticin 481 (5, 29), and thus, initial assessment of the cyclization activities of the mutants focused on the bioactivity of the assay products after removal of the leader peptide. LctA was treated in parallel with wild type LctM and the four mutant proteins, and the assay products were purified by RP-HPLC in order to isolate fully dehydrated peptides. The leader peptide was removed subsequently with endoprotease Lys-C (5) and the antimicrobial activity was tested using an agar diffusion assay against a Lactococcus lactis indicator strain. As shown in Figure 4, clear zones of growth inhibition were observed for the reaction with wild type LctM and H725N, whereas no such activity was detected for C781A, C836A, and the double mutant C836A/C781A. The antimicrobial activity of the H725N-treated LctA product indicates it is both dehydrated and cyclized. The cysteine to alanine mutants of LctM convert LctA to a dehydrated but non-toxic product, indicating both enzymatic and spontaneous cyclizations fail to provide the correct ring structures. Therefore, H725N must retain partial cyclase function. In fact, analysis of the assay products by HPLC showed reproducibly that the assay product formed in the reactions of the three Cys mutants eluted about one min earlier than the product of wild type LctM. The crude assay product obtained with LctM-H725N contained peaks at both retention times. These experiments show that Cys781 and Cys836 are absolutely required for cyclization activity but His725 is important but not essential.
Figure 4.
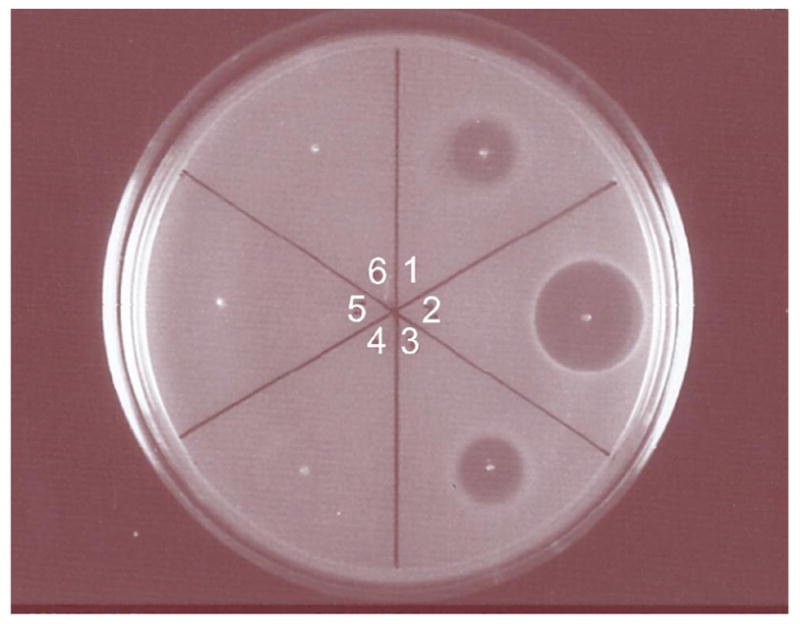
Assessment of antimicrobial activity utilizing L. lactis CNRZ 117 as the indicator strain. All processed samples were treated with LysC prior to application to the plate. Lane 1, lacticin 481 from cell free broth of L. lactis CNRZ 481; lane 2, LctA treated with wild type LctM; lane 3, LctA treated with LctM-H725N; lane 4, LctA treated with LctM-C781A; lane 5, LctA treated with LctM-C836A; lane 6, LctA treated with LctM-C781A/C836A.
Cyclization Activity with Truncated Substrate
The bioactivity assays provided an indirect read-out of cyclization activity and offer insights into the relative cyclization efficiency of the mutant enzymes, but it would be desirable to develop a more direct method of measuring cyclization activity. As mentioned previously, cyclized and uncyclized peptides cannot be distinguished readily by mass spectrometry, but the presence of free cysteines in uncyclized material may be visualized by chemical modification with a thiol-specific reagent. In principle, a myriad of reagents can be utilized to this end (e.g. iodoacetamide or N-ethylmaleimide). For this study, p-hydroxymercuribenzoic acid (PHMB) was chosen due to the large molecular weight shift (+320 Da) that occurs upon adduct formation (28). Individual cysteines react with PHMB to form an arylmercury adduct resulting in a mass shift of +320 Da (Scheme 2). When two Cys are nearby, the initially formed adduct can react with a second Cys to give a bidentate mercury adduct and a 200 Da mass increase (Scheme 2).
Scheme 2.
