

Abstract
We have investigated the reaction of glutamate mutase with the glutamate analog, 2-thiolglutarate. In the standard assay, 2-thiolglutarate behaves as a competitive inhibitor with Ki = 0.05 mM. However, rather than simply binding inertly at the active site, 2-thiolglutarate elicits cobalt-carbon bond homolysis and the formation of 5′-deoxyadenosine. The enzyme exhibits a complicated EPR spectrum in the presence of 2-thiolglutarate that is markedly different from any previously observed with the enzyme. The spectrum was well simulated by assuming that it arises from electron-electron spin coupling between a thioglycolyl radical and low-spin Co2+ in cob(II)alamin. Analysis of the zero-field splitting parameters obtained from the simulations places the organic radical at ∼ 10 Å from the cobalt, and at a tilt angle of ∼ 70° to the normal of the corrin ring. This orientation is in good agreement with that expected from the crystal structure of glutamate mutase complexed with substrate. 2-thiolglutarate appears to react in a manner analogous to glutamate by first forming a thioglutaryl radical at C-4 that then undergoes fragmentation to produce acrylate and the sulfur-stablized thioglycolyl radical. The thioglycolyl radical accumulates on the enzyme suggesting it is too stable to undergo further steps in the mechanism at a detectable rate.
Keywords: enzyme, coenzyme-B12, free radicals, isomerization, substrate analog, EPR spectrometry
Adenosylcobalamin1 (Coenzyme B12, AdoCbl) is the coenzyme for a group of enzymes that catalyze unusual rearrangement or elimination reactions, as well as for class II ribonucleotide reductases. In these enzymes, the coenzyme serves as a masked form of the 5′-deoxyadenosyl radical that is generated through homolytic fission of the AdoCbl cobalt-carbon bond (1-6). An interesting and still poorly understood aspect of these reactions is how these enzymes catalyze homolysis of the coenzyme because the generation of free radicals from stable, closed shell molecules is energetically highly unfavorable.
In all cases, homolysis of AdoCbl is tightly coupled to the formation of substrate-based radicals (or in the case of ribonucleotide reductase, a protein-based thiyl radical). The initially formed 5′-deoxyadenosyl radical is extremely unstable and has never been observed spectroscopically. Where they have been characterized, the organic radical that accumulates in the enzyme active site is situated on the substrate (or protein) and is stabilized by delocalization or inductive effects due to the structure of the substrate molecule (7-11).
AdoCbl-dependent glutamate mutase catalyzes the unusual carbon skeleton isomerization of L-glutamate to L-threo-3-methylaspartate, which is the initial step in the fermentation of glutamate by various Clostridial species. This enzyme has proven a useful system to investigate the principles by which enzymes generate reactive radical intermediates and harness them for catalysis of chemically difficult reactions (12, 13). Both wild-type and mutant enzymes have been studied using a variety of spectroscopic and kinetic methods to gain mechanistic insights into this reaction (14-22).
To better understand how the structure of the substrate influences the energetics of radical formation, we have investigated the reactions of glutamate mutase with several glutamate analogs that are summarized in Figure 1. Although the enzyme is highly specific for L-glutamate and L-threo-3-methylaspartate, we have found that the enzyme will bind molecules that are conservatively substituted at the α-carbon of glutamate. Thus, we have demonstrated that L-2-hydroxyglutarate is a slow substrate for the enzyme (23). On the other hand, 2-ketoglutarate does not appear to undergo rearrangement, but reacts with the coenzyme to induce tritium exchange between the 5′-carbon of AdoCbl and carbon-4 of 2-ketoglutarate, thereby demonstrating this analog's competence to form radicals reversibly (24). Therefore, this analog might be characterized as a “partial” substrate. 2-methyleneglutarate, which is a substrate for the related B12 enzyme 2-methyleneglutarate mutase, reacts quite differently with glutamate mutase. 2-Methyleneglutarate causes homolysis of the coenzyme, but the adenosyl radical is intercepted by the exomethylene group to generate an adduct between adenosine and methyleneglutarate in which a stable tertiary radical is formed (19). This radical addition reaction represents a significant departure from all other known reactions of AdoCbl, which involve hydrogen abstraction.
Figure 1.

Effects of changing the functionality at C-2 of the substrate on the reaction catalyzed by glutamate mutase.
Having investigated the effect of substituting the glutamate nitrogen with oxygen and carbon functionalities, we were interested to examine the reactivity of the enzyme with a sulfur-containing analog, 2-thiolglutarate. We anticipated that this compound might react with the enzyme to generate radical intermediates, which would be stabilized by the sulfur and amenable to spectroscopic characterization. Here we present experiments that demonstrate that 2-thiolglutarate does indeed react with glutamate mutase to generate Cbl(II) and an organic radical, which is probably the thioglycolate radical that arises by fragmentation of the initially formed 2-thiolglutaryl radical.
EXPERIMENTAL PROCEDURES
Materials
The purification from recombinant E. coli of the engineered single subunit glutamate mutase protein, GlmES, which has been used in this and previous mechanistic studies, has been described previously (25). R,S-2-thioglutaric acid was synthesized from glutaric acid using a literature procedure (26). AdoCbl was purchased from Sigma Chemical Company; solutions containing AdoCbl were kept in light-proofed vials and handled in dim light. Tritiated glutamic acid was purchased from Amersham.
