

Abstract
PrrA is a global trancription regulator activated upon phosphorylation by its cognate kinase PrrB in response to low oxygen conditions in Rhodobacter sphaeroides. Here we show by gel filtration, analytical ultracentrifugation and NMR diffusion measurements that treatment of PrrA by a phosphate analogue, BeF3-, results in dimerization of the protein, producing a protein that binds DNA. No dimeric species was observed in the absence of BeF3-. On addition of BeF3-, the inhibitory activity of the N-terminal domain on the C-terminal DNA-binding domain is relieved, after which PrrA becomes capable of binding DNA as a dimer. The interaction surface of the DNA-binding domain with the regulatory domain of PrrA is identified by NMR as being a well conserved region centered on helix α6, which is on the opposite face to the DNA recognition helix. This suggests that there is no direct blockage of DNA binding in the inactive state, but rather that PrrA dimerization promotes a correct arrangement of two adjacent DNA binding domains to recognise specific DNA binding sequences.
Keywords: Two-Component System, PrrA, RegA, dimerization, phosphorylation, DNA recognition
The purple, non-sulfur bacterium Rhodobacter sphaeroides has very versatile metabolic activities including aerobic and anaerobic respiration, photosynthesis, and carbon and nitrogen assimilation. The R. sphaeroides global regulator PrrA coordinately controls a large number of genes involved in the complex switch between aerobic and anaerobic lifestyles and the optimum use of reducing power. It regulates genes necessary for the synthesis of the photosynthetic apparatus, electron transport, nitrogen and carbon fixation, anaerobic respiration, [NiFe] hydrogenase and aerotaxis, as well as the expression of the Prr gene cluster itself (1-6).
The Prr system is a bacterial two-component signal transduction system (TCS) (7), consisting of the two proteins PrrB and PrrA. Two-component systems homologous to Prr have been found in other proteobacteria including other photosynthetic species, like the very similar and well-studied Reg system from R. capsulatus, but also in non-photosynthetic bacteria, and suggest a very conserved transduction mechanism, despite different in vivo functions (8,9). PrrB is a membrane-bound histidine kinase and is activated under low oxygen conditions, probably via a third member of the pathway, PrrC, through formation of an intermolecular disulfide bond using a conserved cysteine (10,11). It then autophosphorylates on a conserved histidine and the phosphate is transferred to the response regulator (RR) PrrA on a conserved aspartate residue.
PrrA is a two-domain protein. The N-terminal receiver domain (residues 1-130) is a CheY-like domain common to all bacterial TCS response regulators, and there are a number of structures of these domains including CheY (12), FixJ (13) and NtrC (14). The phosphorylated aspartate is located in the N-terminal domain, and the C-terminal effector domain runs from residue 141 to residue 184 at the C-terminus and is linked to the N-terminal domain by a short proline-rich fragment. Response regulators have been classified into three main families depending on the sequence similarities of their DNA binding domain: NarL, NtrC, and OmpR/PhoB. PrrA does not belong to any of these families. The solution structure of the C-terminal domain of PrrA, PrrAC, forms a three-helix-bundle with a FIS-like (Factor for Inversion Stimulation) helix-turn-helix DNA binding motif and is the shortest effector domain of known structure found so far (15).
Several DNA sequences in distal or proximal regions of genes regulated by PrrA (and RegA) have been identified to be bound by PrrA (1,4,16-21). In vitro selection experiments with a PrrA homolog from Bradyrhizobium japonicum (22), alignment of the DNA sequences bound by PrrA and RegA (which are 100% identical in the DNA-binding region), and NMR titrations, allowed characterization of the determinants of DNA-binding specificity (15). PrrA binds to an imperfect GCGNC inverted repeat, with the unusual feature of possessing variable half site spacing. Residues on the DNA-binding surface of PrrAC, essentially located on the recognition helix (α8), were proposed to be the main specific DNA recognition elements. The almost palindromic nature of the sequences recognised strongly suggests that PrrA binds as a symmetrical dimer on DNA, as do many HTH containing proteins, and guided the generation of a model of the complex (15).
Although all RR share a common receiver domain, their activation mechanism, i.e. the communication of the phosphorylation signal to the effector domain, differs between different RR. Only four structures of full-length RR have been characterized in their inactive state. In NarL and CheB, activation of the regulatory domain leads to release of the effector domain, whose active site is blocked when unphosphorylated (23,24). In the OmpR/PhoB homologs DrrB and DrrD from Thermotoga maritima, despite different extents of inter-domain interactions, neither is blocking the effector domain’s recognition helix, suggesting that efficient DNA binding is promoted by dimerization (25,26). Several RR with a DNA binding activity have been shown to dimerize upon phosphorylation in order to bind a symmetrically or tandemly arranged DNA sequence (27-31).
Studies of the influence of phosphorylation on PrrA (and RegA) suggest that PrrA DNA-binding and transcription activation activity are increased upon phosphorylation (32,33). PrrAC in isolation is also capable of significant DNA binding (15). Furthermore, PrrA has been shown to be active in regulation of gene expression in vivo in aerobic conditions, when it is supposed to be unphosphorylated (1,16). In conclusion, PrrA DNA-binding activity is inhibited to a certain extent in unphosphorylated PrrA, but the way this inhibition is achieved is still unclear.
We report here that R. sphaeroides PrrA, in complex with BeF3-, a phosphate analogue used in several studies of response regulators (34), forms a stable dimer. PrrA dimer is able to bind strongly to a specific DNA fragment from cycAP2 promoter. The dimeric form of PrrA is thus the fully active form of the response regulator. Because unphosphorylated PrrA has also been shown to be able to activate transcription (32) and bind DNA (21), we have investigated the presence of a dimeric form in the unphosphorylated species by analytical ultracentrifugation (AUC) and NMR diffusion. We could not detect any unphosphorylated dimer.
Comparison of PrrA and PrrAC NMR spectra defines a surface of the PrrA C-terminal domain likely to be involved in interdomain contacts leading to DNA-binding inhibition, demonstrating that inhibition does not involve a direct blockage of the DNA binding site. The region of PrrA proposed to participate in interdomain contacts is particularly well conserved in PrrA homologs.