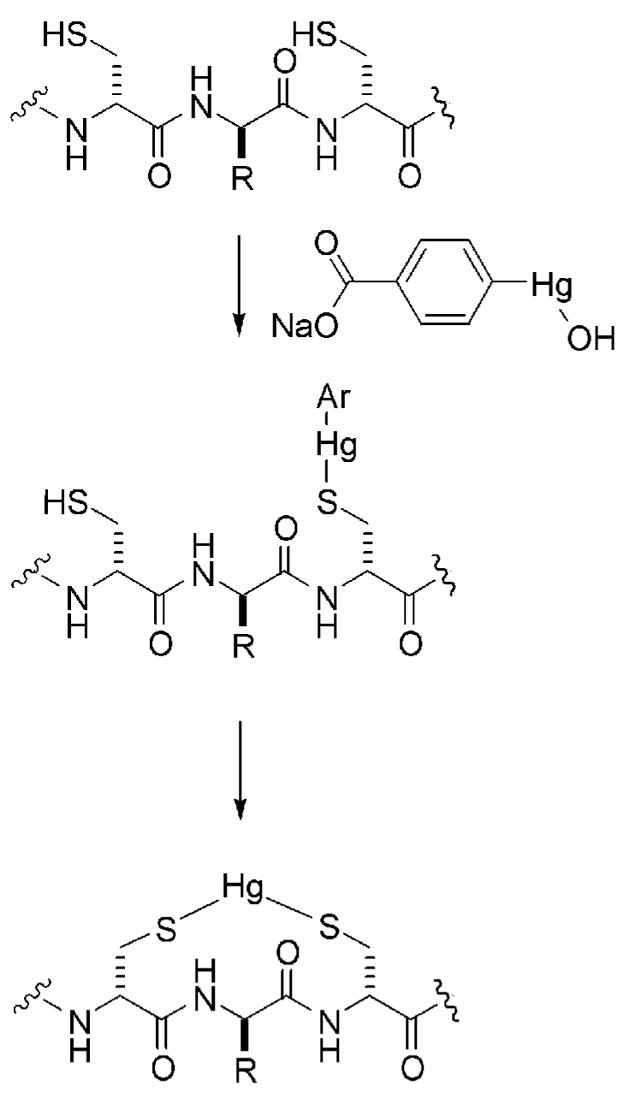
Compared to smaller molecular weight thiol specific reagents, PHMB allowed better distinction between derivatized peptides and unreacted substrates and ion adducts (sodium or potassium) of substrates or dehydrated peptides that were typically observed by MALDI-MS analysis of crude assay mixtures. We initially attempted to use PHMB derivatization on assay products of full length LctA. However, a control in which substrate LctA was subjected to the normal assay conditions in the absence of enzyme followed by incubation with the derivatization mixture under a series of conditions showed that the reaction of PHMB with LctA was never complete (Figure S1). The use of guanidinium hydrochloride to disrupt any protein structure and tris(carboxyethyl)phosphine (TCEP) to reduce any disulfide bonds that may have formed did not change this outcome. This finding severely hampered assessment of the cyclization activity of the LctM mutants using the full length LctA substrate because using lack of derivatization as evidence for cyclization would not be conclusive. Therefore, use of PHMB was investigated with an alternative substrate. First the substrate peptide was truncated to include only the first 38 residues of LctA, which contains only a single Cys at position 38. This truncated substrate has been shown previously to be a substrate for cyclization by wild type LctM generating the A-ring of lacticin 481 (5). In addition, Ser35 was mutated to Ala to generate the substrate LctA(1–38)S35A such that the intermediate after dehydration contained only a single dehydrated amino acid, Dhb33 (see box, Figure 1B). Non-enzymatic cyclization onto dehydrobutyrine residues is much slower than onto dehydroalanines (30–32), and removal of Dha35 by mutation of Ser35 to Ala prevents the latter reaction from occurring.
As intended, treatment of the resulting mutant LctA(1–38)S35A with PHMB, resulted in near quantitative formation of a single arylmercury adduct (Figure S2 in supporting information). When the mutant substrate was treated with wild type LctM or any of the mutants in the presence of ATP and Mg2+, one dehydration was detected corresponding to Thr33 being converted to Dhb. However, only the product of the reaction with wild type LctM was completely cyclized since no arylmercury adduct was detected after incubation with the enzyme for 4 h (Figure 5A). The absence of a free thiol in the enzymatic assay product was not due to intermolecular reaction of a thiol of one peptide with the dehydroamino acid of another peptide because such adducts would have been readily detectable by MS and were not observed.
Figure 5.