Kinetics of Enzyme inhibition by 2-thiolglutarate
Glutamate mutase activity was assayed using the coupled spectrophotometric assay and following the increase in absorption at 240 nm (27, 28). A typical assay solution contained 0.38 nM GlmES, 25 μM AdoCbl, 2 units of β-methylaspartase, 10 mM KCl and 1 mM MgCl2 in 50 mM potassium phosphate, pH 7.0, in a total volume of 1 mL. Reactions were initiated by addition of L-glutamate. The initial rates of reaction were measured in the presence of concentrations of D,L-2-thiolglutarate ranging between 0.0 and 1.0 mM, and L-glutamate concentrations ranging between 0.5 and 10 mM. Plots of kinetic data were generated and curve fitting performed using the KaleidaGraph™ program (Abelbeck Software).
UV-visible spectra of holo-glutamate mutase
1 mL of a 50 μM solution of GlmES in 50mM potassium phosphate, pH 7.0, was introduced into an anaerobic cuvette and made anaerobic by repeated evacuation and flushing with argon gas. Stock solutions of AdoCbl and 2-thiolglutarate, pH 7.0, were prepared separately and purged with Argon gas for at least 30 minutes. The holoenzyme was reconstituted by addition of the stock AdoCbl solution to the cuvette to give a final concentration of 35 μM holoenzyme. After incubation for several minutes the spectrum of the holoenzyme was recorded. D,L-2-thiolglutarate, final concentration 2 − 8 mM, was then added via an airtight syringe to initiate the reaction, and spectra were recorded.
EPR spectroscopy
Samples were prepared in an anaerobic glove box at 4 °C, in dim light. 0.26 mL of 0.35 mM GlmES in 250 mM potassium phosphate buffer, pH 7.0, and 12μL of a 1 M solution of DTT were added to an argon-purged EPR tube containing 1 mg of solid 2-thioglutaric acid. The thioglutaric acid was dissolved by gentle pipetting, and the solution was incubated for about 1 minute. 30μL of a 10 mM solution of AdoCbl was added to initiate the reaction and the solution was gently mixed by pipetting. The sample was frozen in dry ice in the glove box and then removed and stored, protected from light, in liquid nitrogen. The final concentrations of GlmES, thioglutaric acid and AdoCbl were 0. 30mM, 20 mM and 1.0 mM respectively. A sample containing 0.30 mM GlmES, 1 mM AdoCbl and 20 mM L-glutamate was prepared in a similar manner, except that the reaction was initiated by addition of glutamate last.
EPR spectra were recorded in the dark using a Bruker EMX EPR spectrometer equipped with a liquid nitrogen Dewar system. The conditions for obtaining spectra were as follows: temperature, 115 K; microwave power, 20mW; microwave frequency, 9.2GHz; modulation frequency, 100kHz; modulation amplitude, 3 G. The data were analyzed using the Bruker Win-EPR data manipulation program.
Spectral Simulations
The EPR spectrum obtained from a sample of glutamate mutase incubated with 2-thiolglutarate was analyzed using the following spin Hamiltonian:
| (1) |
The first two terms in equation 1 represent the Zeeman interaction of the low-spin Co2+ and the thioglycolyl radical with the external magnetic field, respectively. The third and fourth terms are the isotropic exchange interaction and the electron spin-electron spin dipolar (ZFS) interaction, respectively. The last term represents the nuclear hyperfine interaction of low-spin Co2+ with the 59Co nuclear spin (I = 7/2). All other nuclear spins contribute to the linewidth of the EPR spectrum of the Cbl(II) - thioglycolyl radical pair. In general, the principal axes of each tensor in equation 1 need not be collinear. Euler rotations (29) were therefore used to express each tensor in a common reference frame, in this case the g-axis system of Co2+. The strategy used to diagonalize the energy matrix and simulate the field-swept powder EPR spectrum was described previously (30). In this analysis, the 59Co-hyperfine interaction is treated to first-order, effectively rendering the energy matrix block-diagonal in mI. Initial estimates for the g-tensor of the thioglycolyl radical were obtained from electronic structure calculations. The geometry of the thioglycolyl radical was optimized using Becke-style 3-Parameter Density Functional Theory (DFT) with the Lee-Yang-Parr correlation functional (B3LYP) and Pople's polarized double-ζ 6−31G* basis set with the Gaussian98 software package (31). The g-tensor was then obtained from a single-point calculation on the optimized structure using the B3LYP hybrid functional in combination with the DFT-optimized valence triple-ζ basis, TZVP, with the program ORCA (F. Neese, Max-Planck-Institut fuer Bioanorganische Chemie, Muelheim an der Ruhr, Germany). Estimates for the ZFS parameters and Euler angles were subsequently acquired by modeling the geometry optimized structure of the thioglycolyl radical into the active-site of glutamate mutase using the coordinates obtained from the crystal structure of the substrate bound complex (Protein Data Base entry 1I9C) (32). These parameters were then refined by trial and error until a reasonable fit was obtained.
Analysis of deuterium content of 5′-dA
Stock solutions of GlmES, AdoCbl and 2-thioglutaric acid were made up in 100mM potassium phosphate D2O buffer that had been repeatedly lyophilized from D2O to remove protium. A solution containing 50 μM GlmES, 10 mM (S)-2-thioglutaric acid and 5 mM DTT in 100 mM potassium phosphate D2O buffer (pD 7.0) was made anaerobic by repeated evacuation and flushing with argon gas for 30min. A concentrated (10 mM), anaerobic solution of AdoCbl in 100 mM potassium phosphate D2O buffer was added to the enzyme solution to give a final concentration of 100 μM AdoCbl, and the mixture was allowed to incubate in the dark for 5min under an argon atmosphere. 5′-dA was recovered from the reaction by HPLC using a 25 cm C18 reverse phase column (Spherisorb S5 ODS2). The column was pre-equilibrated in 5 % acetonitrile and 0.1 % TFA; and eluted with 16mL linear gradient of 0 − 40 % solvent B (95 % acetonitrile, 0.1 % TFA); 5′-dA eluted at ∼20 % acetonitrile.
