

Abstract
Voltage-gated potassium channels (VGPCs) play an important role in many physiological functions by controlling the electrical properties and excitability of cells. Changes in tonicity in the peripheral nervous system can activate nociceptors and produce pain. Here, using whole cell patch clamp techniques, we explore how hypo and hypertonicity modulate VGPCs in cultured rat and mouse trigeminal ganglion (TG) neurons. We found that hypo and hypertonicity had different effects on slow-inactivating K+ current (IK) and fast-inactivating K+ current (IA): hypotonicity increased IK but had no effect on IA while hypertonicity depressed both IK and IA. The increase of IK by hypotonicity was mimicked by Transient Receptor Potential Vanilloid 4 (TRPV4) receptor activator (4α-PDD) but hypotonicity did not exhibit increase in TRPV4−/− mice TG neurons, suggesting that TRPV4 receptor was involved in hypotonicity induced response. We also found that inactivation of PKC selectively reversed the increase of IK by hypotonicity, whereas antagonism of G-protein selectively rescued the inhibitions of IK and IA by hypertonicity, indicating that different intracellular signaling pathways were required for the modulation by hypo and hypertonicity. In summary, changes in osmolality have various effects on IK and IA and different receptors and second messenger systems are selective for the modulation of VGPCs induced by hypo versus hypertonicity.
Keywords: voltage-gated potassium channels, hypotonicity, hypertonicity, TRPV4, intracellular signaling pathway, second messenger system
Many studies have shown that osmolarity plays an important role in the regulation of neurons excitability and neurotransmitter release (Andrew et al., 1989; Azouz et al., 1997; Baraban and Schwartzkroin, 1998; Kilb et al., 2006; Saly and Andrew, 1993; Traynelis and Dingledine, 1989). Through this regulation, changes in osmolality can cause many disorders, such as epilepsy (Kilb et al., 2006; Schwartzkroin et al., 1998) in the central nerve system and pain in the peripheral nerve system (Alessandri-Haber et al., 2005; Alessandri-Haber et al., 2004; Alessandri-Haber et al., 2003; Suzuki et al., 2003). Recently, there are evidences that both hypo and hypertonic stimuli can activate C-fiber afferent and induce pain (Alessandri-Haber et al., 2005; Alessandri-Haber et al., 2004; Alessandri-Haber et al., 2003; Suzuki et al., 2003). Besides this, hypo and hypertonicity also facilitate the nociceptive signal transductions induced by pH (Hamamoto et al., 2000), capsaicin (Liu et al., 2007), carrageenan (Alessandri-Haber et al., 2006) and mechanical stimuli (Alessandri-Haber et al., 2004; Alessandri-Haber et al., 2006) and are involved in inflammatory and neuropathic pain (Alessandri-Haber et al., 2006; Alessandri-Haber et al., 2005; Alessandri-Haber et al., 2004). Transient Receptor Potential Vanilloid 4 (TRPV4) is one of the members of Transient Receptor Potential Vanilloid (TRPV) family, which can be activated by hypotonicity, modest heat, mechanical stimulus, synthetic activators like 4α-phorbol-12,13-didecanoate (4α-PDD) and endogenous agonists derived from arachidonic acid and its metabolites (Liedtke and Kim, 2005; Voets et al., 2002; Vriens et al., 2004). Many studies report that TRPV4 receptor is involved in hypo and hypertonicity induced nociception (Alessandri-Haber et al., 2005; Alessandri-Haber et al., 2004; Alessandri-Haber et al., 2003; Suzuki et al., 2003).
Voltage-gated potassium channels (VGPCs) comprise 12 families (named Kv1 to Kv12) and each family consists of several subunits (Gutman et al., 2005; Misonou and Trimmer, 2004). It is reported that more and more α subunits as well as β subunit of VGPCs have been found in rat trigeminal ganglion (TG) (Gutman et al., 2005; Rasband and Trimmer, 2006). VGPCs show significant diversity in their biophysical and pharmacologic properties and there are at least two types of VGPC currents in sensory neurons: slow-inactivating potassium current (IK), displaying slow or sometimes incomplete inactivation and fast-inactivating potassium current (IA) exhibiting fast inactivation kinetics (Catacuzzeno et al., 2003; Gold et al., 1996; Grunewald, 2003; Stewart et al., 2003). VGPCs participate in the action potential (AP) and determine the excitability of cells by regulating the resting membrane potential, the threshold of AP and the frequency of AP firings (Rasband and Trimmer, 2006; Yost, 1999). Studies show that VGPCs are involved in the nociceptive signal transduction induced by inflammatory mediators and nerve injury in primary sensory neurons (Everill and Kocsis, 1999; Harriott et al., 2006; Ishikawa et al., 1999; Rasband et al., 2001; Sculptoreanu et al., 2004; Stewart et al., 2003; Takeda et al., 2006; Xu et al., 2006; Yoshimura and de Groat, 1999) and are considered as important targets in the development of new analgesic (Ocana et al., 2004). In this study we tested whether VGPCs were modulated by hypo and hypertonicity and whether TRPV4 receptor and specific second messenger pathway were involved in these effects.
EXPERIMENTAL PROCEDURES
Cell culture
Trigeminal ganglion (TG) neurons from male Sprague–Dawley rats (180–200 g) were cultured as described previously (Liu et al., 2004). TG neurons from C57BL/6 wild type and TRPV4 knockout (Liedtke and Friedman, 2003) mice were cultured using similar method for adult rats. Briefly, trigeminal ganglions were dissected aseptically and washed with cold (4 °C) modified Hank’s balanced salt solution (mHBSS). The ganglia were diced into small pieces, and then incubated in 3 ml mHBSS with 0.1% collagenase (Type XI-S) for 20–40 min at 37 °C. Individual cells were dissociated by triturating them through a fire-polished glass pipette, followed by a 10 min incubation at 37°C with 10μg/ml DNase I (Type IV) in F-12 medium (Life Technologies, Gaithersburg, MD) and centrifuged for 5 min at 1500 rpm/min. After centrifuging three times, the cells were cultured in F-12 supplemented with 10% fetal bovine serum. The cells were plated on poly-D-lysine coated glass coverslips (15 mm diameter) and cultured 24 h at 37 °C in a water saturated atmosphere with 5% CO2.
Care of animals conformed to standards established by the National Institutes of Health. All animal protocols were approved by the Duke University Institutional Animal Care and Use Committee.
Patch clamp recording
The cells were placed in a recording chamber mounted on the stage of an inverted microscope (Leica Inc. Germany) and perfused continually with extracellular solution at room temperature (21–22 °C) at the rate of 3ml/min. Whole-cell patch recordings were obtained using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA) and the output was digitized with a Digidata 1332A converter (Axon Instruments). Data were acquired at a sampling rate of 10 kHz and filtered at 5 kHz. In the experiments the capacitance was compensated and series resistance was compensated more than 90%. Data obtained from neurons in which uncompensated series resistance resulted in voltage-clamp errors > 5mV were not taken in further analysis. Liquid junction potentials were compensated before patching. When the osmolality of external solutions was changed from isotonicity to hypo or to hypertonicity, measurements of the changes in liquid junction potentials were less than 2 mV and were not corrected. The cell diameter was measured with a calibrated eyepiece under phase contrast illumination. Neurons having projected soma diameters ranging between 15–30 μm were used.
The voltage-dependent activation curve (G–V curve) was measured by a series of depolarizing pulses (500 ms) from −100 mV to +50 mV stepping by 10 mV with interval time of 5 s. The voltage–dependent inactivation curve (inactivation–voltage curve) was measured by double pulses: precondition pulses (2 s for IK and 500 ms for IA) ranging from −120 mV to +50 mV by stepping 10 mV and following +50 mV test pulse (500 ms) with internal time of 5 s. For all experiments the holding potential was −80 mV.
The resistance of the glass pipettes (No. 64-0817(G85150T-3), Warner Instruments Inc., Hamden, CT, USA) was 1–2 MΩ when filled with pipette solution composed of (in mM): K-aspartate 120, KCl 20, CaCl2 1.0, MgCl2 2.0, Tris-ATP 5.0, HEPES 10, EGTA 10 at pH 7.2 and osmolality 300mOsm. The external solution at pH 7.4 was shown in table 1. IK and IA were selectively recorded with 4-AP or TEA-Cl in the extracellular solution. Hypo and hypertonic external solutions were obtained by adjusting the concentration of D-Mannitol. The osmolality was measured using a vapor pressure osmometer (Model 3300, Advanced Instruments, Norwood, MA).