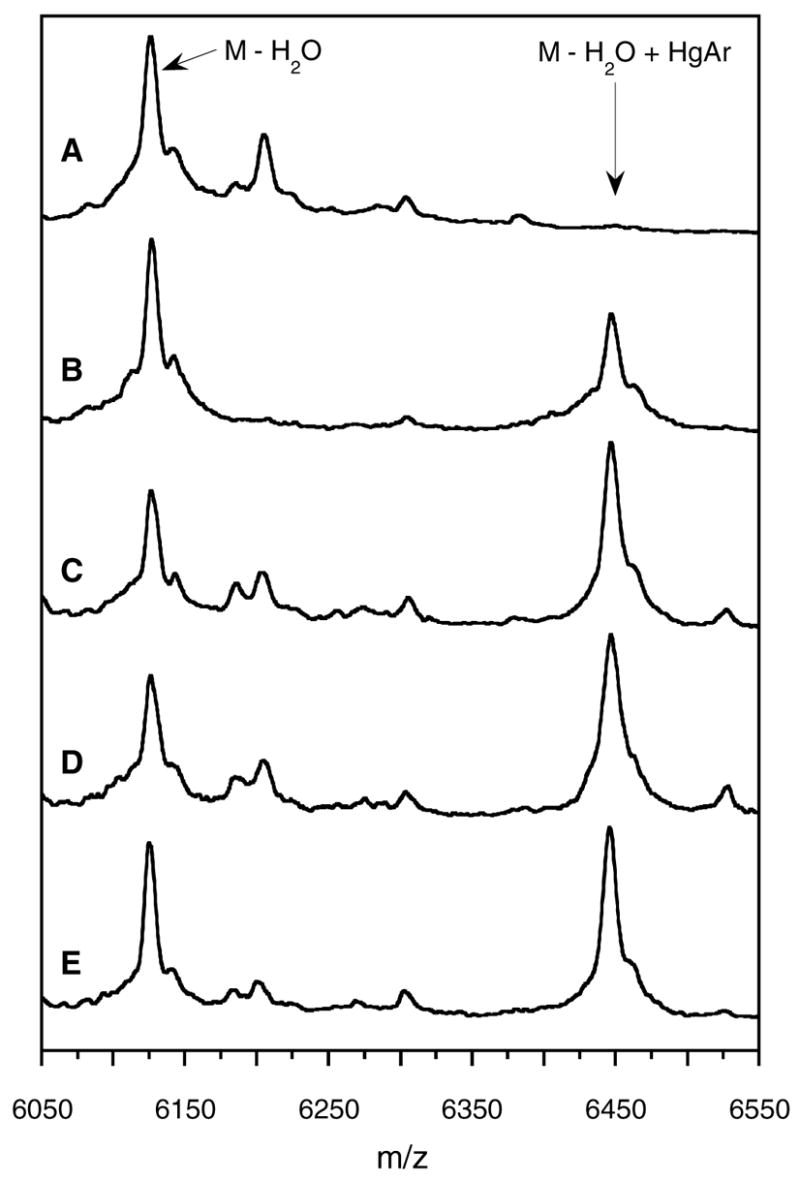
LctA(1-38)S35A following treatment with ATP, Mg2+, and A) wild type LctM, B) LctM-H725N, C) LctM-C781A, D) LctM-C836A, and E) LctM-C781A/C836A. All samples were treated with PHMB. Arylmercury modification of Cys38 is observed for all four LctM mutants (m/z = 6447) whereas only dehydrated and cyclized material (m/z = 6127) is detected for wild type LctM.
On the other hand, the products obtained with the mutant enzymes contained a mixture of cyclized and uncyclized material as indicated by arylmercury adduct formation (Figure 5). Addition of zinc salts to the assay mixtures did not affect the outcome of these experiments. The relative amounts of cyclized compared to uncyclized peptide were approximately the same for the three Cys mutants whereas the H725N mutant appears to generate more cyclized product. This is more clearly seen when parallel cyclization reactions were stopped before completion of the cyclization by the wild type enzyme (Figure 6). Although possible differences in ionization efficiency prevent accurate quantitation, it is clear that the extent of cyclization is greatest for wild type LctM (Figure 6A), decreased for the H725N mutant (Figure 6B), and greatly diminished for the other three mutants as exemplified for the C781A/C836A double mutant in Figure 6C. when cyclization by the wild type enzyme was still incomplete.
Figure 6.