Tritium exchange experiments
5′-tritium-labeled AdoCbl (specific activity ∼5,000 dpm/nmol) was prepared enzymatically through the exchange of tritium from tritium-labeled glutamate as described previously (33). In a septum-sealed vial solution containing 50 μM glutamate mutase, 10 mM (S)-thioglutaric acid and 5 mM DTT in 150 mM potassium phosphate buffer, pH 7.0 was made anaerobic by repeated evacuation and purging with argon. Tritium-labeled AdoCbl, 26 μM final concentration, was introduced into the mixture using an airtight syringe and the solution was incubated in the dark for 30 min at room temperature under argon gas. AdoCbl and 5′-dA were recovered by reverse phase HPLC, as described above, and their tritium contents were determined by scintillation counting. Control reactions were performed in which either glutamate was substituted for 2-thiolglutarate, or the enzyme was omitted.
Mass spectroscopy
Samples collected after HPLC were concentrated to dryness by freeze-drying and re-dissolved in 15 μL of a solution of water/acetonitrile/formic acid, in a ratio of 1:1:0.003. The samples were analyzed by time-of-flight electrospray mass spectrometry (LCT Micromass). The samples were introduced into the spectrometer through an in-line HPLC pump, in a carrier solvent of 90 % methanol, 10 % water at 0.1 mL/min. The desolvation temperature was 150 °C. The spectral data were analyzed using the MassLynx 4.0 software suite.
RESULTS
Inhibition of glutamate mutase activity by 2-thiolglutarate
To determine whether 2-thiolglutarate was capable of binding at the enzyme active site, we first examined whether this substrate analog was a competitive inhibitor of the enzyme. In these and all other experiments 2-thiolglutarate was used as the racemic mixture. Glutamate mutase is specific for the S-enantiomer of its substrates, and the R-enantiomer does not bind to the enzyme. Therefore the presence of the R-enantiomer of 2-thiolglutarate should not interfere, and the concentrations of 2-thiolglutarate referred to here are those of the S-enantiomer only. Initial velocity measurements of glutamate mutase activity were performed at a fixed concentration of AdoCbl (25 μM) in the presence of various substrate concentrations and 2-thiolglutarate concentrations. Plots of 1/[S] versus 1/v (Figure 2) show a clear pattern of intersecting lines, which is diagnostic for competitive inhibition and consistent with the inhibitor binding at the same site as the substrate. Computer fitting of the data yielded values for Ki = 0.055 ± 0.004 mM for (S)-2-thiolglutarate and Km = 0.54 ± 0.05 mM for glutamate.
Figure 2.

Competitive inhibition of glutamate mutase activity by (S)-2-thiolglutarate. Double reciprocal plots of initial velocity versus glutamate concentration at various concentrations of 2-thiolglutarate: 0.0 mM (○), 0.05 mM (•), 0.125 mM (□), 0.25 mM (■), 0.5 mM (△). Inset: a linear plot of the same data.
Homolysis of AdoCbl by 2-thiolglutarate
Our kinetic data indicated that 2-thiolglutarate was binding in the active site of the enzyme. Therefore, we next used UV-visible spectroscopy to examine whether 2-thiolglutarate binding to the holo-enzyme would initiate homolysis of AdoCbl and the formation of Cbl(II). Addition of 1 mM (S)-2-thiolglutarate to holo-glutamate mutase (35 μM) resulted in significant changes to the UV-visible spectrum of the holo-enzyme. In particular, a decrease of absorbance at 530 nm and an increase at 470 nm, indicative of formation of Cbl(II) on the enzyme (Figure 3), were evident. The spectral changes were almost identical to those that occurred when the holo-enzyme was incubated with 10 mM L-glutamate, and were complete within the mixing time of the experiment, ∼ 20s. Assuming Δε530 = 4000 M−1 cm−1 for the homolysis of AdoCbl, the fraction of enzyme with Cbl(II) bound after 30 s incubation with 2-thiolglutarate was 34 %, whereas with L-glutamate it was 37 %. Upon longer incubation times (up to 30 min), with either 2-thiolglutarate or L-glutamate, the slow conversion of the Cbl(II) species to hydroxocobalamin was observed, presumably due to adventitious oxidation of Cbl(II) despite the precautions make to the reaction anaerobic.
Figure 3.

UV-visible spectral changes associated with the binding of 2-thiolglutarate to holo-glutamate mutase. A: Spectrum of holoenzyme (35 μM) in resting state. B: Spectrum of holoenzyme recorded 20 s after adding 2-thiolglutarate, (1 mM final concentration).
In a separate experiment, set up under similar conditions, we also confirmed that 5′-dA was produced in the reaction of 2-thiolglutarate with the holoenzyme. The reaction products were analyzed by reverse-phase HPLC. The peak corresponding to 5′-dA was collected and analyzed by electrospray mass spectrometry to confirm its identity.
Is 2-thiolglutarate a substrate for glutamate mutase?