MATERIALS AND METHODS
Expression and purification of PrrA and PrrAC was performed as described (15). NMR experiments were recorded on a Bruker DRX600 spectrometer at 275 K. All experiments were carried out using the buffer system used in earlier NMR studies (15) and shown to be most conducive to maintaining fully folded PrrAC, namely 200 mM ammonium sulfate, 50 mM sodium phosphate, 50 mM sodium chloride, 10 mM dithiothreitol and 10 mM magnesium chloride, pH 6.0. Gel permeation chromatography was carried out using Superdex G75 (Pharmacia) in a 20 × 300 mm column, in the standard NMR buffer. BeF3- was generated in situ by adding 2 mM beryllium chloride and 6 mM sodium fluoride. The D63A mutant of PrrA (and a wild-type for comparison purposes) was overexpresed in E. coli and purified using an intein/chitin-binding domain fusion system, as described (32).
Velocity analytical centrifugation
A Beckman XL1 ultracentrifuge with an An60-Ti analytical rotor was used. The rotor was allowed to equilibrate to 283 K and centrifugation was performed at 45000 rpm. An optical scan at 280 nm of every cell (OD against radial distance) was taken every 40 seconds. Sedimentation data were analysed using the time-derivative g(s*) method using the dcdt+ program (35). Sedimentation coefficients of the individual species present in the sample were obtained, every single non-interacting species giving a gaussian distribution. In order to calculate apparent molecular weights, ρ (solvent density) and ν (the partial specific volume) were calculated using the sednterp program (http://www.jphilo.mailway.com/download.htm) using the protein amino acid sequence, the temperature and the solvent conditions.
NMR diffusion by pulsed field gradients
NMR experiments were recorded on a Bruker DRX-600 with a single-axis gradient coil at 275 K. The PFG-LED (longitudinal encoding and decoding) pulse sequence was used (36). Different gradient strengths were applied in a series of experiments, using between 0 and 80 % of the maximal gradient strength. Decay of signals when the gradient strength is increased were fitted using a least-squares method to provide the diffusion coefficient. Dioxan was used as an internal standard at a 0.5 % concentration in all samples. Diffusion experiments in the conditions used for PrrA samples were also performed on hen lysozyme (500 μM), as a check on calibration. Lysozyme has a radius of gyration of 15.3 Å (37), which was consistent with the Rh of dioxan being 4.08 Å (38). The apparent PrrA Rh was calculated from the dioxan Rh using their ratios of diffusion coefficients.
Line broadening
To PrrA, PrrA.BeF3- and PrrAC (300 μM starting concentration) was added increasing amounts of a 25-bp section of the cycAP2 promoter from R. sphaeroides, with the sequence 5′-TCGTTGTGCGGCAATCCGTCATATA-3′. DNA was prepared as reported (15). The peak heights of the resulting spectra were measured and corrected for sample dilution.
RESULTS
PrrA forms a dimer upon addition of BeF3-
PrrA dimer was first observed after purification of PrrA following overexpression in the heterologous host Escherichia coli. On gel filtration, PrrA was eluted as two species of significantly different elution times, corresponding to monomer and dimer when compared to standards of known molecular weight. The dimer probably arises from phosphorylation during overexpression in E. coli, since unspecific phosphorylation has been previously reported in the PrrA homolog RegA (22). The two species both ran at approximately the position expected for monomeric PrrA on denaturing SDS-PAGE (data not shown). Dimeric and monomeric species have very different linewidths in 1H 1D NMR spectra. The dimer presents broad linewidths, consistent with the presence of a species of a higher molecular weight compared to the monomer.
In order to demonstrate that PrrA forms a dimer upon phosphorylation, phosphorylation had to be reproduced in vitro. It is known that PrrA possesses autodephosphorylation activity, and that the phosphorylated form has a half-life of around 330 min (32). We have therefore used BeF3- to generate a stable pseudo-phosphorylated PrrA as previously described (15). PrrA.BeF3- and the purified dimer had the same elution time when injected on gel filtration (Figure 1A), and very similar 1D 1H NMR spectra, which are both very different from those of PrrA alone (Figure 1B). These gel filtration results indicate that the protein is at least 90% dimer. They also imply at least 90% conversion to the BeF3- adduct. By mass spectroscopy, there is no trace of the underivatised species, and only species with approximately the expected mass gain are found. The conversion to the adduct therefore appears to be essentially quantitative.
Figure 1.

Dimerisation of PrrA upon addition of BeF3-. A: Superdex G75 gel filtration profile of PrrA and PrrA.BeF3-. PrrA (purified as the monomer) and PrrA.BeF3- were injected onto the column. Monomer and dimer are eluted at their expected molecular weight; the dimer elutes at the same volume as the minor species from the E. coli growth. Some protein aggregates are formed during concentration and are eluted in the void volume (39 ml). The dimer peak has a “tail” suggesting the presence of a low quantity of monomer (<10 %) or an exchange during gel filtration between monomeric and dimeric species. The column was calibrated using standards of known molecular weights: bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (26 kDa), lysozyme (14.3 kDa) and insulin (5.8 kDa).
B: 1H 1D spectra of PrrA and PrrA.BeF3-. Spectra of PrrA (bottom) and PrrA.BeF3- (top) (500 μM) were recorded at 2°C under identical conditions and overlaid. The spectrum of PrrA.BeF3- shows a large broadening of the NMR signal resulting in an important decrease in signal intensity. Dimer purified from E. coli has a very similar NMR spectrum (not shown).
The results therefore demonstrate that the BeF3- adduct is mainly dimeric, but that there is an equilibrium between monomer and dimer. Literature cited above shows that the unphosphorylated form of PrrA retains some activity, suggesting that PrrA might be able to form a dimer when unphosphorylated, but with a weaker affinity constant. Several techniques have therefore been used in order to investigate this equilibrium in both PrrA and the BeF3- adduct.
Effect of BeF-3 on the monomer-dimer equilibrium: Velocity analytical ultracentrifugation
Velocity analytical centrifugation experiments were performed with PrrA and PrrA.BeF3- at different concentrations of protein (30, 100 and 200 μM) and at 10 °C. Analysis of the sedimentation coefficient distributions was performed with dcdt+ (35), and the fitting of the distributions produced PrrA monomer and dimer sedimentation coefficients of about 2.0 S and 3.1 S respectively (Figure 2 and Table 1). The sedimentation coefficients are in a ratio of 1:1.5, as expected for dimerization of a globular protein (35). Sedimentation coefficients were converted to approximate molecular weights after correction for solvent viscosity and temperature, and were 29 kDa for monomer and 48 kDa for dimer, compared to expected molecular weights of 22.5 and 45 kDa. The difference between experimental and calculated masses for the monomer is consistent with its expected prolate shape, which leads to a higher sedimentation coefficient. The sedimentation coefficients are therefore consistent with the presence of a monomeric and a dimeric form of PrrA.