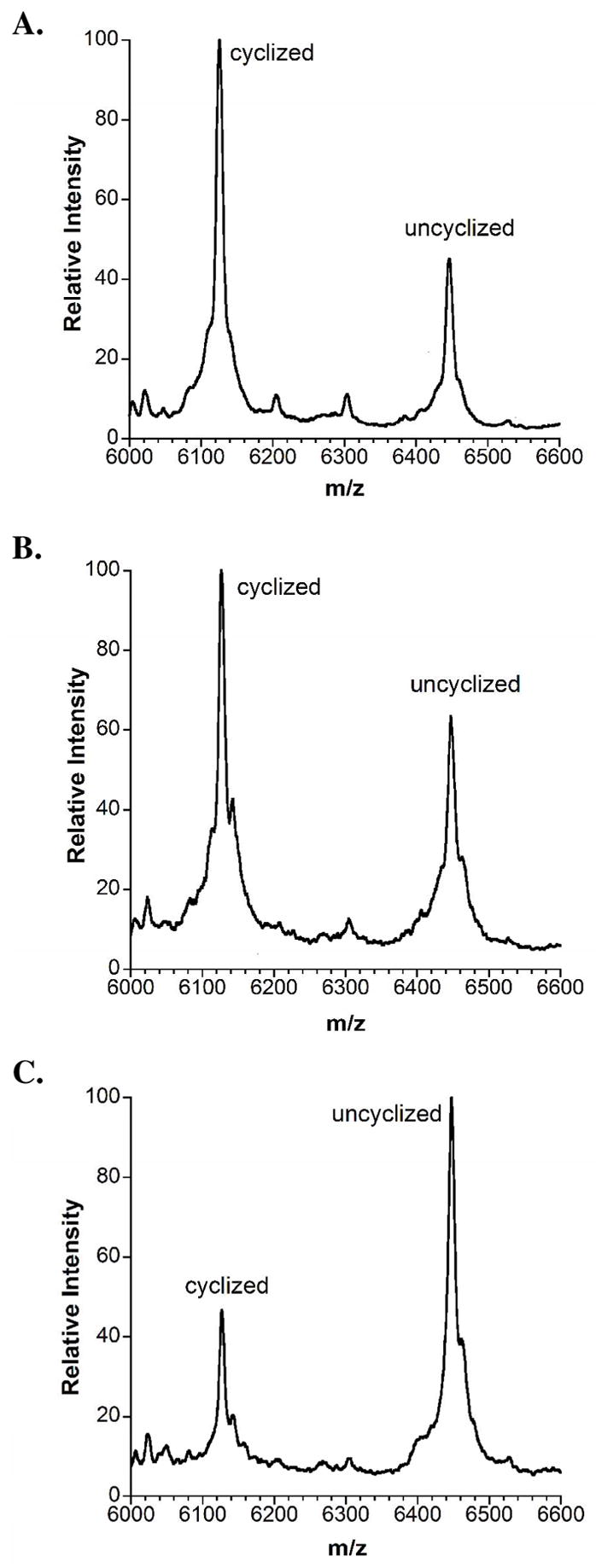
Cyclization assays of LctA(1-38)S35A incubated with ATP, Mg2+, and (A) wild type LctM, (B) LctM-H725N, and (C) LctM-C781A/C836A. All three assays were stopped at a timepoint (3 h)
Discussion
Previous work on SpaC and NisC, the cyclases involved in subtilin and nisin biosynthesis respectively, showed that these LanC enzymes bind a stoichiometric amount of zinc (10). That work also identified two of the ligands to the zinc within the SpaC protein through mutation of Cys303 and Cys349 to non-chelating alanines and subsequent extended X-ray absorbance fine structure (EXAFS) analysis. The identity of the zinc ligands was recently confirmed in the X-ray structure of NisC (9). As shown herein, mutation of the homologous conserved residues in LctM, Cys781, Cys836 as well as the conserved His725 resulted in lacticin synthetase mutants with dehydration activity as evidenced by reaction with native substrate LctA. These findings clearly indicate that the dehydration and cyclization reactions of LctM are independent and that the mutant proteins are correctly folded.
The cyclization activity of the mutant enzymes was assessed in two different assays providing consistent results. The use of an antimicrobial activity assay showed that wild type LctM and the His725Asn mutant were able to support correct formation of all three thioether rings. Similar results were observed with a truncated substrate with the wild type enzyme more efficiently catalyzing the cyclization process than the H725N mutant. On the other hand, the three Cys mutants of LctM were not able to produce bioactive material with full length LctA and all generated about the same low level of cyclization with the truncated LctA analog. The similarity in the level of cyclization achieved by the three Cys mutants coupled with the lack of antimicrobial activity suggests that the cyclization observed with the truncated substrate corresponds to non-enzymatic reaction of Cys38 with Dhb33. Indeed, non-enzymatic intramolecular reaction of cysteine with Dhb to produce methyllanthionine, although significantly slower than the analogous reaction between Cys and Dha (31), has been shown to be complete within 6 h for the B-ring of nisin (30). This conclusion is also supported by the observation of two peaks in the HPLC analysis of the reaction products with full length LctA, an earlier eluting peak without biological activity that was produced as the only product by the three Cys mutants and a later eluting peak that is produced as the only product by wild type LctM. We interpret the earlier eluting peak to correspond to uncyclized and/or non-enzymatic cyclized product in which either the topology of the rings or their stereochemistry is incorrect. The assays of His725Asn with full length LctA produced both peaks, consistent with a competition between nonenzymatic incorrect cyclization and enzymatic cyclization by a mutant with reduced activity.