Table 1.
Extracellular solution (pH=7.4)
| VGPCs | mOsm | Choline-Cl | KCl | MgCl2 | HEPES | Mannitol | TEA-Cl | 4-AP | CaCl2 | CdCl2 |
|---|---|---|---|---|---|---|---|---|---|---|
| IK | 220 | 85 | 5.0 | 3.0 | 10 | 25 | – | 3.0 | ||
| 300 | 85 | 5.0 | 3.0 | 10 | 106 | – | 3.0 | |||
| 350 | 85 | 5.0 | 3.0 | 10 | 130 | – | 3.0 | |||
| 220-Ca | 85 | 5.0 | 1.0 | 10 | 25 | – | 3.0 | 2.0 | 1.0 | |
| 300-Ca | 85 | 5.0 | 1.0 | 10 | 106 | – | 3.0 | 2.0 | 1.0 | |
| 350-Ca | 85 | 5.0 | 1.0 | 10 | 130 | – | 3.0 | 2.0 | 1.0 | |
| IA | 220 | 28 | 5.0 | 3.0 | 10 | 25 | 70 | – | ||
| 300 | 28 | 5.0 | 3.0 | 10 | 106 | 70 | – | |||
| 350 | 28 | 5.0 | 3.0 | 10 | 130 | 70 | – | |||
| 220-Ca | 28 | 5.0 | 1.0 | 10 | 25 | 70 | – | 2.0 | 1.0 | |
| 300-Ca | 28 | 5.0 | 1.0 | 10 | 106 | 70 | – | 2.0 | 1.0 | |
| 350-Ca | 28 | 5.0 | 1.0 | 10 | 130 | 70 | – | 2.0 | 1.0 |
Data analysis
IK and IA were measured at the peak outward current. Data were analyzed using pClamp (Axon Instruments, Foster City, CA) and SigmaPlot software (SPSS Inc., Chicago, IL). All of data were presented as mean ± SEM and the significance was indicated as P<0.05 (*) and P<0.01 (**) tested by paired or unpaired Student’s t tests. G–V curve and inactivation–voltage curve were fitted by Boltzmann functions, which G/Gmax = 1/(1 + exp (V0.5 – Vm)/k) or I/Imax =1/(1 + exp (V0.5 – Vm)/k), with V0.5 being membrane potential (Vm) at which 50% of activation or inactivation was observed and k being the slope of the function. The dose–response curve was fitted by Hill equation, which Ipeak=Ipeakmax/[1+(IC50/C)n], with n as the Hill coefficient, and IC50 as the concentration producing 50% effect.
Chemicals
Cell culture materials were purchased from GIBCO (Life Technologies, Rockville, MD). 4α-PDD (4α-phorbol-12,13-didecanoate), D(-)Mannitol and U73122 (1-[6-((17β-3-Methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione) were purchased from CALBIOCHEM (San Diego, CA) and others, unless stated, all came from Sigma Chemical Company.
8-Br-cAMP (8-Bromoadenosine 3′,5′-cyclic monophosphate ), 8-Br-cGMP (8-Bromoguanosine-3′,5′-cyclomonophosphate sodium salt ), H-89 (N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride), KT5823, PMA (phorbol-12,13-dibutyrate), BIM (Bisindolylmaleimide II), staurosporine, GTP-γs (Guanosine 5′-O-(3-thiotriphosphate) tetralithium salt), GDP-βs(Guanosine 5′-[β-thio]diphosphate trilithium salt), Wortmannin, LY-294002 (2-(4-Morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride), U73122 and 4α-PDD were prepared as stock solutions in DMSO. The final concentrations in external solution or pipette solution were ≤0.1%.
GTP-γs, GDP-βs, H-89, KT5823, U73122, Wortmannin, LY294002 and staurosporine, were present in the pipette solution. Whereas 4α-PDD, 8-Br-cAMP, 8-Br-cGMP, BIM, PMA were applied in the external solution.
RESULTS
Effect of anisotonicity on IK and IA
Hypotonic treatment
In the present study, IK and IA were recorded in TG neurons with diameter between 15–30 μm for it has been reported that the small and medium size “nociceptive” TG and DRG neurons and associated Aδ- and C-fiber afferents are critical for detecting noxious stimuli and initiating pain sensation (Cardenas et al., 1995; Harper and Lawson, 1985; Slugg et al., 2000).
Figure 1A shows that IK was increased reversibly upon exposure to hypotonicity (220mOsm). On the average, IK was increased by 34.52±8.58% from 513.13±51.10 pA/pF to 687.53±38.76 pA/pF (n=37, paired t test, P<0.01). After hypotonicity was washed out the current recovered to 523.34±22.72 pA/pF. We also found that G–V curve did not shift (Fig. 1C) but inactivation–voltage curve significantly shifted (32 mV) to the depolarizing direction when the extracellular solution was changed from 300mOsm to 220mOsm (Fig. 1D) (Table 2).
Figure 1. Effect of hypotonicity on IK.

A. The typical recordings show that IK, evoked by voltage step to +50 mV, was increased from 4.57 nA to 6.38 nA when the external solution was changed from 300mOsm to 220mOsm for 3min and the current recovered to 4.77 nA after washout. B. The peak current-voltage relationship (I–V) was shown before and during hypotonicity treatment. C. In the presence of hypotonicity, G–V curve did not shift significantly. V0.5 was −0.93±1.30 mV and −0.90±0.71 mV (n=8, paired t test, P>0.05) for 300mOsm and 220mOsm, respectively; k was 16.46±1.13 and 16.07±0.41 (n=8, paired t test, P>0.05), respectively. Data were transformed from the I–V data shown in B. D. The typical recordings show that hypotonicity increased IK and inactivation–voltage curve significantly shifted to depolarizing direction. V0.5 was −72.67±1.54 mV and −40.55±5.25 mV (n=8, paired t test, P<0.05) for 300mOsm and 220mOsm, respectively; k was −9.67±1.32 and −10.98±4.61 (n=8, paired t test, P>0.05), respectively.
Table 2.
Effect of anisotonicity and 4α-PDD on G–V and inactivation–voltage curve of VGPCs
| G–V curve | inactivation–voltage curve | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| V0.5 (mV) | k | V0.5 (mV) | k | ||||||
| 300mOsm | Aniso/4α-PDD | 300mOsm | Aniso/4α-PDD | 300mOsm | Aniso/4α-PDD | 300mOsm | Aniso/4α-PDD | ||
| Hypo (220mOsm) | IK | −0.93±1.30 | −0.90±0.71 | 16.46±1.13 | 16.07±0.41 | −72.67±1.54 | −40.55±5.25* | −9.67±1.32 | −10.98±4.61 |
| n=8 | n=8 | ||||||||
| IA | −3.57±0.18 | −3.33±0.72 | 19.47±1.20 | 21.08±0.75 | −68.47±0.23 | −69.42±0.21 | −9.37±0.20 | −9.67±0.15 | |
| n=11 | n=11 | ||||||||
| 4α-PDD (3.0μM) | IK | −0.58±0.60 | −0.11±0.48 | 17.83±0.53 | 18.38±0.42 | −70.44±0.10 | −50.40±0.42* | −10.08±0.08 | −7.60±0.36* |
| n=9 | n=9 | ||||||||
| IA | −3.46±0.17 | −4.42±0.25 | 19.62±0.15 | 20.80±0.23 | −65.93±0.29 | −66.38±0.74 | −11.49±0.25 | −11.58±0.65 | |
| n=8 | n=8 | ||||||||
| Hyper (350mOsm) | IK | −0.92±1.23 | −8.16±1.02* | 18.13±1.08 | 17.63±0.95 | −69.99±1.48 | −83.00±.122* | −11.68±1.28 | −15.19±0.87* |
| n=11 | n=12 | ||||||||
| IA | −3.98±0.25 | −9.43±0.99* | 19.16±0.81 | 20.38±0.94 | −69.45±0.27 | −86.32±0.47* | −9.93±0.24 | −16.37±0.28* | |
| n=10 | n=10 | ||||||||
Paired t test was used when comparing the effect of anisotonicity and 4α-PDD on V0.5 and k in G–V and inactivation–voltage curve.
P<0.05 vs 300mOsm.
Figure 2A shows that hypotonicity (220mOsm) had almost no effect on IA. On the average, IA was 294.46±39.76 pA/pF and 302.84±17.34 pA/pF before and during 220mOsm treatment (n=10, paired t test, P>0.05). As shown in figure 2C and 2D, neither G–V nor inactivation–voltage curve of IA significantly shifted under hypotonic treatment (Table 2).