Given that 2-thiolglutarate appeared to initiate coenzyme cleavage, we decided to investigate whether the enzyme might catalyze the rearrangement of 2-thiolglutarate to the methylaspartate analogue, 2-thiol-3-methylsuccinate. A sample was prepared in D2O buffered with 50mM potassium phosphate, pD 7.0, containing 36 μM glutamate mutase, 30 μM AdoCbl and 20 mM (S)-2-thiolglutarate. The solution was incubated at 25 °C for 30 min in the dark. For comparison, a control sample that omitted AdoCbl was also prepared. The sample was transferred to an NMR tube and the proton NMR spectrum recorded at 400 MHz. The spectrum contained only peaks attributable to 2-thiolglutarate or AdoCbl (data not shown); in particular, there was no evidence for a characteristic doublet at ∼ 0.6 ppm that would correspond to the methyl hydrogens of 2-thio-3-methylsuccinate. However, we cannot exclude the possibility of a very small extent of conversion occurred or that the equilibrium constant for the formation of 2-thiol-3-methylsuccinate is sufficiently unfavorable that it would not accumulate to sufficiently high concentrations to be observed.
Origin of hydrogen in 5′-dA
If 2-thiolglutarate behaved in an exactly analogous fashion to glutamate one would expect a hydrogen atom to be abstracted from the C-4 carbon. However, if the substrate were bound in a slightly different orientation in the active site, it might be feasible for hydrogen to be removed from the thiol group. This reaction has precedent in the generation of a thiyl radical by class II (AdoCbl-dependent) ribonucleotide reductase. We have also observed an unexpected reaction when 2-methyleneglutarate was incubated with the enzyme in which the adenosyl radical undergoes addition to the exo-methylene double bond to give a radical adduct between the coenzyme and substrate analog. This result, again, made us question more closely how 2-thiolglutarate might be reacting.
To determine the origin of the hydrogen transferred to 5′-dA, we conducted the reaction in D2O buffered with potassium phosphate (pD = 7.0). A solution containing 50 μM glutamate mutase, 0.10 mM AdoCbl, 10 mM (S) 2-thiolglutarate and 5 mM DTT was incubated at room temperature in the dark for 5 min. 5′-dA was recovered from the reaction mixture by HPLC and analyzed by electrospray mass spectrometry. If hydrogen abstraction was occurring from C-4 of 2-thiolglutarate, then 5′-dA should contain only protons in the 5′-methyl group resulting in a peak at [m+1] = 252 for 5′-dA. On the other hand, if abstraction occurred from the thiol group, then deuterium should be incorporated into 5′-dA resulting in a peak at [m+1] = 253. The mass spectrum showed no evidence that deuterium was incorporated into 5′-dA. The major peak for 5′-dA was at [m+1] = 252. The peak at [m+1] = 253 was about 13 % of the intensity of the major peak, which is slightly higher than expected for the 13C-content of 5′-dA but probably within the limits of experimental error.
Tritium exchange between AdoCbl and 2-thiolglutarate
To determine whether hydrogen transfer between AdoCbl and 2-thiolglutarate was reversible, we examined the ability of the enzyme to catalyze the transfer of tritium at the 5′-carbon of AdoCbl to 2-thiolglutarate. We have previously examined the transfer of tritium from AdoCbl to glutamate and methylaspartate and shown that it is essentially complete after 15 s (kapp = 0.67 ± 0.05 s−1) (33). Even after prolonged incubation (30 min, after which time the enzyme was essentially inactive0 of 2-thiolglutarate with holo-enzyme reconstituted with tritiated AdoCbl, about 95 % of the radioactivity was accounted for in the peaks corresponding to 5’-dA and AdoCbl. A small number of counts, ∼ 5 % of the total, were found in the flow-through fraction from the HPLC column containing 2-thiolglutarate. However, a control experiment in which the enzyme was omitted resulted in a similar number of counts being present in the flow-through fraction; otherwise radioactivity was only associated with AdoCbl. Thus, we estimate that less than 2 % of the tritium was transferred to 2-thiolglutarate under the conditions of the experiment. A further control in which enzyme, tritiated AdoCbl and L-glutamate were incubated together under similar conditions confirmed that almost all the tritium was, as expected, lost from the coenzyme. Therefore, we conclude that hydrogen transfer from AdoCbl to 2-thiolglutarate occurs either very slowly or not at all.
EPR spectroscopy
To better characterize the radical species generated by the reaction of 2-thiolglutarate with the enzyme, we recorded EPR spectra of glutamate mutase in the presence of 2-thiolglutarate. For comparison, a sample of the enzyme reacted with L-glutamate was prepared in parallel, and its EPR spectrum recorded under the same conditions. The EPR spectrum of the enzyme-glutamate complex is identical to that which has been described and analyzed previously (7). The spectra are shown in Figure 4.
Figure 4.