Based on the recent X-ray structure of NisC, the mutated cysteine residues are both ligands to the Zn, resulting in a coordination environment of two Cys, one His (His837 in LctM), and one water in the wild-type enzyme. Zinc sites containing a water molecule usually signal a catalytic role for the metal (33). Indeed, the similarity of the NisC structure with that of the β-subunit of protein farnesyl transferase (PFTase) (11) as well as the parallels in the reactions they catalyze, alkylation of Cys residues, strongly suggests that the zinc in the lantibiotic cyclases also fulfills a catalytic function. The dehydratase reaction of LctM has been shown to involve phosphorylation of the Ser/Thr residues prior to the elimination step (6, 34) ruling out a role for the zinc of Lewis acid catalysis in water elimination. PFTase is a member of a family of zinc proteins that catalyze reactions of thiols with electrophiles and that have in common a net negatively charged zinc site upon coordination of the deprotonated thiol (12, 13). The zinc decreases the pKa of the thiol (35, 36), and the net negative charge activates the thiolate for nucleophilic attack and also prevents deprotonation of the bound water in the resting state of the enzyme (37, 38). Mutation of Cys ligands in other members of the family such as PFTase (37), the Ada protein (39), cobalamin-dependent Met synthase (40), betaine-homocysteine methyltransferase (41), cobalamin-independent Met synthase (42, 43), and epoxyalkane:CoM transferase (44) also resulted in completely inactive or catalytically severely impaired proteins. In many of these previous studies, the ability to bind zinc was strongly diminished similar to the ICP-MS analysis of the mutants reported here. We therefore also carried out our assays in the presence of additional Zn2+ but this did not restore activity of the mutants involving zinc ligands. The results presented here provide the first experimental support for the role of an intact zinc site of a LanM protein for activation of cysteines towards attack onto dehydro amino acids. These results are consistent with a recent study that showed that mutation of the zinc ligands of the SpaC cyclase abolished production of the class I lantibiotic subtilin in Bacillus subtilis (45).
In NisC, the histidine residue corresponding to His725 in LctM is located in the active site and is close to the water molecule that is liganded to the zinc. Our current working model for the mechanism of cyclization involves displacement of this water by a cysteine from the dehydrated substrate peptide followed by deprotonation of the thiol (Scheme 1). His725 could be the active site base that accepts the thiol proton. In farnesyl transferase, coordination of cysteines to the zinc in short model peptides reduced its pKa by three units (36). Similarly, coordination of a cysteine to a cobalt ion that substituted for the zinc resulted in a lowering of the pKa of the thiol from 8.3 for the free peptide (GCVLS) to approximately 6.4 upon binding to the enzyme (35). Given that the pKa’s of the cysteines in all of these peptides are on the order of 5.8–7.7 upon coordination, an active site base appears not to be absolutely required based on the thermodynamics of the system. Indeed, dedicated general bases do not appear to be present in the X-ray structures of farnesyl transferase, B12-dependent Met synthase (46), betaine-homocysteine methyltransferase (47) or the B12-independent Met synthase (48). Moreover, homocysteine binding has been shown to result in release of a proton to solvent in the B12-dependent Met synthase at pH 7.8 (40, 49). The mutagenesis studies performed here indicate that His725 appears to be important but not mandatory for the cyclization reaction due to its ability to generate biologically active lacticin 481 (Figure 4), currently the most stringent test for an active lantibiotic cyclase because it requires chemo-, regio-, and stereoselective cyclization (Scheme 1B). Although the X-ray structure of NisC suggests this residue is an active site base because it is close to the water that is believed to be displaced by the substrate thiol, no substrate or product bound complexes have been reported to date. Therefore, based on the currently available data it cannot be ruled out that His725 serves as the general acid that protonates the enolate or that it perhaps contributes to stabilization of the enolate through hydrogen bonding. We previously suggested that the general acid that protonates the enolate in NisC and other LanC proteins could be the conserved Arg280 that is present in the NisC active site and that is positioned correctly for an anti addition to Z-Dhb to produce (2S,3S,6R)-3-methyllanthionine. However, this residue is not conserved in the C-terminal domains of the LanM proteins (Figure 2) and mutation of this residue in NisC does not abolish cyclization activity (Li & van der Donk, unpublished results).
In summary, this study shows that mutation of the Cys ligands to the zinc in LctM abolishes correct macrocyclization of the LctA substrate. These mutants do retain their dehydration activity. The results presented also illustrate a potential complication regarding the use of LanC or LanM proteins to engineer novel lanthionine containing peptides. As demonstrated with the truncated LctA substrate, observed cyclization as judged by lack of derivatization of a Cys by a thiol specific probe does not guarantee that the product was obtained via enzymatic catalysis, especially if the substrate has been incubated for prolonged time periods. Finally, we show in this study that the fully conserved His725 is important but not required for correct enzymatic cyclization of LctA.