Figure 2.
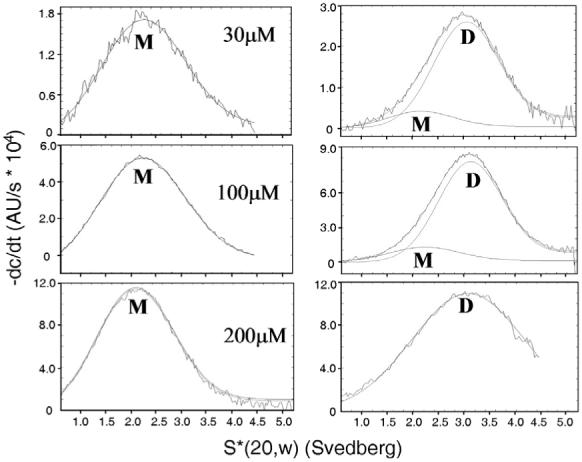
Velocity analytical centrifugation of PrrA and PrrA.BeF3- at different protein concentrations. Fitting of sedimentation coefficients was calculated with dcdt+ at the different protein concentrations indicated. Left: PrrA. Right: PrrA.BeF3-. The overall fitted functions and the individual functions corresponding to monomeric and dimeric species are indicated. The difference between the fitted function and original data (residual) showed good fits in all cases.
Table 1.
Sedimentation Coefficients of PrrA and PrrA.BeF3- Determined by AUC
| PrrA M | PrrA.BeF3- M | % M | PrrA.BeF3- D | % D | |
|---|---|---|---|---|---|
| 30 μM | 2.096 ± 0.007 | 2.05 ± 0.01 | 13 | 3.06 ± 0.01 | 87 |
| 100 μM | 2.055 ± 0.002 | 2.092 ± 0.004 | 14 | 3.033 ± 0.005 | 86 |
| 200 μM | 1.925 ± 0.002 | NA | - | 3.062 ± 0.006 | - |
M: monomer
D: dimer
In PrrA.BeF3- samples the majority of the protein is dimeric, as expected, although a small amount of monomer is observable at protein concentrations of 30 and 100 μM. At 200 μM, only a dimeric form was observed (Figure 2). By contrast, in unphosphorylated PrrA, only a monomeric form was observed. Furthermore, the variation of the sedimentation coefficients upon concentration, in the unphosphorylated form, does not suggest any significant self-association. The amounts of monomer and dimer observed at 30 and 100 μM PrrA.BeF3- permit an estimation of the dimer dissociation constant, of 1.2 μM and 4.6 μM respectively from the two measurements, giving an average estimate of approximately 3 μM. Using this value at 200 μM protein, the amount of monomer is calculated at 8% of the total, consistent with observations.
Effect of BeF3- on the monomer-dimer equilibrium: NMR diffusion measurements
Pulsed field gradient (PFG) NMR is a technique of choice to investigate oligomerization of macromolecules by examining changes in diffusion coefficients (38), which can be used to calculate apparent hydrodynamic radii (Rh). It is also one of the few methods that can be used to study proteins at the high concentrations used in NMR experiments. The aim of the experiment was to measure a change in Rh of PrrA with concentration, in order to further characterize the dimerization process. Peaks in the amide and methyl area of PrrA and PrrA.BeF3- at 100 and 500 μM were integrated and the hydrodynamic radii calculated relative to dioxan, the probe used as a reference (37). From the results of ref. 37, one would expect PrrA to have a monomeric Rh of around 22 Å. Results are presented in Table 2. The apparent hydrodynamic radii of PrrA and PrrA.BeF3- are significantly different, about 23 Å and 32.6 Å, consistent with monomeric and dimeric species respectively.
Table 2.
NMR Diffusion Experiments on PrrA and PrrA.BeF3-a
| PrrA | PrrA.BeF3- | |
|---|---|---|
| Rh 500 μM (Å) | 24 ± 2 | 32.6 ± 0.6 |
| Rh 100 μM (Å) | 22 ± 1 | 27.0 ± 1 |
| Decrease in Rh (%) | 4 ± 6 | 16 ± 5 |
The decrease of the hydrodynamic radius in the PrrA sample is not significant because of a large error on the measurement.
A concentration-dependent change in dimer-monomer equilibrium was observed in PrrA.BeF3- (Table 2) in the same way as previously shown by AUC, again suggesting a dimerization constant in the low micromolar range in the presence of BeF3-. There was no significant change in the hydrodynamic radius of PrrA alone upon dilution, again indicating that the unphosphorylated PrrA is essentially completely monomeric.
In order to confirm that BeF3- acts at the active site aspartate, and is therefore forming a close analog of the true phosphorylated form, the addition of BeF3- was carried out in parallel for 200 μM solutions of wild-type protein and a D63A mutant, which is not expected to be affected since it lacks the crucial aspartate sidechain. As above, the apparent hydrodynamic radius of wild-type PrrA increased significantly, but the radius of the mutant changed from 23 to 25 Å, a difference that is not statistically different from zero.
Treatment of PrrA by BeF3- increases DNA binding
Samples of full-length PrrA were titrated with a 25-bp sequence corresponding to the R. sphaeroides cycAP2 promoter, which has been shown to be protected against DNase I digestion by activated PrrA (19). Addition of DNA had no effect on the spectrum of PrrA, but caused line broadening and consequent loss of peak height in the spectrum of PrrA.BeF3-, due to the increased overall molecular weight and slower rotational diffusion of the PrrA/DNA complex (Figure 3). This result demonstrates that PrrA has no significant binding to the cycAP2 promoter in the absence of BeF3-, but binds in the presence of BeF3-.
Figure 3.