Figure 2. Effect of hypotonicity on IA.

A. After exposure to hypotonicity (220mOsm), IA was not affected and the peak amplitude was 5.87 nA, 5.91 nA and 5.93 nA before, during and after hypotonicity application, respectively. B. I–V curve did not change before and during hypotonicity treatment. C. G–V curve did not shift in the presence of hypotonicity. V0.5 was −3.57±0.18 mV and −3.33±0.72 mV (n=11, paired t test, P>0.05) for 300mOsm and 220mOsm, respectively; k was 19.47±1.20 and 21.08±0.75 (n=11, paired t test, P>0.05), respectively. Data were transformed from the I–V data shown in B. D. Hypotonicity had no effect on inactivation–voltage curve. V0.5 was −68.47±0.23 mV and −69.42±0.21 mV (n=11, paired t test, P>0.05) for 300mOsm and 220mOsm, respectively; k was −9.37±0.20 and −9.67±0.15 (n=11, paired t test, P>0.05), respectively.
Hypertonic treatment
Different from the effect of hypotonicity, both IK and IA were inhibited irreversibly in the presence of hypertonicity (350mOsm) (Figs. 3 and 4). On the average, IK was reduced by 34.54±8.21% from 495.18±30.18 pA/pF to 309.11±28.57 pA/pF (n=18, paired t test, P<0.01) when the extracellular solution was changed from 300mOsm to 350mOsm (Fig. 3A). IA was inhibited by 27.55±6.17% from 294.29±18.22 pA/pF to 210.47±13.25 pA/pF (n=17, paired t test, P<0.01) in the presence of 350mOsm (Fig. 4A). Neither IK (318.59±54.26 pA/pF) nor IA (214.98±11.29 pA/pF) recovered to the control level after hypertonicity was washed out. It was also shown that for IK and IA, G–V curve as well as inactivation–voltage curve significantly shifted to the hyperpolarizing direction when exposed to hypertonicity (Figs. 3D and 4D) (Table 2).
Figure 3. Effect of hypertonicity on IK.

A. The typical recordings show that IK was reduced from 8.16 nA to 5.32 nA when the external solution was changed from 300mOsm to 350mOsm for 3 min but the current recovered only slightly (5.84 nA) after washout. B. I–V curve was shown before and during hypertonicity treatment. C. In the hypertonic solution G–V curve shifted to the hyperpolarization direction. V0.5 was −0.92±1.23 mV and −8.16±1.02 mV (n=11, paired t test, P<0.05) for 300mOsm and 350mOsm, respectively; k was 18.13±1.08 and 17.63±0.95 (n=11, paired t test, P>0.05), respectively. Data were transformed from the I–V data shown in B. D. The typical recordings show that the amplitude of IK was reduced in the presence of hypertonic solution and inactivation–voltage curve shifted to hyperpolarizing direction with V0.5 being −69.99±1.48 mV and −83.00±1.22 mV (n=12, paired t test, P<0.05) for 300mOsm and 350mOsm, respectively; k being −11.68±1.28 and −15.19±0.87 (n=12, paired t test, P<0.05), respectively.
Figure 4. Effect of hypertonicity on IA.
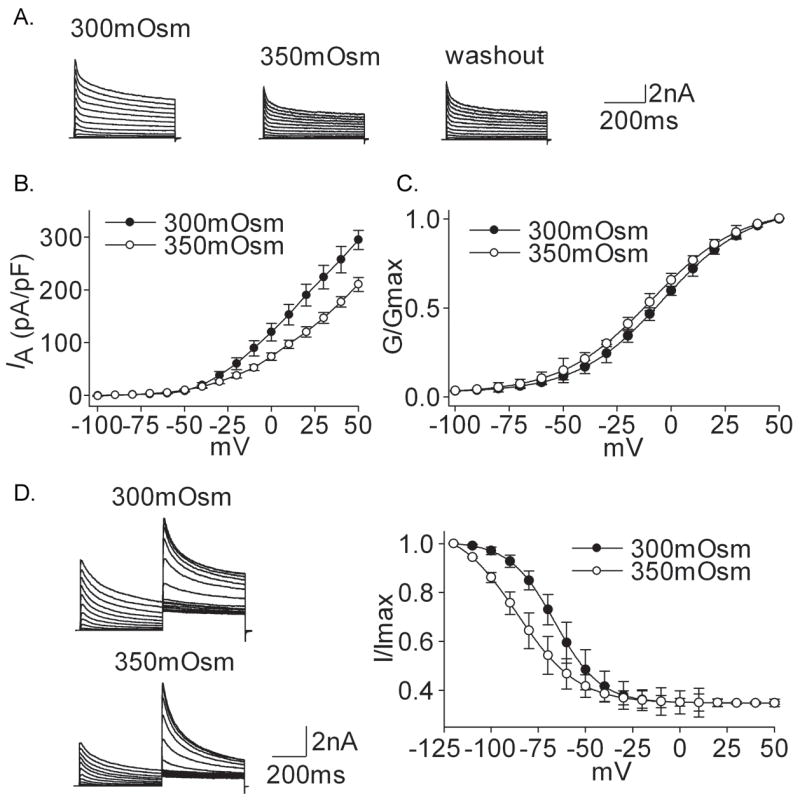
A. The typical recordings show that IA was reduced from 8.34 nA to 6.01 nA when the external solution was changed from 300mOsm to 350mOsm but the current almost did not recover (6.75 nA) after washout. B. I–V curve was shown before and during hypertonicity treatment. C. In the hypertonic solution G–V curve shifted to the hyperpolarization direction. V0.5 was −3.98±0.25 mV and −9.43±0.99 mV (n=10, paired t test, P<0.05) for 300mOsm and 350mOsm, respectively; k was 19.16±0.81 and 20.38±0.94 (n=11, paired t test, P>0.05), respectively. Data were transformed from the I–V data shown in B. D. The typical recordings show that the amplitude of IA was inhibited upon exposure to hypertonic stimulus and inactivation–voltage curve shifted to the hyperpolarizing direction. V0.5 was −69.45±0.27 mV and −86.32±0.47 mV (n=12, paired t test, P<0.05) for 300mOsm and 350mOsm, respectively; k was −9.93±0.24 and −16.37±0.28 (n=10, paired t test, P<0.05), respectively.
The dose–dependent modulations of VGPCs by anisotonic treatment are presented in figure 5, in which hypertonic stimuli had the similar phenotype on IK and IA whereas hypotonicity had different effects by increasing IK and leaving IA unaffected. Since 220mOsm and 350mOsm produced significant effects on VGPCs: 220mOsm increased IK by 34.52% while 350mOsm decreased IK and IA by 34.54% and 27.55% respectively, these concentrations were used in all subsequent experiments.
Figure 5. Effect of anisotonicity on IK and IA.

A. IK was enhanced by hypotonic stimuli and inhibited by hypertonic stimuli. B. IA was not significantly affected when exposed to hypotonicity but inhibited by hypertonic stimuli.
Effect of 4α-PDD on IK and IA
TRPV4 receptor is regarded as an important osmotic cellular sensor (Liedtke and Friedman, 2003; Liedtke and Kim, 2005). To test the hypothesis that TRPV4 receptors may be involved in the effect of anisotonicity on VGPCs, in rat TG neurons we tested the effect of TRPV4 receptor agonist, 4α-PDD, on IK and IA.
Similar to the effect of hypotonicity, IK was enhanced reversibly by 4α-PDD. On the average, application of 3.0 μM 4α-PDD increased IK by 43.58±8.05% from 513.26±30.69 pA/pF to 736.93±22.71 pA/pF (n=9, paired t test, P<0.01). The increase was recoverable (>90%) after 4α-PDD was washed out. In the presence of 4α-PDD, G–V curve, representing the voltage dependence of activation, did not shift (n=9, paired t test, P>0.05) but inactivation–voltage curve significantly shifted to the depolarizing direction (Fig 6, B and C) (Table 2). The concentration–dependent increase of IK by 4α-PDD is shown in figure 6A. IK was increased 6.20±3.60% and 81.40±8.80% by 0.03 μM and 30 μM 4α-PDD, respectively. The dose–response curve was fitted by Hill equation with IC50 being 4.01 μM.
Figure 6. Effect of 4α-PDD on IK and IA.