Supplementary Material
Supporting Information Available. Sequences of oligonucleotide primers used for site-directed mutagenesis, and additional mass spectrometric data on PHMB assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. Champak Chatterjee for cloning of the His6-LctA(1-37)S35A construct.
Abbreviations
- EXAFS
extended X-ray absorbance fine structure
- FPLC
fast protein liquid chromatography
- Gdn-HCl
guanidinium chloride
- Hcys
homocysteine
- IMAC
immobilized metal affinity chromatography
- Lan B
class I lantibiotic dehydratase enzymes
- LanC
class I lantibiotic cyclase enzymes
- LanM
class II lantibiotic synthetase enzymes
- MALDI-TOF MS
matrix assisted laser desorption ionization – time of flight mass spectrometry
- OE-PCR
overlap extension polymerase chain reaction
- PFTase
protein farnesyl transferase
- PHMB
p-hydroxymercuribenzoic acid
- RP-HPLC
reverse phase high performance liquid chromatography
- TCEP
tris(carboxyethyl)phosphine
- TFA
trifluoroacetic acid
Footnotes
This work was supported by the National Institutes of Health (GM58822).
References
- 1.Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and Mode of Action of Lantibiotics. Chem Rev. 2005;105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 2.Cotter PD, Hill C, Ross RP. Bacterial lantibiotics: strategies to improve therapeutic potential. Curr Protein Pept Sci. 2005;6:61–75. doi: 10.2174/1389203053027584. [DOI] [PubMed] [Google Scholar]
- 3.Delves-Broughton J, Blackburn P, Evans RJ, Hugenholtz J. Applications of the bacteriocin, nisin. Antonie van Leeuwenhoek. 1996;69:193–202. doi: 10.1007/BF00399424. [DOI] [PubMed] [Google Scholar]
- 4.Pag U, Sahl HG. Multiple activities in lantibiotics--models for the design of novel antibiotics? Curr Pharm Des. 2002;8:815–833. doi: 10.2174/1381612023395439. [DOI] [PubMed] [Google Scholar]
- 5.Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Lacticin 481: in vitro reconstitution of lantibiotic synthetase activity. Science. 2004;303:679–681. doi: 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee C, Miller LM, Leung YL, Xie L, Yi M, Kelleher NL, van der Donk WA. Lacticin 481 Synthetase Phosphorylates its Substrate during Lantibiotic Production. J Am Chem Soc. 2005;127:15332–15333. doi: 10.1021/ja0543043. [DOI] [PubMed] [Google Scholar]
- 7.Miller LM, Chatterjee C, van der Donk WA, Kelleher NL. The Dehydration Activity of Lacticin 481 Synthetase is Highly Processive. J Am Chem Soc. 2006;128:1420–1421. doi: 10.1021/ja057203d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McClerren AL, Cooper LE, Quan C, Thomas PM, Kelleher NL, van der Donk WA. Discovery and in vitro biosynthesis of haloduracin, a new two-component lantibiotic. Proc Natl Acad Sci USA. 2006;103:17243–17248. doi: 10.1073/pnas.0606088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, Yu J-PJ, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Structure and Mechanism of the Lantibiotic Cyclase Involved in Nisin Biosynthesis. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 10.Okeley NM, Paul M, Stasser JP, Blackburn N, van der Donk WA. SpaC and NisC, the Cyclases Involved in Subtilin and Nisin Biosynthesis, are Zinc Proteins. Biochemistry. 2003;42:13613–13624. doi: 10.1021/bi0354942. [DOI] [PubMed] [Google Scholar]
- 11.Park HW, Boduluri SR, Moomaw JF, Casey PJ, Beese LS. Crystal structure of protein farnesyltransferase at 2.25 angstrom resolution. Science. 1997;275:1800–1804. doi: 10.1126/science.275.5307.1800. [DOI] [PubMed] [Google Scholar]
- 12.Hightower KE, Fierke CA. Zinc-catalyzed sulfur alkylation: insights from protein farnesyltransferase. Curr Opin Chem Biol. 1999;3:176–181. doi: 10.1016/s1367-5931(99)80030-1. [DOI] [PubMed] [Google Scholar]
- 13.Matthews RG, Goulding CW. Enzyme-catalyzed methyl transfers to thiols: the role of zinc. Curr Opin Chem Biol. 1997;1:332–339. doi: 10.1016/s1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]
- 14.Dufour A, Hindré T, Haras D, Le Pennec JP. The biology of the lantibiotics of the lacticin 481 subgroup is coming of age. FEMS Microbiol Rev. 2006 doi: 10.1111/j.1574-6976.2006.00045.x. [DOI] [PubMed] [Google Scholar]