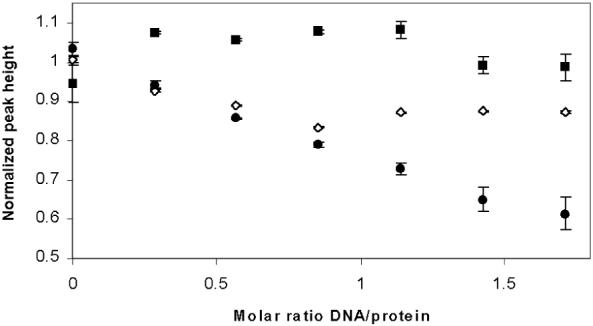
Changes in peak height in the methyl region of the NMR spectrum (0.5 - 0.9 ppm) of full-length PrrA (squares), PrrA.BeF-3 (open diamonds) and PrrAC (circles) on binding to the cycAP2 promoter. The changes are a result of line broadening, and have been normalized to an approximately equal starting height. Because free PrrAC has narrower lines than does PrrA.BeF3-, its changes in height on DNA binding are expected to be greater for the same degree of binding. Thus, the absolute changes in height are not readily interpretable, and the apparent similarity of the slopes for PrrAC and PrrA.BeF3- is coincidental.
The C-terminal domain of PrrA also bound to DNA (Figure 3). Addition of DNA to PrrA.BeF3- caused an increase in linewidth up to approximately a 1:1 molar ratio, after which no further increase in linewidth was observed. By contrast, the linewidth of PrrAC continued to increase up to at least 1.8 molar equivalents, implying that binding saturation was not reached until well after a 1:1 DNA:protein ratio. This suggests a strong binding interaction with PrrA.BeF3-, but rather weaker binding for PrrAC.
Differences in the PrrAC NMR spectrum in full-length PrrA and in PrrAC
Phosphorylation of the N-terminal regulatory domain of PrrA by PrrB increases the DNA binding activity of the C-terminal domain. This function is common to many TCS response regulators, but the manner in which it is achieved is variable. In our experimental conditions, PrrAC and full-length PrrA in complex with a phosphate mimic, BeF3-, are able to significantly bind specific DNA while PrrA alone is not, suggesting that the regulatory domain has an inhibitory function when unphosphorylated. In order to understand how PrrA is activated, we have investigated the interface between its N- and C-terminal domains by comparison of PrrA and PrrAC NMR spectra.
The 1D 1H NMR spectra of PrrA and PrrAC are almost superimposable, suggesting a very low contribution from the N-terminal domain to the spectrum. In the 15N-HSQC spectrum of full-length PrrA, only approximately ten or twenty amide peaks can be assigned to the N-terminal domain while most of the remaining resonances match those observed on PrrAC (Figure 4A). Despite considerable efforts, we have been unable to find conditions in which signals from the N-terminal domain can be seen. Experiments reported above do not suggest extensive oligomerization or aggregation of the full-length protein. The lack of signals from the N-terminal domain could therefore be a consequence of partial unfolding or a chemical exchange process. Although partial unfolding is possible, PrrA is at least partially functional in the conditions used in this study, in that it can dimerize and bind DNA. The same construct, but in different solution conditions, is also able to dephosphorylate PrrB-P (39). Thus, the N-terminal domain is concluded to be in dynamic equilibrium between the active folded form and one or more other forms.
Figure 4.

Comparison of PrrA C-terminal domain spectra in the full-length protein and in PrrAC. A: Overlaid 15N-HSQC spectra of PrrA (black) and PrrAC(red), obtained in identical solution conditions. Signals from the C-terminal domain comprise most of the signals observed in full-length PrrA NMR spectra. PrrAC residue differences in NH peak positions from the backbone but also sidechain resonances from asparagines, glutamines and arginines can be observed on the 15N HSQCs. Most PrrAC resonances are easily identifiable in full-length PrrA and suggest the C-terminal domain is in a very similar conformation to that in the structure of PrrAC determined previously. A few examples are shown on the figure. Several peaks from the C-terminal domain could not be found without ambiguity, presumably because they experience large chemical shift variation.
B: Backbone NH chemical shift differences of PrrAC between the isolated domain and full-length PrrA. Chemical shift differences are represented, as a function of residue, from the overlaid 15N-HSQC in A. Differences are weighted as [(δH)2 + (δN/10)2]1/2. In purple are represented PrrAC signals that unambiguously could not be found in the PrrA spectrum. The red and orange lines represent the lower limits defined for large and medium chemical shift differences respectively. These differences are represented in Fig. 5 on the PrrAC structure.
The consequence of this unusual behavior of the N-terminal domain is that most PrrAC peaks are easily identified in the PrrA spectrum, allowing a comparison of the resonances of the C-terminal domain in the full-length protein and PrrAC (Figure 4B).
The most dramatic perturbations in chemical shift are located at residue R147 in the middle of helix α6, and around R172, a key residue in sequence-specific DNA binding located in the recognition helix α8 (15). Twelve PrrAC resonances in the inter-domain linker (125, 127-129, 137), in α6 (145, 148-150, 152,153) and in the α6-α7 loop (157) cannot be found in PrrA. It is very likely that they experience large chemical shift variations between the full length PrrA and the C-terminal domain alone. Some sidechains also show significant changes in chemical shift, mainly R143 but also R172 and R181 Hεs (data not shown). Some milder backbone perturbations are also observed (residues 131, 136, 139-141, 151, 158, 167, 170, 171, 173, 174, 178, 179). These differences occur mainly in the interdomain linker, which was found to have no definite conformation in the PrrAC structure (15) and presumably changes conformation in the full-length protein, and on the internal face of the α7 and α8 helices, in particular L167, L174 and L178 belonging to the hydrophobic core. These changes could result from perturbations of core residues I149 and I152 from the α6 helix.
Whereas R172 exhibits large backbone and sidechain chemical shift changes, the rest of the recognition helix seems globally only mildly affected by the presence of the N-terminal domain, with many resonances on the surface of α8 found unchanged in full-length PrrA. The perturbations around R172 could result from a slight change of conformation due to a rearrangement of the hydrophobic core, as observed with the chemical shift variations of several core residues, itself possibly as a consequence of contacts with the N-terminal domain via α6. It is interesting to observe perturbations to what is presumed to be one of the key specificity-determining residues involved in DNA binding (15) caused by the interaction with the N-terminal domain.