A. The plot shows the percentage in increase of IK by 4α-PDD at concentrations of 0.03, 0.1, 0.3, 1.0, 3.0, 10 and 30 μM. The dose–response curve fits to Hill equation with IC50 being 4.01 μM and n being 0.80. B. G–V curve of IK did not shift upon exposed to 3.0 μM4α-PDD. V0.5 was −0.58±0.60 mV and −0.11±0.48 mV (n=9, paired t test, P>0.05) before and during 4α-PDD treatment, respectively; k was 17.83±0.53 and 18.38±0.42 (n=9, paired t test, p>0.05), respectively. C. After application of 3.0 μM 4α-PDD, inactivation–voltage curve of IK significantly shifted to the depolarizing direction. V0.5 was −70.44±0.10 mV and −50.40±0.42 mV (n=9, paired t test, P<0.05) before and during 4α-PDD treatment, respectively; k was −10.08±0.08 and −7.60±0.36 (n=9, paired t test, P<0.05), respectively. D. Application of 3.0 μM 4α-PDD had no effect on G–V curve of IA (300mOsm: V0.5=−3.46±0.17 mV, k=19.62±0.15; 4α-PDD: V0.5=−4.42±0.25 mV, k=20.80±0.23, n=8, paired t test, P>0.05). E. Application of 4α-PDD had no effect on inactivation–voltage curve of IA either (300mOsm: V0.5=−65.93±0.29 mV, k=−11.49±0.25; 4α-PDD: V0.5=−66.38±0.74 mV, k=−11.58±0.65, n=8, paired t test, P>0.05).
We also tested the effect of 3.0 μM 4α-PDD on IA. This concentration was chosen because in our previous studies and here, 4α-PDD at this concentration had significant effects on voltage-gated sodium channels (VGSCs), voltage-gated calcium channels (VGCCs) (unpublished results) and IK. We found that IA was 290.17±20.04 pA/pF and 287.13±25.14 pA/pF before and during 3.0 μM 4α-PDD treatment, respectively (n=8, paired t test, P>0.05). Moreover, 4α-PDD had no effect on G–V curve or inactivation–voltage curve of IA (Fig. 6, D and E) (Table 2). These data indicated that 4α-PDD mimicked the effect of hypotonicity on VGPCs.
Hypo- and hypertonicity induced modulations on VGPCs in TG neurons from trpv4 wild type (TRPV4+/+) and trpv4 knockout (TRPV4−/−) mice
We further explored the possible role of TRPV4 receptor by measuring the effect of changing the osmolality on VGPCs in TG neurons from TRPV4+/+ and TRPV4−/−mice with soma diameters ranging between 10–25 μm.
Hypotonic treatment
Figure 7A shows the effects of hypotonicity (220mOsm) on IK obtained from TRPV4+/+ and TRPV4−/− mice. Similar to the results found in rat TG neurons, IK was increased reversibly from 523.22±10.32 pA/pF to 675.30±16.13 pA/pF after hypotonicity treatment in TRPV4+/+ mice TG neurons (n=15, paired t test, P<0.05). In contrast, IK was 486.42±35.52 pA/pF and 493.75±31.43 pA/pF before and during hypotonicity application in TRPV4−/− mice TG neurons (n=17, paired t test, P>0.05). Significant difference was found in the increase of IK by hypotonicity between TRPV4+/+ (29.21±7.80%) and TRPV4−/− mice (1.41±2.72%) (unpaired t test, P<0.01).
Figure 7. Effects of anisotonicity on IK and IA in TG neurons from TRPV4+/+ and TRPV4−/− mice.

A. Upon exposure to hypotonicity (220mOsm), IK was increased by 29.21±9.81% (n=15) in TRPV4+/+ mice TG neurons, which was significantly different from that in TRPV4−/− mice (1.41±2.72%, n=17) (unpaired t test, P<0.01). B. After exposure to hypotonicity (220mOsm), IA was not significantly affected. On the average, in TRPV4+/+ mice TG neurons, the amplitude was 253.75±31.43 pA/pF and 243.19±33.99 pA/pF (n=14, paired t test, P>0.05) before and during hypotonicity treatment, respectively, and in TRPV4−/− mice TG neurons IA was 246.42±35.52 pA/pF and 232.13±32.58 pA/pF (n=13, paired t test, P>0.05), respectively. C. After exposure to hypertonicity (350mOsm), IK was reduced by 38.82±7.25% (n=14) and 38.52±6.38% (n=16) in TRPV4+/+ and TRPV4−/− TG neurons respectively (unpaired t test, P>0.05). D. After exposure to hypertonicity (350mOsm), the inhibition of IA by hypertonicity was 30.68±7.16% (n=14) and 33.31±6.61% (n=14) in TRPV4+/+ and TRPV4−/− mice TG neurons respectively (unpaired t test, P>0.05).
Figure 7B shows that hypotonicity had no effect on IA in either TRPV4+/+ (n=14, paired t test, P>0.05) or TRPV4−/− mice TG neurons (n=13, paired t test, P>0.05).
Hypertonic treatment
IK was inhibited irreversibly when the extracellular solution was changed from 300mOsm to 350mOsm. In TRPV4+/+ and TRPV4−/− TG neurons, IK was reduced by 38.82±7.25% (n=14, paired t test, P<0.01) and 38.52±6.38% (n=16, paired t test, P<0.01) respectively, which was not statistically different (unpaired t test, P>0.05) (Fig 7C).
Similar to the inhibition of IK, in the presence of hypertonicity, IA was inhibited irreversibly by 30.68±7.16% (n=14, paired t test, P<0.05) and 33.31±6.61% (n=14, paired t test, P<0.05) in TRPV4+/+ and TRPV4−/− TG neurons, respectively (Fig 7D). No statistical difference was found in the reduction of IA by hypertonicity between TRPV4+/+ and TRPV4−/− mice (unpaired t test, P>0.05).
In summary, these data indicated that anisotonicity had similar effect on VGPCs in TG neurons from rats and TRPV4+/+ mice. However, it was obvious that the increase of IK by hypotonicity seen in TRPV4+/+ mice was markedly reduced in TRPV4−/− mice, suggesting the involvement of TRPV4 receptor in the increase of IK by hypotonic stimulus.
Second messenger systems involved in the modulation of anisotonicity on IK and IA
It is known that VGPCs, as well as VGCCs and VGSCs, can be modulated by many intracellular pathways through phosphorylation or dephosphorylaion. In this study, we tested several intracellular pathways in rat TG neurons to determine which, if any, of them takes part in the modulation of IK and IA by hypo and hypertonicity.
PKC system
We firstly tested whether PKC system was involved in the action of anisotonicity on IK and IA. In this study, after exposure to1.0 μM phorbol-12, 13-dibutyrate (PMA, the agonist of PKC) for 3 min, IK was inhibited by 10.01±3.12% (n=19, paired t test, P<0.05) but IA was not apparently affected (n=12, paired t test, P>0.05). Application of 1.0 μM Bisindolylmaleimide II (BIM, PKC antagonist) and 1.0 μM staurosporine (PKC antagonist) increased IK by 8.17±3.11% (n=17, paired t test, P<0.05) and 9.11±4.39% (n=9, paired t test, P<0.05) respectively, leaving IA unchanged. As shown in figure 8, application of BIM and staurosporine significantly blocked the increase of IK by hypotonicity whereas unaffected the inhibitions of IK or IA by hypertonicity. These results suggested the selective involvement of PKC system in the increase of IK by hypotonicity.
Figure 8. Modulation of PKC system on the effects induced by hypo and hypertonicity.

A. Pre-incubation with PKC antagonists, BIM and staurosporine, significantly reversed the increase of IK by hypotonicity from 34.52±8.58% to 17.48±5.59% (n=12) (unpaired t test, P<0.05) and 15.11±6.17% (n=10) (unpaired t test, P<0.05) respectively. B. Neither BIM nor staurosporine statistically affected the inhibitions of VGPCs by hypertonicity (unpaired t test, P>0.05). In the presence of BIM and staurosporine, IK was inhibited by 37.88±7.38% (n=11) and 35.43±6.01% (n=8), respectively and IA by 25.03±6.12% (n=12) and 27.13±5.05% (n=8), respectively.