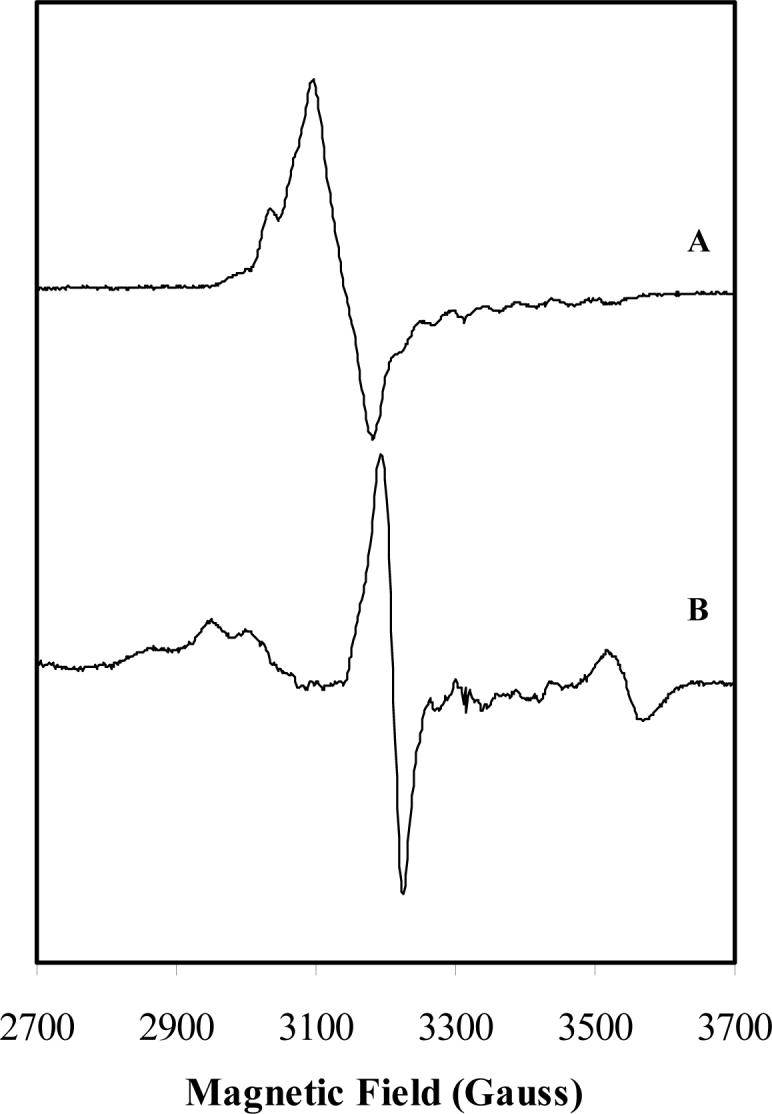
EPR spectra of glutamate mutase. Spectrum A: Enzyme reacted with 10 mM L-glutamate, which is characteristic of Cbl(II) strongly coupled to an organic radical. Spectrum B: Enzyme reacted with 10 mM (S)-2-thiolglutarate, which shows features characteristic of Cbl(II) coupled to a sulfur containing organic radical.
The overall appearance of the EPR spectrum of the enzyme reacted with 2-thiolglutarate suggests the presence of a cob(II)alamin species interacting with an organic radical. The octet hyperfine splitting feature arising from the cobalt nuclear spin of 7/2, which is clearly seen in the enzyme-glutamate complex, appears also to be present in the spectrum. Clearly evident are additional features in the lower magnetic field region (2600−3100 G) and one at higher field strength with g = 1.85, which do not occur in the spectrum of the enzyme reacted with glutamate.
To examine whether this very complicated EPR spectrum arises from a single species or is the result of a mixture of species, a power saturation study was undertaken. A series of spectra recorded over a wide range of microwave powers indicated the presence of a single species with identical power dependence (spectra not shown). (The sharp feature at g = 1.98 showed different power saturation dependence, confirming it as a contaminating species.)
Simulation of the EPR spectrum
The unusual appearance of the spectrum lead us to hypothesize that the organic radical component might be strongly influenced by the sulfur atom, and might plausibly be the thioglycolyl radical derived from the fragmentation of thiolglutaryl radical. We undertook simulation of the spectrum, assuming a spin-coupled interaction between low-spin Co2+ in cob(II)alamin and a thioglycolyl radical. As shown in Figure 5, the features of this complex spectrum are reproduced remarkably well by the simulation. The simulation parameters that best reproduce the experimental spectrum are given in Table 1. Because the pKa of the sulfur atom is close to that of the pH at which the spectrum was recorded, the spectrum was simulated assuming either that the sulfur atom was protonated or deprotonated. Although parameters vary slightly for the two cases (Table 1), the simulations of the spectrum are equally good. Therefore, we could not conclude what the protonation state of sulfur was from the spectrum. Attempts to simulate the spectrum assuming a thiyl radical did not provide a good fit.
Figure 5.

Comparison of the simulated and experimental EPR spectra for glutamate mutase reacted with 10 mM (S)-2-thiolglutarate. The simulation assumes the sulfur is protonated; a very similar fit is obtained if the sulfur is assumed to be deprotonated. The parameters used to simulate the spectrum are given in Table 1.
Table 1.
Parameters used in the simulation of the EPR spectrum of glutamate mutase reacted with 2-thiolglutarate.
| Parameter | Values (sulfur protonated) | Values (sulfur deprotonated) |
|---|---|---|
| Co2+ g-tensor values, g(Co2+) | 2.33, 2.23, 2.01 | 2.32, 2.23, 2.00 |
| Radical g-tensor values, g(radical) | 2.009, 2.004, 2.002 | 2.016, 2.010, 2.002 |
| Exchange coupling constant, J | 345 G | 350 G |
| Zero-field splitting (ZFS) parameters | D = −27 G, E = 8 G | D = −27 G, E = 7 G |
| 59Co hyperfine tensor, (A) | 8 G, 5 G, 107 G | 7 G, 12 G, 107 G |
| Euler angles between g(Co2+) and g(radical) | 65, 50, −90 | 80, 80, 180 |
| Euler angles between g(Co2+) and ZFS-tensor | 70, 45, −85 | 60, 75, −70 |
From the zero-field splitting parameters and the Euler angles aligning the zero-field splitting tensor with the Co2+ g-tensor, the cobalt atom and the thioglycolyl radical are calculated to be ∼ 10 Å apart, with the thioglycolate radical lying at an angle of ∼ 70° (sulfur protonated) or ∼ 60° (sulfur deprotonated) from the normal to the plane of the corrin ring or. Inspection of the crystal structure of the enzyme-substrate complex (32, 34) suggests that these geometrical constraints for the Co2+ - thioglycolyl radical pair are quite consistent with the enzyme active site.