- 15.Siezen RJ, Kuipers OP, de Vos WM. Comparison of lantibiotic gene clusters and encoded proteins. Antonie van Leeuwenhoek. 1996;69:171–184. doi: 10.1007/BF00399422. [DOI] [PubMed] [Google Scholar]
- 16.Rincé A, Dufour A, Le Pogam S, Thuault D, Bourgeois CM, Le Pennec JP. Cloning, expression, and nucleotide sequence of genes involved in production of lactococcin DR, a bacteriocin from Lactococcus lactis subsp. lactis. Appl Environ Microbiol. 1994;60:1652–1657. doi: 10.1128/aem.60.5.1652-1657.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altena K, Guder A, Cramer C, Bierbaum G. Biosynthesis of the lantibiotic mersacidin: organization of a type B lantibiotic gene cluster. Appl Environ Microbiol. 2000;66:2565–2571. doi: 10.1128/aem.66.6.2565-2571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Widdick DA, Dodd HM, Barraille P, White J, Stein TH, Chater KF, Gasson MJ, Bibb MJ. Cloning and engineering of the cinnamycin biosynthetic gene cluster from Streptomyces cinnamoneus cinnamoneus DSM 40005. Proc Natl Acad Sci USA. 2003;100:4316–4321. doi: 10.1073/pnas.0230516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung YJ, Steen MT, Hansen JN. The subtilin gene of Bacillus subtilis ATCC 6633 is encoded in an operon that contains a homolog of the hemolysin B transport protein. J Bacteriol. 1992;174:1417–1422. doi: 10.1128/jb.174.4.1417-1422.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnell N, Engelke G, Augustin J, Rosenstein R, Ungermann V, Götz F, Entian KD. Analysis of genes involved in the biosynthesis of the lantibiotic epidermin. Eur J Biochem. 1992;204:57–68. doi: 10.1111/j.1432-1033.1992.tb16605.x. [DOI] [PubMed] [Google Scholar]
- 21.Engelke G, Gutowski-Eckel Z, Hammelmann M, Entian KD. Biosynthesis of the lantibiotic nisin: genomic organization and membrane localization of the NisB protein. Appl Environ Microbiol. 1992;58:3730–3743. doi: 10.1128/aem.58.11.3730-3743.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer C, Bierbaum G, Heidrich C, Reis M, Süling J, Iglesias-Wind MI, Kempter C, Molitor E, Sahl HG. Nucleotide Sequence of the Lantibiotic Pep5 Biosynthetic Gene Cluster and Functional Analysis of PepP and PepC. Eur J Biochem. 1995;232:478–489. doi: 10.1111/j.1432-1033.1995.tb20834.x. [DOI] [PubMed] [Google Scholar]
- 23.Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 25.Senanayake SD, Brian DA. Precise large deletions by the PCR-based overlap extension method. Mol Biotechnol. 1995;4:13–15. doi: 10.1007/BF02907467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans TC, Benner J, Xu MQ. Semisynthesis of Cytotoxic Proteins Using a Modified Protein Splicing Element. Protein Sci. 1998;7:2256–2264. doi: 10.1002/pro.5560071103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci USA. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pitts KE, Summers AO. The roles of thiols in the bacterial organomercurial lyase (MerB) Biochemistry. 2002;41:10287–10296. doi: 10.1021/bi0259148. [DOI] [PubMed] [Google Scholar]
- 29.Chatterjee C, Patton GC, Cooper L, Paul M, van der Donk WA. Engineering Dehydro Amino Acids and Thioethers into Peptides using Lacticin 481 Synthetase. Chem Biol. 2006;13:1109–1117. doi: 10.1016/j.chembiol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 30.Zhou H, van der Donk WA. Biomimetic Stereoselective Formation of Methyllanthionine. Org Lett. 2002;4:1335–1338. doi: 10.1021/ol025629g. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, Gieselman M, Zhou H, Averin O, van der Donk WA. Biomimetic studies on the mechanism of stereoselective lanthionine formation. Org Biomol Chem. 2003;1:3304–3315. doi: 10.1039/b304945k. [DOI] [PubMed] [Google Scholar]
- 32.Okeley NM, Zhu Y, van der Donk WA. Facile Chemoselective Synthesis of Dehydroalanine-Containing Peptides. Org Lett. 2000;2:3603–3606. doi: 10.1021/ol006485d. [DOI] [PubMed] [Google Scholar]
- 33.Vallee BL, Auld DS. Zinc: biological functions and coordination motifs. Acc Chem Res. 1993;26:543–551. [Google Scholar]