G-protein system
We then studied G-protein system using GTP-γs (non-hydrolyzable GTP analog) and GDP-βs (non-hydrolyzable GDP analog). With 0.3 mM GTP-γs or 0.3 mM GDP-βs in the pipette solution, IK was reduced by 9.90±1.54% (n=27, paired t test, P<0.05) and 10.49±2.60% (n=24, paired t test, P<0.05) respectively, whilst IA by 19.00±3.02% (n=10, paired t test, P<0.01) and 19.36±5.74% (n=10, paired t test, P<0.01), respectively. Neither GTP-γs nor GDP-βs apparently affected the increase of IK by hypotonicity (Fig. 9A). However, both GTP-γs and GDP-βs reversed the inhibitions of IK and IA by hypertonicity (Fig. 9B). This result suggested that G-protein system was selectively required for the inhibitions of VGPCs by hypertonicity.
Figure 9. Modulation of G-protein system on the effects induced by hypo and hypertonicity.

A. Neither GTP-γs nor GDP-βs had significant effect on the increase of IK by hypotonicity (unpaired t test, P>0.05). In hypotonic solution (220mOsm) IK was enhanced by 36.00±7.91% (n=12) and 38.23±8.01% (n=13) with GTP-γs and GDP-βs in the pipette solution, respectively. B. Both GTP-γs and GDP-βs markedly reversed the inhibitions of IK (unpaired t test, P<0.05) and IA (unpaired t test, P<0.05) by hypertonicity. In hypertonic solution (330mOsm) IK was reduced by 18.73±5.90% (n=12) and 19.29±8.02% (n=12) in the presence of GTP-γs and GDP-βs, respectively and IA by 11.53±4.19% (n=11) and 13.17±5.26% (n=12), respectively.
PKA system
We next explored PKA system by using 8-Br-cAMP (a kind of membrane permeable analogue of cAMP, agonist of PKA) and H-89 (antagonist of PKA). After exposure to 1.0 mM 8-Br-cAMP, IK and IA were decreased by 9.16±2.16% (n=11, paired t test, P<0.05) and 10.25±4.47% (n=8, paired t test, P<0.05), respectively. After pre-application of 10 μM H-89 for 10 min, IK and IA were enhanced by 9.60±2.39% (n=13, paired t test, P<0.05) and 10.26±5.85% (n=13, paired t test, P<0.05), respectively. As shown in figure 10A, pre-application of H-89 had no significant effect on the increase of IK by hypotonicity when compared with that in normal pipette solution (unpaired t test, P>0.05). Also, H-89 did not affect the inhibitions of IK or IA by hypertonicity (unpaired t test, P>0.05). These data indicated that PKA system was not necessary for the actions of anisotonic stimuli on VGPCs.
Figure 10. Modulation of PKA, lipid cascade and PKG systems on the effects induced by hypo and hypertonicity.

A. For PKA system, pre-incubation with PKA inhibitor, H-89, had no significant effect on the modulations of VGPCs by hypo or hypertonicity (unpaired t test, P>0.05). In the presence of 10 μM H-89, IK was increased 38.04±8.64% (n=10) by hypotonicity, while IK and IA were decreased 29.46±7.68% (n=10) and 30.00±6.64% (n=14) by hypertonicity respectively. B. For lipids cascade, in hypotonic solution, IK was increased by 38.32±7.62% (n=11), 36.61±6.83% (n=14) and 37.86±8.59% (n=12) with Wortmannin, LY294002 and U73122 in the pipette solution, respectively. Compared with the inhibition of hypotonicity with normal pipette solution (34.52±8.58%, n=37), none of them was significantly different (unpaired t test, P>0.05). Similarly, pre-incubation with Wortmannin, LY294002 and U73122 did not affect the inhibition of IK or IA by hypertonicity (unpaired t test, P<0.05). In the presence of Wortmannin, LY294002 and U73122, the inhibition of IK by hypertonicity was 34.71±6.72% (n=11), 30.73±7.29% (n=10) and 33.23±7.55% (n=12) respectively and the inhibition of IA was 27.25±8.02% (n=11), 29.50±6.61% (n=11) and 30.16±8.79% (n=11) respectively. C. For PKG system, KT5823 did not statistically alter the modulation of VGPCs by hypo or hypertonicity (unpaired t test, P>0.05). In the presence of KT5823, IK was increased 38.33±7.15% (n=13) by hypotonicity, while IK and IA were inhibited 31.12±6.12% (n=15) and 25.17±5.67% (n=14) by hypertonicity, respectively.
Lipids cascade
Phosphatidylinositol 3-kinase (PI3K) is a kind of lipid kinase and reported to modulate potassium current amplitude and its development (Oliver et al. 2004). It has been reported that phosphatidylinositol-4′5′-bisphosphate (PIP2) can modulate the function of VGPCs (Wu et al. 1998). Here, LY294002 (PI3K inhibitor) and Wortmannin (PI3K and PI4K inhibitor) were used to test whether the actions on IK or IA at different tonicities could be affected. With 2.0 μM Wortmannin or 50 μM LY294002 in the pipette solution, IK was reduced by 27.81±2.39% (n=22, paired t test, P<0.01) and 14.94±6.94% (n=18, paired t test, P<0.05) respectively, while IA by 22.61±7.06% (n=22, paired t test, P<0.01) and 27.84±5.64% (n=14, paired t test, P<0.01) respectively. We found that neither LY294002 nor Wortmannin reversed the increase of IK by hypotonicity (unpaired t test, P>0.05). Similarly, neither of them blocked the inhibitions of IK or IA by hypertonicity (unpaired t test, P>0.05) (Fig. 10B).
U73122 is the PLC inhibitor which can increase the concentration of PIP2 and decrease the production of IP3 and DAG. With 10 μM U73122 in the pipette solution, IK and IA were reduced by 16.78±6.82% (n=22, paired t test, P<0.05) and 20.09±6.97% (n=12, paired t test, P<0.01), respectively. Figure 10B shows that pre-application of U73122 didn’t affect the actions on IK or IA when treated with anisotonicity. These data indicated that lipids cascade was not involved in the actions on VGPCs under hypo or hypertonic conditions.
PKG system
We also performed the experiment to investigate whether PKG system was responsible for the modulations of IK or IA by changes in osmolality. Exposure to 1.0 mM 8-Br-cGMP (a kind of membrane permeable analogue of cGMP, agonist of PKG) for 3 min had no effect on IK (n=12, paired t test, P>0.05) but decreased IA by 15.03±3.33% (n=10, paired t test, P<0.05). After pre-incubation with 10 μM KT5823 (antagonist of PKG), IA was increased by 9.77±2.09% (n=14, paired t test, P<0.05), while IK was unaffected (n=24, paired t test, P>0.05). Figure 10C shows that KT5823 did not markedly affect the actions on IK or IA when the extracellular osmolality deviated from isotonicity, indicating that PKG system was not required for the effects of anisotonic stimuli on VGPCs.
DISCUSSION
Modulation of VGPCs by anisotonicity
TG neurons are primary afferent neurons that carry sensory signals from the face, oral cavity, and nasal cavity to the medulla (Sessle, 1999). Many nociceptive stimuli are known to change the neuronal excitability at peripheral nerve endings of the TG neurons (Davies, 1988; Lazarov, 2002), so in the present study, TG neurons were used as a pain model to study the tonicity induced tuning of VGPCs. We demonstrate that VGPCs, including IK and IA, could be modulated by the changes of osmolality in cultured TG neurons. The modulation of anisotonicity on VGPCs is type-dependent. Both IK and IA were inhibited by hypertonic solutions, but after exposure to hypotonicity IK was markedly increased whilst IA was left unaffected. In this study, we also performed the experiment by using the calcium buffer and found the increase of IK by hypotonicity and inhibition of IK and IA by hypertonicity (data not shown). So it was indicated that the modulation of VGPCs by anisotonicity was calcium independent. We then explored the role of TRPV4 receptor and found that TRPV4 might underlie the modulation of IK by hypotonicity. Considering the downstream signaling pathways, different intracellular messenger systems were selective for the modulation of VGPCs induced by hypo versus hypertonicity.