In summary, the middle part of the α6 helix, between residues 145 and 153, is the most perturbed area of the C-terminal domain in full-length PrrA (Figure 5), which suggests that this region of PrrAC is in contact with the N-terminal domain in unphosphorylated PrrA, since these contacts are absent when PrrAC is expressed separately. We have previously published a structure of PrrAC and a model of its interaction with DNA (15). We have also attempted to model the N-terminal domain into this model, guided by the chemical shift changes that suggest that it is in contact with the center of α6, and using crystal structures of homologous receiver domains. The modelling shows that PrrAN does not block binding of PrrAC to DNA, in any possible interface arrangement consistent with the data. It does however prevent binding of PrrA to DNA as a dimer. We therefore conclude that PrrAN functions by preventing assembly of a dimeric PrrA onto adjacent DNA binding sites (rather than by a direct steric blockage of binding), and that phosphorylation releases PrrAC, allowing rearrangement of the linker between the two domains, and thus permits both dimerization of PrrA and cooperative binding of PrrA dimer to DNA.
Figure 5.

Chemical shift differences between PrrAC in isolation and in full length PrrA. In red are represented large HN backbone chemical shift changes and in purple peaks that could not be found in the PrrA spectrum. Medium backbone chemical shift changes are shown in orange. R143, R172 and R181 Hε experience large chemical shift changes and their sidechains are represented in blue. The end of the PrrA N-terminal domain (end of helix α5) is shown in cyan. The W146 peak was too weak to follow its variation.
On addition of BeF3- to full-length PrrA, almost all the NMR signals broaden and cannot be seen in 2D 15N HSQC spectra, which can at least partially be explained by dimerization and the consequent increase in molecular weight. We were therefore unable to measure chemical shift changes for the DNA-binding domain upon addition of BeF3-.
Residues involved in contacts with the N-terminal domain are conserved in PrrAC homologs
Homologs of the Prr system are found in members of several divisions of the proteobacteria, including the α-proteobacteria R. capsulatus, Roseobacter denitrificans, Rhodovulum sulphidophilum, B. japonicum and Sinorhizobium melitoti (3,8,9); the β-proteobacteria (hypothetical homologs in Burkholderia fungorum, Ralstonia metallidurans and Nitrosomonas europeae), and the γ-proteobacteria (Pseudomonas aeruginosa Rox (40) and hypothetical homologues in Ps. syringae, Ps. putida, Ps. fluorescens, Xanthomonas axonopodis as well as Vibrio cholerae). The homologs of the α-proteobacteria are particularly highly conserved. The important homologies between the proteins, and the fact that some of them can replace each other in vitro and in vivo, suggest that they are functionally very similar (8,9). The C-terminal domain of PrrA, and particularly the helix-turn-helix motif, is very well conserved (∼100 %) in the α-proteobacteria, which suggests that these PrrA homologs adopt similar modes of DNA binding and specificity. An alignment of the most conserved PrrAC homologs has been performed (Figure 6). Hydrophobic residues important to maintain the topology of the three-helix bundle are conserved (144, 149, 152, 153, 160, 164, 167, 169, 174, 178) and correspond, except for V160, to hydrophobic positions found in the structurally similar E. coli FIS and S. typhimurium NtrC (28,41).
Figure 6.

Alignment of PrrAC with homologous RR effector domains and hypothetical proteins. Conservation >70% is outlined by boxes. PrrAC secondary structure (helices α6-8) is indicated at the bottom of the figure. The conservation occurs mainly in the first loop and the helix-turn-helix motif. Residues shown to be involved in DNA binding in the first loop (binding to phosphate backbone) and at the beginning of α8 (specific binding to bases) are highly conserved. Hyp: hypothetical protein. Species not otherwise given in the text are: Mesorhizobium loti, Rhodopseudomonas loti, Brucella suis, Brucella melitensis, Rhizobium leguminosarium, Agrobacterium tumefaciens, Caulobacter crescentus, Magnetospirillum magnetotacticum, Azotobacter vinelandii, Ralstonia metallidurans, Ralstonia solanacearum, Microbulbifer degradans, Shewanella oneidensis, Xylella fastidiosa, Eubacterium acidaminophilum, Xanthomonas campestris, Oceanobacillus iheyensis, Leishmania major.
The helix-turn-helix motif is conserved even in the less homologous proteins. Residues on the recognition helix α8 that are involved in contacts with specific DNA (R171, R172, T173, Q175, R176 and K180) (15) are conserved, as well as residues involved in phosphate backbone binding like N159 and R143 from the α6-α7 loop and α6 respectively. In the N-terminal part of PrrAC, P138 and S140 are also conserved in more than 70% of sequences. S140 is in good position to provide an N-cap for helix α6. It would involve a hydrogen bond between S140 OH1 and R143 NH and could stabilise the folding of the domain.
More unusual is the conservation of a patch of residues in the middle part of the α6 helix, since these residues cannot be required for DNA recognition. Residues 146 to 151 show an overall conservation of more than 70% for residues 146 to 150, 151 being either an arginine or a glutamine. In the WEHIQR conserved motif, W146 and I149 are involved in the folding of the domain; I149 is completely buried in the hydrophobic core and W146 is partially buried and “closes” the hydrophobic core. However, the conserved residues E147, H148, and Q150 form a hydrophilic patch on the surface of the domain and are suggested by our NMR data to form the core of the interaction surface with the N-terminal domain. Furthermore, the observed chemical shift variations of R143 and R181 Hε support the hypothesis of interdomain contacts occurring at the level of the conserved patch on α6. Thus, sequence similarities suggest that the contact surface is functionally important.
DISCUSSION
DNA binding or transcription activation activity of PrrA in both unphosphorylated and phosphorylated states has been observed in previous studies of R. sphaeroides and in the homologous RegA from R. capsulatus (1,32,42). Evaluations of the difference in activity between the two forms of the response regulator have produced conflicting results partly due to the inefficiency of phosphorylation by small phosphodonors like acetyl phosphate (≤ 20 % efficiency) and the possibility of purifying PrrA phosphorylated during overexpression in E. coli. Nevertheless, Comolli and colleagues demonstrated significant differences in transcription activation between PrrA and PrrA-P, after ensuring that PrrA was not purified in a phosphorylated state (32).