- 34.You YO, van der Donk WA. Mechanistic Investigations of the Dehydration Reaction of Lacticin 481 Synthetase Using Site-Directed Mutagenesis. Biochemistry. 2007 doi: 10.1021/bi602663x. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hightower KE, Huang CC, Casey PJ, Fierke CA. H-Ras peptide and protein substrates bind protein farnesyltransferase as an ionized thiolate. Biochemistry. 1998;37:15555–15562. doi: 10.1021/bi981525v. [DOI] [PubMed] [Google Scholar]
- 36.Rozema DB, Poulter CD. Yeast protein farnesyltransferase. pKas of peptide substrates bound as zinc thiolates. Biochemistry. 1999;38:13138–13146. doi: 10.1021/bi990794y. [DOI] [PubMed] [Google Scholar]
- 37.Harris CM, Derdowski AM, Poulter CD. Modulation of the zinc(II) center in protein farnesyltransferase by mutagenesis of the zinc(II) ligands. Biochemistry. 2002;41:10554–10562. doi: 10.1021/bi020349u. [DOI] [PubMed] [Google Scholar]
- 38.Wilker JJ, Lippard SJ. Modeling the DNA Methylphosphotriester Repair Site in Escherichia coli Ada. Why Zinc and Four Cysteines? J Am Chem Soc. 1995;117:8682–8683. [Google Scholar]
- 39.Sun LJ, Yim CK, Verdine GL. Chemical communication across the zinc tetrathiolate cluster in Escherichia coli Ada, a metalloactivated DNA repair protein. Biochemistry. 2001;40:11596–11603. doi: 10.1021/bi011001m. [DOI] [PubMed] [Google Scholar]
- 40.Goulding CW, Matthews RG. Cobalamin-dependent methionine synthase from Escherichia coli: involvement of zinc in homocysteine activation. Biochemistry. 1997;36:15749–15757. doi: 10.1021/bi971988l. [DOI] [PubMed] [Google Scholar]
- 41.Breksa AP, 3rd, Garrow TA. Recombinant human liver betaine-homocysteine S-methyltransferase: identification of three cysteine residues critical for zinc binding. Biochemistry. 1999;38:13991–13998. doi: 10.1021/bi991003v. [DOI] [PubMed] [Google Scholar]
- 42.González JC, Peariso K, Penner-Hahn JE, Matthews RG. Cobalamin-Independent Methionine Synthase from Escherichia coli: A Zinc Metalloenzyme. Biochemistry. 1996;35:12228–12234. doi: 10.1021/bi9615452. [DOI] [PubMed] [Google Scholar]
- 43.Zhou ZZ, Peariso K, Penner-Hahn JE, Matthews RG. Identification of the zinc ligands in cobalamin-independent methionine synthase (MetE) from Escherichia coli. Biochemistry. 1999;38:15915–15926. doi: 10.1021/bi992062b. [DOI] [PubMed] [Google Scholar]
- 44.Krum JG, Ellsworth H, Sargeant RR, Rich G, Ensign SA. Kinetic and microcalorimetric analysis of substrate and cofactor interactions in epoxyalkane:CoM transferase, a zinc-dependent epoxidase. Biochemistry. 2002;41:5005–5014. doi: 10.1021/bi0255221. [DOI] [PubMed] [Google Scholar]
- 45.Helfrich M, Entian K-D, Stein T. Structure-Function Relationships of the Lanthionine Cyclase SpaC Involved in Biosynthesis of the Bacillus subtilis Peptide Antibiotic Subtilin. Biochemistry. 2007 doi: 10.1021/bi062124f. on-line document prior to publication, Feb 17 2007. [DOI] [PubMed] [Google Scholar]
- 46.Evans JC, Huddler DP, Hilgers MT, Romanchuk G, Matthews RG, Ludwig ML. Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc Natl Acad Sci USA. 2004;101:3729–3736. doi: 10.1073/pnas.0308082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans JC, Huddler DP, Jiracek J, Castro C, Millian NS, Garrow TA, Ludwig ML. Betaine-homocysteine methyltransferase: zinc in a distorted barrel. Structure. 2002;10:1159–1171. doi: 10.1016/s0969-2126(02)00796-7. [DOI] [PubMed] [Google Scholar]
- 48.Pejchal R, Ludwig ML. Cobalamin-independent methionine synthase (MetE): a face-to-face double barrel that evolved by gene duplication. PLoS Biol. 2005;3:e31. doi: 10.1371/journal.pbio.0030031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jarrett JT, Choi CY, Matthews RG. Changes in protonation associated with substrate binding and Cob(I)alamin formation in cobalamin-dependent methionine synthase. Biochemistry. 1997;36:15739–15748. doi: 10.1021/bi971987t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. Sequences of oligonucleotide primers used for site-directed mutagenesis, and additional mass spectrometric data on PHMB assays. This material is available free of charge via the Internet at http://pubs.acs.org.