Participation of VGPCs in hypertonicity-induced nociception
VGPCs play fundamental roles, such as regulation of membrane repolarization, resting membrane potential, and frequency of firing and neurotransmitter release (Mathie et al., 1998; Yellen, 2002). Studies show that the inhibition of IK can depolarize the cell membrane, prolong AP duration, reduce the amplitude of afterhyperpolarization and lower the threshold for AP firing (Birinyi-Strachan et al., 2005; Safronov et al., 1996), while the inhibition of IA can induce repetitive firing (Amir et al., 2002; Storm, 1988). It is widely accepted that a decrease of VGPCs, including IK and IA, can increase the excitability of cells (Birinyi-Strachan et al., 2005; Yost, 1999). Recent evidences suggest that VGPCs participate in nociceptive signal transduction induced by inflammatory mediators (Harriott et al., 2006; Sculptoreanu et al., 2004; Stewart et al., 2003; Takeda et al., 2006; Yoshimura and de Groat, 1999; Xu et al., 2006) and nerve injury (Everill and Kocsis, 1999; Ishikawa et al., 1999; Rasband et al., 2001) in primary sensory neurons. For example, it is found that in temporomandibular joint inflammation the excitability of rat trigeminal root ganglion neurons is enhanced via the decrease of IA (Takeda et al., 2006) and the excitability of masseter muscle afferents is increased by inhibiting VGPCs in inflammation (Harriott et al., 2006). In the present study, we found that both IK and IA were blocked by hypertonicity and larger response was found in the presence of larger osmotic gradient (Fig. 5). In addition to the reduction in current amplitude, inactivation–voltage curves of both IK and IA significantly shifted to the hyperpolarizing direction, which could contribute to the observed decreases of IK and IA (Figs. 3 and 4). In contrast, no reduction of IK or IA was found under hypotonic treatment (Figs. 1 and 2). So it was suggested that the inhibitions of IK and IA selectively participated in the nociception induced by hyper but not hypotonicity.
Involvement of TRPV4 receptor in the modulation of IK by hypotonicity
In the peripheral nervous system, C-fiber afferents can be activated by both hypo and hypertonic stimuli (Alessandri-Haber et al., 2005; Alessandri-Haber et al., 2003; Matran et al., 1989; Viana et al., 2001). Moreover, changes in osmolality can facilitate the nociceptive signal transduction induced by many stimuli (Alessandri-Haber et al., 2006; Alessandri-Haber et al., 2004; Hamamoto et al., 2000; Liu et al., 2007), so it is proposed that anisotonicity plays an important role in the process of nociception. Knockdown of TRPV4 either by gene-disruption or antisense oligonucleotides has proved that TRPV4 is involved in mechanical stimulus as well as hypo and hypertonicity-induced nociception in rodents (Alessandri-Haber et al., 2005; Alessandri-Haber et al., 2004; Alessandri-Haber et al., 2003; Suzuki et al., 2003). In the present study, we found that the increase of IK by hypotonicity was well confirmed by TRPV4 agonist (4α-PDD), that is, the increase is reversible and accompanied with inactivation–voltage curve depolarization shifting (Figs. 1 and 6). Furthermore, we found that hypotonicity did not exhibit increase of IK in TRPV4-/- mice TG neurons. By contrast, the inhibitory effect of IK or IA by hypertonicity was unaffected in TRPV4−/− mice TG neurons (Fig. 7). These data indicated that TRPV4 receptor selectively participated in the increase of IK by hypotonicity. In this study, we found that IA was unaffected by hypotonicity in TRPV4−/− mice TG neurons, but we were also aware that IA was not affected by hypotonicity in TRPV4+/+ mice and was not changed by 4α-PDD or hypotonicity in rat TG neurons. These results therefore indicated that TRPV4 had no modulatory effect on IA.
Besides the hypotonic solution, TRPV4 has been shown to be activated by other stimuli. Many studies report the role of TRPV4 as the thermosensitive receptor and TRPV4 can be activated by warm temperature (27–35 ºC) (Gao et al., 2003; Guler et al, 2002; Todaka et al., 2004). Given the fact that TRPV4 can be activated at a temperature of 35°C, it is possible that TRPV4 might become sensitized in the brain because the body core temperature is ~37 °C. Therefore we suggest that the hypotonicity induced tuning of IK might be facilitated at physiological condition (37 °C) or sometimes at pathological conditions (for instance, at a temperature of 40 °C during a fever).
In the present study, it was noted that effects of hypotonicity and TRPV4 receptor were complicated. On one hand, hypotonicity and TRPV4 could induce the inward current (Alessandri-Haber et al., 2003; Vriens et al., 2004) and enhance the capsaicin-induced current (Liu et al., 2007) which facilitate the depolarization of AP and increase the excitability of neurons, resulting in algetic effect. On the other hand, they facilitate IK and inhibit VGCCs and VGSCs (unpublished results), which decreases the excitability of neurons and leads to the analgesic effect. If the hypotonicity induced algetic effect is more profound than the analgesic effect, nociception will be induced as an integrated result. As is known that the abnormal excitability of nociceptor is mediated by changes in function of many ion channels including voltage-gated channels and receptors, therefore the nociception induced by hypertonicity just as that by hypotonicity is very likely an integrative result.
Here, it was also noteworthy that although both hypo and hypertonic stimuli can induce pain behavior, the mechanism may be different. Considering the modulation of VGPCs, first, hypo and hypertonicity exhibit different phenotype: hypotonicity increased IK but had no effect on IA, whereas hypertonicity decreased both IK and IA and second, the increase of IK by hypotonicity was TRPV4 dependent, while the inhibitions of IK and IA by hypertonicity was TRPV4 independent.
Given that TRPV4 was not involved in the inhibition of VGPCs by hypertonicity, there might be other receptors that mediate hypertonic response. In fact, there are other possibilities underlying the inhibition induced by hypertonic solution. For instance, the shrinking membrane by hypertonicity might exert lateral pressure on VGPCs, which causes incomplete opening of channels. However, in many cases, hypertonic shrinkage is partially rescued by regulatory volume increase (RVI), so in order to properly define the role of membrane mechanism, actual membrane tension will have to be measured during the experiment. Another possibility is that the inhibition of VGPCs by hypertonicity was mediated through intracellular messengers or regulator substance. Next we will discuss some candidates accounting for the modulation of VGPCs by anisotonic treatment.
Involvement of specific intracellular pathways in the modulations of VGPCs by hypo versus hypertonicity
VGPCs are modulated by numerous second messenger pathways, such as G-protein (ffrench-Mullen et al., 1994; Galeotti et al., 1999; Galeotti et al., 2001; Ocana et al., 2004), cAMP-PKA (Akins and McCleskey, 1993; Hampson et al., 1995; Jonas and Kaczmarek, 1996; Mason et al., 2002; Mu et al., 2000), cGMP-PKG (Liu and Simon, 2003; Moreno et al., 2001; Ocana et al., 2004), PKC (Doerner et al., 1988; Jonas and Kaczmarek, 1996; Lotan et al., 1990; Sun et al., 2003) and lipids cascade (Sun et al., 2003; Sun et al., 2004). Our present study shows that the activities of IK and IA are modulated by the above intracellular pathways.
G-protein is an important modulator for VGPCs. Intracellular dialysis with G-protein antagonist (GDP-βs) can significantly diminish the Carbachol-induced inhibition of IK (ffrench-Mullen et al., 1994). Recently it is reported that the modulation of G-protein on VGPCs underlies the antinociceptive effects for tricycles antidepressants and H1-antihistamines (Galeotti et al., 1999; Galeotti et al., 2001; Ocana et al., 2004). In this study, we found that the inhibitions of IK and IA by hypertonicity were significantly reversed by pretreatment with G-protein antagonist (GDP-βs) but the increase of IK by hypotonicity was unaffected, indicating the selective involvement of G-protein in hypertonic induced response (Fig. 8).
PKC also plays an important role in modulating the activities of VGPCs. For example, PKC is involved in the inhibition of angiotensin II on delayed rectifier potassium current (Du et al., 2004; Pan et al., 2001) and the modulation of delayed rectifier potassium channels by alpha1-adrenergic activation (Kim et al., 2005). Consisting with the report in hippocampus neurons (Doerner et al., 1988), activation of PKC selectively reduced IK but had no effect on IA. Here we found that the increase of IK by hypotonicity was markedly reversed by PKC antagonists (BIM and staurosporine), whereas the inhibitions of IK and IA by hypertonicity were not affected, implying the selective participation of PKC system in hypotonic induced response. Collectively, these data demonstrated that specific and different intracellular pathways were involved in the modulation of VGPCs by hypo versus hypertonicity (Fig. 9). However, we are also aware that antagonism of G-protein and PKC did not reverse the modulation of VGPCs by hypo or hypertonicity completely, so there might be other mechanisms, such as some factors that can be affected by changes in the cell volume, contributing to the modulations.
CONCLUSION
Our present study demonstrated that changes in osmolality were one of the important regulators of VGPCs. TRPV4 receptor was selectively involved in the increase of IK by hypotonicity and different intracellular pathways were required for the actions on IK and IA induced by hypo versus hypertonicity. We also found that both IK and IA were inhibited by hypertonicity and this probably contributes to hypertonicity-induced nociception. Given the obvious tonicity-induced tuning of VGPCs, it is suggested that besides the anisotonicity-induced nociception, changes in osmolality may participate in many physiological and pathological functions via the modulation of VGPCs.