In this study, we have made use of the observation that BeF3- is an efficient method of activation of response regulators (32,43). We have shown that a large majority of PrrA is converted to dimer, consistent with earlier observations on other RR, and also that (consistent with earlier studies) the BeF3- is acting at the active site aspartate. Furthermore, the dimer binds DNA. We have shown by two different methods that PrrA.BeF3- forms a dimer with a moderately strong dimerisation constant of approximately 3 μM, which means that only the dimeric form of PrrA can be observed at concentrations of 200 μM or greater.
Because DNA binding of unphosphorylated PrrA has been reported in several studies, and it has been observed that PrrA is able to regulate several genes even under aerobic conditions when the Prr pathway is inactive and PrrA is supposed to be unphosphorylated (1), we have investigated the possible presence of a PrrA dimer in unphosphorylated PrrA at different protein concentrations. It was not possible to demonstrate the presence of a dimer in unphosphorylated PrrA with the methods used in this study, and attempted observation of binding of PrrA to a specific DNA fragment was inconclusive because binding was too weak to be measured by NMR (15). The differences in transcription activation activities of PrrA and PrrA-P, measured by Comolli and colleagues (32), were dependent on PrrA concentration as well as the phosphorylation state. The difference in activation was 15.6 fold at 0.15 μM but only 2.5 at 1.5 μM PrrA concentration. These results would be compatible with a dimerization process of PrrA that we were unable to demonstrate. It is possible that the sensitivity of transcription activation assays and the experimental conditions used by Comolli and colleagues allowed dimerization to occur. Additional elements present in the preparation of the RNA polymerase extracts (RNA polymerase, transcription factors), and the presence of the DNA itself could explain the difference of results. The presence of consensus-like PrrA DNA binding sites (15) in promoter regions of genes regulated by R. capsulatus RegA, for genes regulated under anaerobic conditions like ccoNOPQ and cycA (1,16), suggests that RegA and PrrA bind as dimers in all cases. RegA and PrrA might be able to form a dimer when unphosphorylated in the presence of a co-regulator, or might be activated by other kinases than their cognate histidine kinase (44).
It is relevant to consider the binding affinities of the different proteins studied here. The shape of the NMR titrations (Fig. 3) suggests that monomeric PrrAC binds tothe DNA consensus sequence with an affinity of the order of 100 μM, under our assay conditions. By comparison, full-length unactivated PrrA binds much weaker (weak enough not to be able to observe binding by NMR), whereas PrrA.BeF3- binds much more tightly. Glutaraldehyde cross-linking experiments (unpublished data) suggest that the difference in affinity between PrrA and PrrA.BeF3- is approximately 200-fold (a relatively small difference, but consistent with other biological switches). Taking these results together implies approximate affinities of PrrA.BeF3- ≈ 5 μM, PrrAC 100 μM, and PrrA ≈ 1 mM. This is consistent with all of the results presented here, and also with recent DNAse I footprinting (45), but is too weak an affinity to explain the activity and specificity of activated PrrA in vivo. It is therefore likely that additional factors are involved in stabilising the complex, possibly sigma factors from RNA polymerase (3,32).
The similarity of the NMR spectra of the C-terminal domain in isolation or within the full-length PrrA allowed a comparison at the residue level between the two states of the DNA binding domain. This comparison allowed the definition of an area in the middle part of helix α6 which is the most affected by the presence of the N-terminal regulatory domain. The conservation of a patch of residues on the surface of α6 supports the proposition of this surface as being mainly responsible for the interaction with the N-terminal domain. Unlike NarL and CheB, the mode of inhibition by the PrrA effector domain does not involve a direct blockage of the DNA binding site. PrrA dimerizes upon phosphorylation, and following a change of conformation probably releases the C-terminal domain, presenting two C-terminal domains in a conformation where they can bind DNA, in a symmetrical manner on two adjacent DNA major grooves. Thus, the mode of PrrA activation resembles that of the two OmpR/PhoB homologs DrrB and DrrD, for which dimerization is proposed to be the main determinant for activation (26).
The very large chemical shift changes of R172 between full-length PrrA and PrrAC are interesting, because they suggest that there is a conformational change propagated through to R172 from the contact of PrrAC with PrrAN. Our model of the PrrA/DNA complex based on NMR titrations (15) identifies R172 as a key determinant of binding specificity. This suggests a possible further level of regulation of DNA binding: it may be that full-strength binding of the recognition helix to specific DNA sequences can only occur once the contact with PrrAN is broken, further reducing the level of DNA binding in the unactivated state. The converse would also be true: that binding of PrrAC to cognate DNA sequences may weaken the binding to PrrAN and thus prevent loss of signal. These consequences merit further study.
PrrAC could not be studied by NMR in the PrrA dimer. A complete loss of NMR signal from PrrAC occurs upon dimerization, suggesting a drastic conformational and/or dynamic change which is not due simply to aggregation or high order oligomerization, since AUC and NMR diffusion measurements do not suggest any oligomers of higher order than dimers, and the loss of signal is too big an effect to reflect only a broadening of signal upon doubling of the molecular weight (46). It is probable that the C-terminal domain is in some kind of exchange between several conformations in the phosphorylated PrrA, resulting in signal broadening, a phenomenon already observed for the N-terminal domain in PrrA, with or without BeF3-.
The Prr system has recently been shown to regulate over 860 genes in R. sphaeroides (S. Kaplan, personal communication), and is becoming established as a paradigm for bacterial regulation (3,47). Genetically, PrrA and its counterpart RegA in R. capsulatus are probably the best understood bacterial TCS systems. This study has elucidated the structural determinants of phosphorylation-dependent activation of PrrA, and has therefore substantially increased our understanding of this important regulatory system.
Acknowledgements
We thank Steve Harding and Chris Walters (National Centre for Hydrodynamics, University of Nottingham) for assistance with the analytical ultracentrifugation studies; Richard Tunnicliffe, Sam Weber and Teresa Collins for their help with the NMR diffusion and titration experiments; Jeremy Craven for help with fitting of the diffusion data; and Chris Potter for assistance in cloning. The authors are members of the North of England Structural Biology Centre (NESBIC).
Footnotes
This work was supported by an equipment grant from the Wellcome Trust and by grants from the NIH (GM37509) and DOE (ER63232-1018220-0007203).