DISCUSSION
From the results presented above, we may summarize the reaction of 2-thiolglutarate with glutamate mutase as follows (Figure 6). 2-thiolglutarate binds to the enzyme active site and triggers homolysis of AdoCbl. The adenosyl radical then reacts to abstract hydrogen from C-4 of 2-thiolglutarate, to generate a radical at that position. The thioglutaryl radical next undergoes fragmentation to generate acrylate and a thioglycolyl radical. These steps mimic the reaction with the natural substrate, glutamate. However, abstraction of hydrogen from 2-thiolglutarate appears to be irreversible, or nearly so, as judged by the low rate of tritium exchange with the coenzyme. This is most likely because upon forming the thioglutaryl radical, fragmentation occurs immediately to form the thioglycolyl radical and acrylate, which appears to be the most stable radical complex. This intermediate presumably accumulates because the thioglycolyl radical is stabilized due to delocalization of the unpaired electron on sulfur.
Figure 6.

Proposed mechanism for the reaction of 2-thiolglutarate with glutamate mutase, resulting in the formation of Cbl(II), 5′-dA and the thioglycolyl radical.
The selective stabilization of radical intermediates by sulfur has been used to probe the mechanism of the radical-SAM-dependent enzyme, lysine-2,3-aminomutase. Here the sulfur-containing lysine analog 4-thia-lysine was used to stabilize the radical initially formed by abstraction of hydrogen at C-3 of lysine, allowing it to be observed by EPR (35). In this case 4-thia-lysine is still turned over by the enzyme, albeit at only about 1 % of the rate of lysine. We were unable to detect the formation of 2-thio-3-methylsuccinate, the expected product of the rearrangement of 2-thiolglutarate. However, the equilibrium constant most likely disfavors the rearrangement of 2-thiolglutarate to 2-thio-3-methylsuccinate, and the product may be prone to decompositions. Therefore we cannot rule out that 2-thiolglutarate is turned over by the enzyme very slowly.
We have previously recorded EPR spectra of glutamate mutase reacted with the slow substrate hydroxyglutarate (23), and with 2-methyleneglutarate, which is a mechanism-based inactivator (19). These spectra are very similar to the Co2+ : C4 glutamyl radical triplet spectrum obtained with glutamate. However, the EPR spectrum of the enzyme : 2-thiolglutarate complex is very different from any that have been described for glutamate mutase previously, suggesting that the organic radical component is not an analog of the glutamyl radical. The fact that the complex spectral features are well simulated by assuming a spin-coupled interaction between a thioglycolyl radical with low spin Co2+ provide strong support for the formation of the thioglycolyl radical in the enzyme active site.
The EPR spectrum of the enzyme reacted with glutamate has been well-characterized and simulated by assuming the exchange-coupled interaction between the C-4 glutamyl radical and Co2+ in Cbl(II) (7, 36). From this simulation, it was concluded that the two unpaired electrons were 6.6 ± 0.9 Å apart, a distance latter found to be in good agreement with that expected from analysis of the crystal structure of the enzyme:substrate complex (32). Assuming that 2-thiolglutarate binds to the enzyme in the same orientation as glutamate, fragmentation of the initially formed 2-thioglutaryl radical to produce the thioglycolyl radical and acrylate would move the radical ∼ 3 Å further from the cobalt atom. Therefore, a ∼ 10 Å separation between the cobalt and thioglycolyl radical that we derive from the simulation appears quite reasonable.
Most of our experimental data are well rationalized by the mechanistic scheme laid out in Figure 6. However, one aspect of the reaction of 2-thiolglutarate with the enzyme remains unclear. If the formation of the thioglycolyl radical is sufficiently favorable for it to effectively be irreversible, as the lack of tritium exchange suggests, then one might expect that all of the enzyme active sites would be converted to this radical form. However, it appears that only ∼ 35 % of the enzyme active sites undergo homolysis upon reaction with 2-thiolglutarate, a proportion similar to that observed with glutamate. Indeed, none of the substrate analogues we have investigated with the enzyme seem to elicit more than 50 % cleavage of the coenzyme.
It is possible that the lack of tritium exchange observed is simply a consequence of very slow exchange kinetics coupled with a large isotope effect discriminating against the transfer of tritium to 2-thiolglutarate. Another possibility is that ‘half-of-sites’ or negative cooperativity between the two active sites of the glutamate mutase dimer may contribute to the lower than expected extent of reaction by effectively halving the active sites available for reaction. In which case ∼ 70 % of the reactive sites would contain cob(II)alamin. It is, of course, also possible that a fraction of the protein is inactive, although previous binding measurements indicate a 1:1 stoichiometry for AdoCbl binding to the enzyme (25) and the protein used in these studies possessed normal levels of activity.