Acknowledgments
We thank Dr. Wolfgang Liedtke for providing TRPV4−/− mice. This work was supported by National Institute of General Medical Sciences Grant GM-63577 and National Natural Science Foundation of China (30571537 and 30271500).
Abbreviations used in this manuscript
- AP
action potential
- BIM
Bisindolylmaleimide II
- DRG
dorsal root ganglion
- 4α-PDD
4α-phorbol-12,13-didecanoate
- 8-Br-cAMP
8-Bromoadenosine 3′,5′-cyclic monophosphate
- 8-Br-cGMP
8-Bromoguanosine-3′,5′-cyclomonophosphate sodium salt
- GDP-βs
Guanosine 5′-[β-thio]diphosphate trilithium salt
- GTP-γs
Guanosine 5′-O-(3-thiotriphosphate) tetralithium salt
- G–V curve
voltage dependent activation curve
- H-89
N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride
- IA
fast-inactivating K+ current
- IK
slow-inactivating K+ current
- inactivation–voltage curve
voltage dependent inactivation curve
- KT5720, (9S,10S,12R)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-e poxy-1H-diindolo[1,2,3-fg
3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid hexyl ester
- LY290022
2-(4-Morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride
- mHBSS
modified Hank’s Balanced Salt solution
- PMA
Phorbol 12-myristate 13-acetate
- RVI
regulatory volume increase
- TG
trigeminal ganglion
- TRPV4
Transient Receptor Potential Vanilloid 4
- TRPV
Transient Receptor Potential Vanilloid
- U73122
1-[6-((17β-3-Methoxyestra-1,3,5(10)-trien-17-yl) amino) hexyl]-1H-pyrrole-2,5-dione)
- VGPCs
voltage-gated potassium channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akins PT, McCleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience. 1993;56:759–769. doi: 10.1016/0306-4522(93)90372-m. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci. 2006;26:3864–3874. doi: 10.1523/JNEUROSCI.5385-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci. 2004;24:4444–4452. doi: 10.1523/JNEUROSCI.0242-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Joseph E, Dina OA, Liedtke W, Levine JD. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain. 2005;118:70–79. doi: 10.1016/j.pain.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- Amir R, Liu CN, Kocsis JD, Devor M. Oscillatory mechanism in primary sensory neurones. Brain. 2002;125:421–435. doi: 10.1093/brain/awf037. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Fagan M, Ballyk BA, Rosen AS. Seizure susceptibility and the osmotic state. Brain Res. 1989;498:175–180. doi: 10.1016/0006-8993(89)90417-4. [DOI] [PubMed] [Google Scholar]
- Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol. 1997;502:175–187. doi: 10.1111/j.1469-7793.1997.175bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Schwartzkroin PA. Effects of hyposmolar solutions on membrane currents of hippocampal interneurons and mossy cells in vitro. J Neurophysiol. 1998;79:1108–1112. doi: 10.1152/jn.1998.79.2.1108. [DOI] [PubMed] [Google Scholar]
- Birinyi-Strachan LC, Gunning SJ, Lewis RJ, Nicholson GM. Block of voltage-gated potassium channels by Pacific ciguatoxin-1 contributes to increased neuronal excitability in rat sensory neurons. Toxicol Appl Pharmacol. 2005;204:175–186. doi: 10.1016/j.taap.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP, Scroggs RS. Variation in serotonergic inhibition of calcium currents in four types of rat sensory neurons differentiated by membranes properties. J Neurophysiol. 1995;74:1870–1879. doi: 10.1152/jn.1995.74.5.1870. [DOI] [PubMed] [Google Scholar]
- Catacuzzeno L, Fioretti B, Franciolini F. Voltage-gated outward K currents in frog saccular hair cells. J Neurophysiol. 2003;90:3688–3701. doi: 10.1152/jn.00308.2003. [DOI] [PubMed] [Google Scholar]
- Davies AM. The trigeminal system: an advantageous experimental model for studying neuronal development. Development. 1988;103:175–183. doi: 10.1242/dev.103.Supplement.175. [DOI] [PubMed] [Google Scholar]
- Doerner D, Pitler TA, Alger BE. Protein kinase C activators block specific calcium and potassium current components in isolated hippocampal neurons. J Neurosci. 1988;8:4069–4078. doi: 10.1523/JNEUROSCI.08-11-04069.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du JQ, Sun CW, Tang JS. Effect of angiotensin II type 1 receptor on delayed rectifier potassium current in catecholaminergic CATH.a cells. Acta Pharmacol Sin. 2004;25:1145–1150. [PubMed] [Google Scholar]
- Everill B, Kocsis JD. Reduction in potassium currents in identified cutaneous afferent dorsal root ganglion neurons after axotomy. J Neurophysiol. 1999;82:700–708. doi: 10.1152/jn.1999.82.2.700. [DOI] [PubMed] [Google Scholar]
- ffrench-Mullen JM, Plata-Salaman CR, Buckley NJ, Danks P. Muscarine modulation by a G-protein alpha-subunit of delayed rectifier K+ current in rat ventromedial hypothalamic neurones. J Physiol. 1994;474:21–26. doi: 10.1113/jphysiol.1994.sp019998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti N, Ghelardini C, Bartolini A. The role of potassium channels in antihistamine analgesia. Neuropharmacology. 1999;38:1893–1901. doi: 10.1016/s0028-3908(99)00068-4. [DOI] [PubMed] [Google Scholar]
- Galeotti N, Ghelardini C, Bartolini A. Involvement of potassium channels in amitriptyline and clomipramine analgesia. Neuropharmacology. 2001;40:75–84. doi: 10.1016/s0028-3908(00)00097-6. [DOI] [PubMed] [Google Scholar]
- Gao X, Wu L, O’Neil RG. Temperature-modulated Diversity of TRPV4 Channel Gating: activation by physical stresses and phorbol ester derivatives through protein kinase c-dependent and -independent pathways. J Biol Chem. 2003;278:27129–27137. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Grunewald B. Differential expression of voltage-sensitive K+ and Ca2+ currents in neurons of the honeybee olfactory pathway. J Exp Biol. 2003;206:117–129. doi: 10.1242/jeb.00053. [DOI] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina MJ. Heat-evoked activation of the ion channel TRPV4. J Neurosci. 2002;22:6408–6414. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- Hamamoto DT, Forkey MW, Davis WL, Kajander KC, Simone DA. The role of pH and osmolarity in evoking the acetic acid-induced wiping response in a model of nociception in frogs. Brain Res. 2000;862:217–229. doi: 10.1016/s0006-8993(00)02138-7. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Evans GJ, Mu J, Zhuang SY, King VC, Childers SR, Deadwyler SA. Role of cyclic AMP dependent protein kinase in cannabinoid receptor modulation of potassium “A-current” in cultured rat hippocampal neurons. Life Sci. 1995;56:2081–2088. doi: 10.1016/0024-3205(95)00192-9. [DOI] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol. 1985;359:31–46. doi: 10.1113/jphysiol.1985.sp015573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriott AM, Dessem D, Gold MS. Inflammation increases the excitability of masseter muscle afferents. Neuroscience. 2006;141:433–442. doi: 10.1016/j.neuroscience.2006.03.049. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Tanaka M, Black JA, Waxman SG. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve. 1999;22:502–507. doi: 10.1002/(sici)1097-4598(199904)22:4<502::aid-mus12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Kaczmarek LK. Regulation of potassium channels by protein kinases. Curr Opin Neurobiol. 1996;6:318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- Kilb W, Dierkes PW, Sykova E, Vargova L, Luhmann HJ. Hypoosmolar conditions reduce extracellular volume fraction and enhance epileptiform activity in the CA3 region of the immature rat hippocampus. J Neurosci Res. 2006;84:119–129. doi: 10.1002/jnr.20871. [DOI] [PubMed] [Google Scholar]