References
- 1.Swem LR, Elsen S, Bird TH, Swem DL, Koch HG, Myllykallio H, Daldal F, Bauer CE. The RegB/RegA two-component regulatory system controls synthesis of photosynthesis and respiratory electron transfer components in Rhodobacter capsulatus. J. Mol. Biol. 2001;309:121–138. doi: 10.1006/jmbi.2001.4652. [DOI] [PubMed] [Google Scholar]
- 2.Oh JI, Kaplan S. Redox signaling: globalization of gene expression. EMBO J. 2000;19:4237–4247. doi: 10.1093/emboj/19.16.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elsen S, Swem LR, Swem DL, Bauer CE. RegB/RegA, a highly conserved redox-responding global two- component regulatory system. Microbiol. Mol. Biol. Rev. 2004;68:263–279. doi: 10.1128/MMBR.68.2.263-279.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elsen S, Dischert W, Colbeau A, Bauer CE. Expression of uptake hydrogenase and molybdenum nitrogenase in Rhodobacter capsulatus is coregulated by the RegB-RegA two- component regulatory system. J. Bact. 2000;182:2831–2837. doi: 10.1128/jb.182.10.2831-2837.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laratta WP, Choi PS, Tosques IE, Shapleigh JP. Involvement of the PrrB/PrrA two-component system in nitrite respiration in Rhodobacter sphaeroides 2.4.3: evidence for transcriptional regulation. J. Bact. 2002;184:3521–3529. doi: 10.1128/JB.184.13.3521-3529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romagnoli S, Packer HL, Armitage JP. Tactic responses to oxygen in the phototrophic bacterium Rhodobacter sphaeroides WS8N. J. Bact. 2002;184:5590–5598. doi: 10.1128/JB.184.20.5590-5598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson VL, Buckler DR, Stock AM. A tale of two components: a novel kinase and a regulatory switch. Nature Struct. Biol. 2000;7:626–633. doi: 10.1038/77915. [DOI] [PubMed] [Google Scholar]
- 8.Masuda S, Matsumoto Y, Nagashima KVP, Shimada K, Inoue K, Bauer CE, Matsuura K. Structural and functional analyses of photosynthetic regulatory genes regA and regB from Rhodovulum sulfidophilum, Roseobacter denitrificans, and Rhodobacter capsulatus. J. Bact. 1999;181:4205–4215. doi: 10.1128/jb.181.14.4205-4215.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emmerich R, Hennecke H, Fischer HM. Evidence for a functional similarity between the two-component regulatory systems RegSR, ActSR, and RegBA (PrrBA) in alpha-proteobacteria. Arch. Microbiol. 2000;174:307–313. doi: 10.1007/s002030000207. [DOI] [PubMed] [Google Scholar]
- 10.Swem LR, Kraft BJ, Swem DL, Setterdahl AT, Masuda S, Knaff DB, Zaleski JM, Bauer CE. Signal transduction by the global regulator RegB is mediated by a redox-active cysteine. EMBO J. 2003;22:4699–4708. doi: 10.1093/emboj/cdg461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McEwan AG, Lewin A, Davy SL, Boetzel R, Leech A, Walker D, Wood T, Moore GR. PrrC from Rhodobacter sphaeroides, a homologue of eukaryotic Sco proteins, is a copper-binding protein and may have a thiol- disulfide oxidoreductase activity. FEBS Letts. 2002;518:10–16. doi: 10.1016/s0014-5793(02)02532-2. [DOI] [PubMed] [Google Scholar]
- 12.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. Crystal structure of activated CheY. J. Biol. Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 13.Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama JP. Conformational changes induced by phosphorylation of the FixJ receiver domain. Structure. 1999;7:1505–1515. doi: 10.1016/s0969-2126(00)88341-0. [DOI] [PubMed] [Google Scholar]
- 14.Volkman BF, Nohaile MJ, Amy NK, Kustu S, Wemmer DE. 3-Dimensional solution structure of the N-terminal receiver domain of NtrC. Biochemistry. 1995;34:1413–1424. doi: 10.1021/bi00004a036. [DOI] [PubMed] [Google Scholar]
- 15.Laguri C, Phillips-Jones MK, Williamson MP. Solution structure and DNA binding of the effector domain from the global regulator PrrA (RegA) from Rhodobacter sphaeroides: insights into DNA binding specificity. Nucleic Acids Res. 2003;31:6778–6787. doi: 10.1093/nar/gkg891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swem DL, Bauer CE. Coordination of ubiquinol oxidase and cytochrome cbb3 oxidase expression by multiple regulators in Rhodobacter capsulatus. J. Bact. 2002;184:2815–2820. doi: 10.1128/JB.184.10.2815-2820.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du SY, Bird TH, Bauer CE. DNA binding characteristics of RegA - A constitutively active anaerobic activator of photosynthesis gene expression in Rhodobacter capsulatus. J. Biol. Chem. 1998;273:18509–18513. doi: 10.1074/jbc.273.29.18509. [DOI] [PubMed] [Google Scholar]
- 18.Dubbs JM, Bird TH, Bauer CE, Tabita FR. Interaction of CbbR and RegA* transcription regulators with the Rhodobacter sphaeroides cbbI promoter-operator region. J Biol. Chem. 2000;275:19224–19230. doi: 10.1074/jbc.M002125200. [DOI] [PubMed] [Google Scholar]
- 19.Karls RK, Wolf JR, Donohue TJ. Activation of the cycA P2 promoter for the Rhodobacter sphaeroides cytochrome c2 gene by the photosynthesis response regulator. Mol. Microbiol. 1999;34:822–835. doi: 10.1046/j.1365-2958.1999.01649.x. [DOI] [PubMed] [Google Scholar]
- 20.Dubbs JM, Tabita FR. Interactions of the cbbII promoter-operator region with CbbR and RegA (PrrA) regulators indicate distinct mechanisms to control expression of the two cbb operons of Rhodobacter sphaeroides. J. Biol. Chem. 2003;278:16443–16450. doi: 10.1074/jbc.M211267200. [DOI] [PubMed] [Google Scholar]
- 21.Hemschemeier SK, Kirndorfer M, Hebermehl M, Klug G. DNA binding of wild type RegA protein and its differential effect on the expresion of pigment binding proteins in Rhodobacter capsulatus. J. Mol. Microbiol. Biotechnol. 2000;2:235–243. [PubMed] [Google Scholar]
