

Summary
Amyotrophic lateral sclerosis (ALS) is primarily a motor neuron disorder. Intriguingly, early muscle denervation preceding motor neuron loss is observed in mouse models of ALS. Enhanced muscle vulnerability to denervation process has been suggested by accelerated muscle deterioration following peripheral nerve injury in an ALS mouse model. Here we provide evidence of biochemical changes in the hindlimb muscle of young, presymptomatic G93A hSOD1 transgenic mice. In this report, we demonstrate that cdk5 activity is reduced in hindlimb muscle of 27-day-old G93A hSOD1 transgenic mice. In vitro analysis revealed mutant hSOD1-mediated suppression of cdk5 activity. Furthermore, the decrease in muscle cdk5 activity was accompanied by a significant reduction in MyoD and cyclin D1 levels. These early muscle changes raise the possibility that the progressive deterioration of muscle function is potentiated by altered muscle biochemistry in these mice at a very young, presymptomatic age.
Keywords: amyotrophic lateral sclerosis, ALS, cyclin-dependent kinase 5, cdk5, G93A hSOD1, transgenic mice, muscle, MyoD, cyclin D1, presymptomatic
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder affecting the upper and lower motor neurons. Motor neuron degeneration is accompanied by muscle weakness and paralysis with an inevitably fatal outcome. Approximately 90% of ALS cases are sporadic, with mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene accounting for 20% of the familial cases [1–3]. Overexpression of various mutant isoforms of human SOD1 (hSOD1) in mice simulates the major pathological hallmarks of ALS, confirming the significant role of mutant hSOD1 in ALS pathology, and resulting in one of the most convincing mouse models of a human disease [4–6].
Accumulating evidence highlights the influence of non-neuronal neighboring cells and local microenvironment on motor neuron degeneration [7]. A study examining chimeric mice that are mixtures of normal and mutant hSOD1-expressing cells has shown that neighboring wildtype non-neuronal cells are capable of rescuing motor neurons expressing mutant hSOD1 [8]. Recently, the deleterious influence of mutant hSOD1 expressing astrocyte and microglia on motor neuron survival have been reported [9–11]. In contrast, siRNA-mediated knock down and Cre recombinase-mediated knockout of mutant hSOD1 in the skeletal muscles of transgenic mice did not affect the disease progression, leading the authors to conclude that the muscle does not play a primary role in mutant hSOD1 mediated motor neuron toxicity [12]. However, even without a direct involvement in mutant hSOD1-mediated motor neuron toxicity, could there be an underlying muscle condition that contributes to and/or enhances the muscle function deterioration resulting from denervation in these mice? Intact muscle and nerve connection is important for the muscle and motor neuron function and their survival [13]. It is unclear whether there is an underlying intrinsic muscle impairment that contributes to the muscle function loss in ALS.
While a significant loss of motor neurons is not observed until approximately 90 days of age in G93A hSOD1 transgenic mice (G93A hSOD1 mice) [14], denervation of neuromuscular junctions is detected as early as 47 days of age [15–19]. Interestingly, Bax deletion-mediated rescue of motor neurons in G93A hSOD1 mice fails to prevent this early neuromuscular denervation [20] suggesting that the denervation in these animals precedes and is independent of motor neuron loss. Additionally, peripheral nerve injury results in an accelerated appearance of disease symptoms in hindlimb muscle of G93A hSOD1 mice, suggestive of increased muscle vulnerability in response to denervation in this model [21].
The potential role of muscle in ALS is highlighted by the therapeutic benefit of muscle-targeted expression of either IGF-1 or GDNF in G93A hSOD1 mice, which significantly delays disease onset and progression [22–24]. IGF-1 is a classic mitogen and its muscle expression has been shown to facilitate muscle and motor neuron regeneration [25] and muscle hypertrophy [26]. The mechanism underlying disease amelioration mediated by muscle-targeted IGF-1 in the ALS mouse model is not known. Although the muscle mass increase resulting from myostatin inhibition treatment initiated at 28 days of age did not have an effect on animal survival, the treatment led to increased motor neuron survival around 84–90 days of age and delayed muscle atrophy in G93A hSOD1 mice [27]. On the other hand, induction of muscle hypertrophy via virally-mediated overexpression of follistatin in the hindlimbs of G93A hSOD1 mice improved muscle strength but did not improve rotarod performance [12].
To determine whether early biochemical changes are present in the muscle of G93A hSOD1 mice, we examined cyclin dependent kinase 5 (cdk5) activity in triceps surae samples from young, presymptomatic mice. Cdk5 is a proline-directed serine/threonine kinase that is involved in numerous cellular functions in the central nervous system [28, 29]. It is also implicated in various aspects of muscle function such as acetylcholine receptor clustering [30–32] and myogenesis [33, 34]. Cdk5 activity is also upregulated in the muscle following denervation [35].