- Kim Y, Park MK, Uhm DY, Shin J, Chung S. Modulation of delayed rectifier potassium channels by alpha1-adrenergic activation via protein kinase C zeta and p62 in PC12 cells. Neurosci Lett. 2005;387:43–48. doi: 10.1016/j.neulet.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Lazarov NE. Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog Neurobiol. 2002;66:19–59. doi: 10.1016/s0301-0082(01)00021-1. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA. 2003;100:13698–13703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62(24):2985–3001. doi: 10.1007/s00018-005-5181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen L, Liedtke W, Simon SA. Changes in Osmolality Sensitize the Response to Capsaicin in Trigeminal Sensory Neurons. J Neurophysiol. 2007;97:2001–2015. doi: 10.1152/jn.00887.2006. [DOI] [PubMed] [Google Scholar]
- Liu L, Simon SA. Modulation of IA Currents by Capsaicin in Rat Trigeminal Ganglion Neurons. J Neurophysiol. 2003;89:1387–1401. doi: 10.1152/jn.00210.2002. [DOI] [PubMed] [Google Scholar]
- Liu L, Yang T, Simon SA. The protein tyrosine kinase inhibitor, genistein, decreases excitability of nociceptive neurons. Pain. 2004;112:131–141. doi: 10.1016/j.pain.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Lotan I, Dascal N, Naor Z, Boton R. Modulation of vertebrate brain Na+ and K+ channels by subtypes of protein kinase C. FEBS Lett. 1990;267:25–28. doi: 10.1016/0014-5793(90)80279-r. [DOI] [PubMed] [Google Scholar]
- Mason HS, Latten MJ, Godoy LD, Horowitz B, Kenyon JL. Modulation of Kv1.5 currents by protein kinase A, tyrosine kinase, and protein tyrosine phosphatase requires an intact cytoskeleton. Mol Pharmacol. 2002;61:285–293. doi: 10.1124/mol.61.2.285. [DOI] [PubMed] [Google Scholar]
- Mathie A, Wooltorton JR, Watkins CS. Voltage-activated potassium channels in mammalian neurons and their block by novel pharmacological agents. Gen Pharmacol. 1998;30:13–24. doi: 10.1016/s0306-3623(97)00034-7. [DOI] [PubMed] [Google Scholar]
- Matran R, Martling CR, Lundberg JM. Inhibition of cholinergic and non-adrenergic, non-cholinergic bronchoconstriction in the guinea pig mediated by neuropeptide Y and alpha 2-adrenoceptors and opiate receptors. Eur J Pharmacol. 1989;163:15–23. doi: 10.1016/0014-2999(89)90390-7. [DOI] [PubMed] [Google Scholar]
- Misonou H, Trimmer JS. Determinants of voltage-gated potassium channel surface expression and localization in Mammalian neurons. Crit Rev Biochem Mol Biol. 2004;39:125–145. doi: 10.1080/10409230490475417. [DOI] [PubMed] [Google Scholar]
- Moreno H, Vega-Saenz dM, Nadal MS, Amarillo Y, Rudy B. Modulation of Kv3 potassium channels expressed in CHO cells by a nitric oxide-activated phosphatase. J Physiol. 2001;530:345–358. doi: 10.1111/j.1469-7793.2001.0345k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Zhuang SY, Hampson RE, Deadwyler SA. Protein kinase-dependent phosphorylation and cannabinoid receptor modulation of potassium A current (IA) in cultured rat hippocampal neurons. Pflugers Arch. 2000;439:541–546. doi: 10.1007/s004249900231. [DOI] [PubMed] [Google Scholar]
- Ocana M, Cendan CM, Cobos EJ, Entrena JM, Baeyens JM. Potassium channels and pain: present realities and future opportunities. Eur J Pharmacol. 2004;500:203–219. doi: 10.1016/j.ejphar.2004.07.026. [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- Pan SJ, Zhu M, Raizada MK, Sumners C, Gelband CH. ANG II-mediated inhibition of neuronal delayed rectifier K+ current: role of protein kinase C-alpha. Am J Physiol Cell Physiol. 2001;281:C17–C23. doi: 10.1152/ajpcell.2001.281.1.C17. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci USA. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS. Voltage-gated potassium channels in sensory neurons. Nociceptive Membrane. 2006;57:323–351. [Google Scholar]
- Safronov BV, Bischoff U, Vogel W. Single voltage-gated K+ channels and their functions in small dorsal root ganglion neurones of rat. J Physiol. 1996;493:393–408. doi: 10.1113/jphysiol.1996.sp021391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saly V, Andrew RD. CA3 neuron excitation and epileptiform discharge are sensitive to osmolality. J Neurophysiol. 1993;69:2200–2208. doi: 10.1152/jn.1993.69.6.2200. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Baraban SC, Hochman DW. Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res. 1998;32:275–285. doi: 10.1016/s0920-1211(98)00058-8. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Yoshimura N, de Groat WC. KW-7158 [(2S)-(+)-3,3,3-trifluoro-2-hydroxy-2-methyl-N-(5,5,10-trioxo-4,10-dihydro thieno[3,2-c][1]benzothiepin-9-yl)propanamide] enhances A-type K+ currents in neurons of the dorsal root ganglion of the adult rat. J Pharmacol Exp Ther. 2004;310:159–168. doi: 10.1124/jpet.104.065409. [DOI] [PubMed] [Google Scholar]
- Sessle BJ. Neural mechanisms and pathways in craniofacial pain. Can J Neurol Sci. 1999;26:S7–S11. doi: 10.1017/s0317167100000135. [DOI] [PubMed] [Google Scholar]
- Slugg RM, Meyer RA, Campbell JN. Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. J Neurophysiol. 2000;83:2179–2191. doi: 10.1152/jn.2000.83.4.2179. [DOI] [PubMed] [Google Scholar]
- Stewart T, Beyak MJ, Vanner S. Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons. J Physiol. 2003;552:797–807. doi: 10.1113/jphysiol.2003.046409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature. 1988;336:379–381. doi: 10.1038/336379a0. [DOI] [PubMed] [Google Scholar]
- Sun C, Du J, Raizada MK, Sumners C. Modulation of delayed rectifier potassium current by angiotensin II in CATH.a cells. Biochem Biophys Res Commun. 2003;310:710–714. doi: 10.1016/j.bbrc.2003.09.069. [DOI] [PubMed] [Google Scholar]
- Sun H, Oudit GY, Ramirez RJ, Costantini D, Backx PH. The phosphoinositide 3-kinase inhibitor LY294002 enhances cardiac myocyte contractility via a direct inhibition of Ik, slow currents. Cardiovasc Res. 2004;62:509–520. doi: 10.1016/j.cardiores.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Ikeda M, Nasu M, Kadoi J, Yoshida S, Matsumoto S. Enhanced excitability of rat trigeminal root ganglion neurons via decrease in A-type potassium currents following temporomandibular joint inflammation. Neuroscience. 2006;138:621–630. doi: 10.1016/j.neuroscience.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M. Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem. 2004;279:35133–35138. doi: 10.1074/jbc.M406260200. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol. 1989;61:927–938. doi: 10.1152/jn.1989.61.5.927. [DOI] [PubMed] [Google Scholar]
- Viana F, de la PE, Pecson B, Schmidt RF, Belmonte C. Swelling-activated calcium signalling in cultured mouse primary sensory neurons. Eur J Neurosci. 2001;13:722–734. doi: 10.1046/j.0953-816x.2000.01441.x. [DOI] [PubMed] [Google Scholar]
- Voets T, Prenen J, Vriens J, Watanabe H, Janssens A, Wissenbach U, Bodding M, Droogmans G, Nilius B. Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem. 2002;277:33704–33710. doi: 10.1074/jbc.M204828200. [DOI] [PubMed] [Google Scholar]
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA. 2004;101:396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RL, Butler DM, Barish ME. Potassium current development and its linkage to membrane expansion during growth of cultured embryonic mouse hippocampal neurons: sensitivity to inhibitors of phosphatidylinositol 3-kinase and other protein kinases. J Neurosci. 1998;18:6261–6278. doi: 10.1523/JNEUROSCI.18-16-06261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Winston JH, Shenoy M, Yin H, Pasricha PJ. Enhanced excitability and suppression of A-type K+ current of pancreas-specific afferent neurons in a rat model of chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G424–G431. doi: 10.1152/ajpgi.00560.2005. [DOI] [PubMed] [Google Scholar]
- Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, de Groat WC. Increased excitability of afferent neurons innervating rat urinary bladder after chronic bladder inflammation. J Neurosci. 1999;19:4644–4653. doi: 10.1523/JNEUROSCI.19-11-04644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost CS. Potassium channels: basic aspects, functional roles, and medical significance. Anesthesiology. 1999;90:1186–1203. doi: 10.1097/00000542-199904000-00035. [DOI] [PubMed] [Google Scholar]