- 22.Emmerich R, Strehler P, Hennecke H, Fischer HM. An imperfect inverted repeat is critical for DNA binding of the response regulator RegR of Bradyrhizobium japonicum. Nucleic Acids Res. 2000;28:4166–4171. doi: 10.1093/nar/28.21.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baikalov I, Schroder I, KaczorGrzeskowiak M, Grzeskowiak K, Gunsalus RP, Dickerson RE. Structure of the Escherichia coli response regulator NarL. Biochemistry. 1996;35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 24.Djordjevic S, Goudreau PN, Xu QP, Stock AM, West AH. Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proc. Natl Acad. Sci. USA. 1998;95:1381–1386. doi: 10.1073/pnas.95.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckler DR, Zhou YC, Stock AM. Evidence of intradomain and interdomain flexibility in an OmpR/PhoB homolog from Thermotoga maritima. Structure. 2002;10:153–164. doi: 10.1016/s0969-2126(01)00706-7. [DOI] [PubMed] [Google Scholar]
- 26.Robinson VL, Wu T, Stock AM. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J. Bacteriol. 2003;185:4186–4194. doi: 10.1128/JB.185.14.4186-4194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maris AE, Sawaya MR, Kaczor-Grzeskowiak M, Jarvis MR, Bearson SMD, Kopka ML, Schroder I, Gunsalus RP, Dickerson RE. Dimerization allows DNA target site recognition by the NarL response regulator. Nature Structural Biol. 2002;9:771–778. doi: 10.1038/nsb845. [DOI] [PubMed] [Google Scholar]
- 28.Pelton JG, Kustu S, Wemmer DE. Solution structure of the DNA-binding domain of NtrC with three alanine substitutions. J. Mol. Biol. 1999;292:1095–1110. doi: 10.1006/jmbi.1999.3140. [DOI] [PubMed] [Google Scholar]
- 29.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure. 2002;10:701–713. doi: 10.1016/s0969-2126(02)00761-x. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H, Msadek T, Zapf J, Madhusudan, Hoch JA, Varughese KI. DNA complexed structure of the key transcription factor initiating development in sporulating bacteria. Structure. 2002;10:1041–1050. doi: 10.1016/s0969-2126(02)00803-1. [DOI] [PubMed] [Google Scholar]
- 31.Toro-Roman A, Mack TR, Stock AM. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: A symmetric dimer mediated by the α4-β5-α5 face. J. Mol. Biol. 2005;349:11–26. doi: 10.1016/j.jmb.2005.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comolli JC, Carl AJ, Hall C, Donohue T. Transcriptional activation of the Rhodobacter sphaeroides cytochrome c2 gene P2 promoter by the response regulator PrrA. J. Bacteriol. 2002;184:390–399. doi: 10.1128/JB.184.2.390-399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bird TH, Du SY, Bauer CE. Autophosphorylation, phosphotransfer, and DNA-binding properties of the RegB RegA two-component regulatory system in Rhodobacter capsulatus. J. Biol. Chem. 1999;274:16343–16348. doi: 10.1074/jbc.274.23.16343. [DOI] [PubMed] [Google Scholar]
- 34.Yan D, Cho HS, Hastings CA, Igo MM, Lee SY, Pelton JG, Stewart V, Wemmer DE, Kustu S. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl Acad. Sci. USA. 1999;96:14789–14794. doi: 10.1073/pnas.96.26.14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philo JS. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal. Biochem. 2000;279:151–163. doi: 10.1006/abio.2000.4480. [DOI] [PubMed] [Google Scholar]
- 36.Altieri AS, Hinton DP, Byrd RA. Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 1995;117:7566–7567. [Google Scholar]
- 37.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 38.Jones JA, Wilkins DK, Smith LJ, Dobson CM. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR. 1997;10:199–203. [Google Scholar]
- 39.Potter CA, Ward A, Laguri C, Williamson MP, Henderson PJF, Phillips-Jones MK. Expression, purification and characterisation of full-length histidine protein kinase RegB from Rhodobacter sphaeroides. J. Mol. Biol. 2002;320:201–213. doi: 10.1016/S0022-2836(02)00424-2. [DOI] [PubMed] [Google Scholar]
- 40.Comolli JC, Donohue TJ. Pseudomonas aeruginosa RoxR, a response regulator related to Rhodobacter sphaeroides PrrA, activates expression of the cyanide-insensitive terminal oxidase. Mol. Microbiol. 2002;45:755–768. doi: 10.1046/j.1365-2958.2002.03046.x. [DOI] [PubMed] [Google Scholar]
- 41.Yuan HS, Finkel SE, Feng JA, Kaczorgrzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant FIS protein - relationship between mutational changes and recombinational enhancer function or DNA binding. Proc. Natl Acad. Sci. USA. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hemschemeier SK, Ebel U, Jager A, Balzer A, Kirndorfer M, Klug G. In vivo and in vitro analysis of RegA response regulator mutants of Rhodobacter capsulatus. J. Mol. Microbiol. Biotechnol. 2000;2:291–300. [PubMed] [Google Scholar]
- 43.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim S-H, Wemmer D, Kustu S, Yan D. BeF3- acts as a phosphate analog in proteins phosphorylated on aspartate: Structure of a BeF3- complex with phosphoserine phosphatase. Proc. Natl. Acad. Sci. USA. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomelsky M, Kaplan S. Isolation of regulatory mutants in photosynthesis gene expression in Rhodobacter sphaeroides 2.4.1 and partial complementation of a PrrB mutant by the HupT histidine kinase. Microbiology. 1995;141:1805–1819. doi: 10.1099/13500872-141-8-1805. [DOI] [PubMed] [Google Scholar]
- 45.Jones DF, Stenzel RA, Donohue TJ. Mutational analysis of the C-terminal domain of the Rhodobacter sphaeroides response regulator PrrA. Microbiology. 2005;151:4103–4110. doi: 10.1099/mic.0.28300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laguri C. PhD Thesis. University of Sheffield; 2003. Structural characterisation of PrrA from Rhodobacter sphaeroides. [Google Scholar]
- 47.Roh JH, Smith WE, Kaplan S. Effects of oxygen and light intensity on transcriptome expression in Rhodobacter sphaeroides 2.4.1: Redox active gene expression profile. J. Biol. Chem. 2004;279:9146–9155. doi: 10.1074/jbc.M311608200. [DOI] [PubMed] [Google Scholar]