In conclusion, although glutamate mutase is very selective of its substrates, we have found that the enzyme tolerates conservative substitutions at the amino group of glutamate, and this has allowed us to examine the effects of modulating the electronic structure of the substrate on catalysis. The experiments presented here, together with our previous studies, highlight the importance of the structure of the substrate in optimizing the stability of radical intermediates for efficient catalysis. The functional group at C-2 of the substrate plays a critical role in the fragmentation-recombination pathway by which the carbon skeleton undergoes rearrangement by stabilizing the transiently formed radical on C-2. When the substrate is glutamate, the amino group stabilizes the intermediate glycyl radical such that it is only 1 − 2 kcal/mol higher in energy than the initially formed glutamyl radical (21). If the amino group is substituted by a hydroxyl group, the energy of the intermediate glycolyl radical is higher, and catalysis is expected to be slower, as we have shown to be the case for the rearrangement of 2-hydroxyglutarate (23). In contrast, with a thiol group at C-2 it appears that the thioglycolyl radical accumulates in the active site, suggesting that this radical is too stable for the reaction to proceed onward towards products or back to substrate at a significant rate.
ACKNOWLEDGEMENT
We thank Christel Fox for help with enzyme preparation and Prof. Leo Radom for insightful comments and discussions during the preparation of this manuscript.
Footnotes
This research was supported by grants from the National Institutes of Health, GM 59227 to E.N.G.M. and GM 35752 to G.H.R.
The abbreviations used are: AdoCbl, adenosylcobalamin; Cbl(II), cob(II)alamin; 5′-dA, 5′-deoxyadenosine
REFERENCES
- 1.Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem. Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R, Ragsdale SW. The many faces of vitamin B12: Catalysis by cobalamin-dependent enzymes. Ann. Rev. Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee R. Radical carbon skeleton rearrangements: Catalysis by coenzyme B12-dependent mutases. Chem. Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 4.Marsh ENG, Drennan CL. Adenosylcobalamin-dependent isomerases: new insights into structure and mechanism. Curr. Opin. Chem. Biol. 2001;5:499–505. doi: 10.1016/s1367-5931(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee R. Radical Peregrinations Catalyzed by Coenzyme B12-Dependent Enzymes. Biochemistry. 2001;40:6191–96198. doi: 10.1021/bi0104423. [DOI] [PubMed] [Google Scholar]
- 6.Frey PA. Radical mechanisms of enzymatic catalysis. Ann. Rev. Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 7.Bothe H, Darley DJ, Albracht SP, Gerfen GJ, Golding BT, Buckel W. Identification of the 4-glutamyl radical as an intermediate in the carbon skeleton rearrangement catalyzed by coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. Biochemistry. 1998;37:4105–13. doi: 10.1021/bi971393q. [DOI] [PubMed] [Google Scholar]
- 8.Licht SS, Booker S, Stubbe JA. Studies on the catalysis of carbon-cobalt bond homolysis by ribonucleoside triphosphate reductase: Evidence for concerted carbon-cobalt bond homolysis and thiyl radical formation. Biochemistry. 1999;38:1221–1233. doi: 10.1021/bi981885i. [DOI] [PubMed] [Google Scholar]
- 9.Bandarian V, Reed GH. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonia-lyase: Determination of the number of exchangeable hydrogens in the enzyme-cofactor complex. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- 10.Mansoorabadi SO, Padmakumar R, Fazliddinova N, Vlasie M, Banerjee R, Reed GH. Characterization of a succinyl-CoA radical-cob(II)alamin spin triplet intermediate in the reaction catalyzed by adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry. 2005;44:3153–3158. doi: 10.1021/bi0482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warncke K. Characterization of the product radical structure in the Co-II-product radical pair state of coenzyme B12-dependent ethanolamine deaminase by using three-pulse H-2 ESEEM spectroscopy. Biochemistry. 2005;44:3184–3193. doi: 10.1021/bi048196t. [DOI] [PubMed] [Google Scholar]
- 12.Marsh ENG. Coenzyme B12-dependent glutamate mutase. Bioorg. Chem. 2000;28:176–189. doi: 10.1006/bioo.2000.1168. [DOI] [PubMed] [Google Scholar]
- 13.Gruber K, Kratky C. Coenzyme B12 dependent glutamate mutase. Curr. Opin. Chem. Biol. 2002;6:598–603. doi: 10.1016/s1367-5931(02)00368-x. [DOI] [PubMed] [Google Scholar]
- 14.Chih H-W, Roymoulik I, Huhta MS, Madhavapeddi P, Marsh ENG. Adenosylcobalamin-dependent glutamate mutase: pre-steady-state kinetic methods for investigating the reaction mechanism. Methods Enzymol. 2002;354:380–399. doi: 10.1016/s0076-6879(02)54030-1. [DOI] [PubMed] [Google Scholar]
- 15.Sension RS, Cole AG, Harris AD, Fox CC, Woodbury N, Lin S, Marsh ENG. Photolysis and Recombination of Adenosylcobalamin Bound to Glutamate Mutase. J. Am. Chem. Soc. 2004;126:1598–1599. doi: 10.1021/ja0396910. [DOI] [PubMed] [Google Scholar]
- 16.Madhavapeddi P, Ballou DP, Marsh ENG. Pre-Steady State Kinetic studies on the Gly171Gln Active Site Mutant of Adenosylcobalamin-dependent Glutamate Mutase. Biochemistry. 2002;41:15802–15809. doi: 10.1021/bi020596y. [DOI] [PubMed] [Google Scholar]
- 17.Xia l., Ballou DP, Marsh ENG. The role of Arg100 in the active site of adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2004;43:3238–3245. doi: 10.1021/bi0357558. [DOI] [PubMed] [Google Scholar]
- 18.Cheng M-C, Marsh ENG. Pre-steady state measurement of intrinsic secondary tritium isotope effects associated with the homolysis of adenosylcobalamin and the formation of 5'-deoxyadensosine in glutamate mutase. Biochemistry. 2004;43:2155–2158. doi: 10.1021/bi036122w. [DOI] [PubMed] [Google Scholar]
- 19.Huhta MS, Ciceri D, Golding BT, Marsh ENG. A novel reaction between adenosylcobalamin and 2-methyleneglutarate catalyzed by glutamate mutase. Biochemistry. 2002;41:3200–3206. doi: 10.1021/bi011965d. [DOI] [PubMed] [Google Scholar]
- 20.Chih H-W, Marsh ENG. Tritium partitioning and isotope effects in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2001;40:13060–13067. doi: 10.1021/bi011298o. [DOI] [PubMed] [Google Scholar]
- 21.Chih H-W, Marsh ENG. Mechanism of glutamate mutase: identification and kinetic competence of acrylate and glycyl radical as intermediates in the rearrangment of glutamate to methylaspartate. J. Am. Chem. Soc. 2000;122:10732–10733. [Google Scholar]
- 22.Marsh ENG, Ballou DP. Coupling of cobalt-carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 1998;37:11864–72. doi: 10.1021/bi980512e. [DOI] [PubMed] [Google Scholar]
- 23.Roymoulik I, Moon N, Dunham WR, Ballou DP, Marsh ENG. Rearrangement of L-2-hydroxyglutarate to L-threo-3-methylmalate catalyzed by adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2000;39:10340–10346. doi: 10.1021/bi000121b. [DOI] [PubMed] [Google Scholar]
- 24.Roymoulik I, Chen H-P, Marsh ENG. The reaction of the substrate analog 2-ketoglutarate with adenosylcobalmin-dependent glutamate mutase. J. Biol. Chem. 1999;274:11619–11622. doi: 10.1074/jbc.274.17.11619. [DOI] [PubMed] [Google Scholar]
- 25.Chen HP, Marsh ENG. Adenosylcobalamin-dependent glutamate mutase: examination of substrate and coenzyme binding in an engineered fusion protein possessing simplified subunit structure and kinetic properties. Biochemistry. 1997;36:14939–45. doi: 10.1021/bi971374g. [DOI] [PubMed] [Google Scholar]
- 26.Majer P, Jackson PF, Delahanty G, Grella BS, Ko YS, Li WX, Liu Q, Maclin KM, Polakova J, Shaffer KA, Stoermer D, Vitharana D, Wang EY, Zakrzewski A, Rojas C, Slusher BS, Wozniak KM, Burak E, Limsakun T, Tsukamoto T. Synthesis and biological evaluation of thiol-based inhibitors of glutamate carboxypeptidase II: Discovery of an orally active GCP II inhibitor. J. Med. Chem. 2003;46:1989–1996. doi: 10.1021/jm020515w. [DOI] [PubMed] [Google Scholar]
- 27.Barker HA. beta-Methylaspartate-glutamate mutase from Clostridium tetanomorphum. Methods Enzymol. 1985;113:121–33. doi: 10.1016/s0076-6879(85)13026-0. [DOI] [PubMed] [Google Scholar]
- 28.Holloway DE, Marsh ENG. Adenosylcobalamin-dependent glutamate mutase from Clostridium tetanomorphum. Overexpression in Escherichia coli, purification, and characterization of the recombinant enzyme. J Biol Chem. 1994;269:20425–30. [PubMed] [Google Scholar]
- 29.Goldstein H, Poole CP, Jr., Safko JL. Classical Mechanics. 3rd ed. Addison Wesley; San Francisco: 2002. [Google Scholar]
- 30.Bandarian V, Reed GH. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: Mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry. 1999;38:12394–12402. doi: 10.1021/bi990620g. [DOI] [PubMed] [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery J, J. A., Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Head-Gordon M, Replogle ES, Pople JA. Gaussian. Vol. 98. A.9, Gaussian, Inc.; Pittsburg, PA: 1998. [Google Scholar]
- 32.Gruber K, Reitzer R, Kratky C. Radical shuttling in a protein: Ribose pseudorotation controls alkyl-radical transfer in the coenzyme B-12 dependent enzyme glutamate mutase. Angew. Chem. Intl. Edn. 2001;40:3377–+. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Marsh ENG. Tritium isotope effects in adenosylcobalamin-dependent glutamate mutase: implications for the mechanism. Biochemistry. 1995;34:7542–7. doi: 10.1021/bi00022a030. [DOI] [PubMed] [Google Scholar]
- 34.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W, Kratky C. Structure of coenzyme B12 dependent enzyme glutamate mutase from Clostridium cochlearium. Structure. 1999;7:891–902. doi: 10.1016/s0969-2126(99)80116-6. [DOI] [PubMed] [Google Scholar]
- 35.Wu WM, Lieder KW, Reed GH, Frey PA. Observation of a 2nd Substrate Radical Intermediate in the Reaction of Lysine 2,3-Aminomutase - a Radical Centered On the Beta-Carbon of the Alternative Substrate, 4-Thia-L-Lysine. Biochemistry. 1995;34:10532–10537. doi: 10.1021/bi00033a027. [DOI] [PubMed] [Google Scholar]
- 36.Leutbecher U, Albracht SP, Buckel W. Identification of a paramagnetic species as an early intermediate in the coenzyme B12-dependent glutamate mutase reaction. A cob(II)amide? FEBS Lett. 1992;307:144–6. doi: 10.1016/0014-5793(92)80754-5. [DOI] [PubMed] [Google Scholar]