Here we report decreased cdk5 activity in the hindlimb muscle of 27-day-old G93A hSOD1 mice. This decreased activity was accompanied by increased interaction of cdk5 with mutant hSOD1 in the muscle. In vitro analysis revealed that the presence of mutant hSOD1 is sufficient for eliciting the suppression of cdk5 activity. Further analysis of the muscle revealed a concomitant decrease in both MyoD and cyclin D1 levels. These results suggest the presence of altered muscle function in very young, presymptomatic G93A hSOD1 mice.
Materials and Methods
Mice
G93A hSOD1 mice, wildtype hSOD1 trasgenic mice (WT hSOD1 mice), and non-transgenic mice (Non-Tg mice) were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice were maintained on a 12 hour light/dark cycle with free access to food and water. Genotyping was performed using genomic DNA, as suggested by the Jackson Laboratory genotyping protocol. Mice were killed by decapitation at various ages and spinal cord and triceps surae were quickly dissected. Dissected samples were either flash frozen for biochemical analysis or immersion fixed in 4% paraformaldehyde for immunohistological analysis. The following numbers of animals were used for the study: G93A mice at 27 days (n=7), 47 days (n=3), 67 days (n=3) of age; non-transgenic age-matched controls at 27 days (n=7), 47 days (n=3), 67 days (n=3) of age; wildtype hSOD1 overexpressing transgenic mice at 27 days (n=5) of age. All animal protocols were approved by the Animal Care and Use Committee at the University of Washington (where the mice were generated and initial experiments performed) and the Animal Care Committee at the University of British Columbia.
Antibodies and Reagents
Antibodies for cdk5 (J-3 and DC-17), SOD1 (G-11), cyclin D1 (DCS-6), MyoD (M-318), p35 (c-19), PCNA (PC10), and cyclin B1 (GNS1) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody for SOD1 (NCL-SOD1) was obtained from Vector Laboratory (Burlingame, CA). Isotype-specific secondary antibodies conjugated to HRP or biotin, and streptavidin-HRP were from Southern Biotechnology (Birmingham, AL); and Streptavdin-Alexa 488 and 594 were from Invitrogen (Carlsbad, CA).
Immunofluorescence
Immunofluorescence was performed on frozen muscle sample sections. Briefly, dissected muscle samples were immersion-fixed for 20 minutes at 4°C in 4% paraformaldehyde, embedded in Tissue-Tek O.C.T. compound (Sakura Finetek USA, Torrance, CA) and stored in −80°C until processing. Tissue blocks were cut at 10μm thickness on a cryostat, mounted on glass slides, and stored at −80°C. Indirect double immunofluorescence staining on sections was performed as follows. Briefly, sections were thawed and the area around the section was outlined using PAP pen (Research Products International, Mount Prospect, IL). Sections were permeabilized using 0.25% Triton X-100 in TBS, and blocked in 5% non-fat dry milk. Sections were incubated overnight at 4°C with the first primary antibody, followed by a one-hour incubation in biotin-conjugated secondary antibody at room temperature. The antigen was visualized by using streptavidin conjugated with Alexa Fluor. This process was repeated for the second antibody. Co-localization was determined using three different fluorescents (Alexa 488, Alexa 594, and DAPI). Images were acquired using an Axioplan2 fluorescence microscope (Carl Zeiss Canada Ltd., Toronto, ON) equipped with a Cool Snap HQ CCD camera (Photometrics, Tucson, AZ) and interfaced with a Dimension E520 computer (Dell Canada Inc., North York, ON) using Metamorph image acquisition software (Version 6.1, Molecular Devices, Downingtown, PA). The figures were assembled using Photoshop (Version 7.0.1, Adobe Canada, Ottawa, ON).
Immunoblotting and Immunoprecipitation
Western blotting was performed as reported [36] using supernatants from spinal cord and triceps surae homogenates. In all experiments, protein levels were normalized to levels of actin. For immunoprecipitations, supernatants from spinal cord and triceps surae homogenates (100μg and 400μg, respectively) were incubated with 1.2μg of cdk5 antibody (J-3) overnight at 4°C and incubated with protein G-sepharose at 4 °C for one hour. After removing the supernatants and washing three times with TBS, samples were electrophoresed, transferred to nitrocellulose (Protran, Schleicher and Schuell, Dassel, Germany), and subjected to immunoblotting. Band density was analyzed using Quantity One software (Biorad, Mississauga, ON).
Cdk5 Immunoprecipitation Kinase Activity Assay
Immunoprecipitation kinase activity assays were performed as described previously for cdk5 [37], with minor changes. Following protein G-sepharose incubation, samples were washed twice with 700μl of cold TBS and once with 700μl of cold kinase buffer. A reaction mix containing HEPES kinase buffer, 10μM ATP, 10μg of histone H1, and 0.5μCi of γ-32P-ATP was added to the samples and incubated at room temperature for 30 minutes. Reactions were halted by adding loading buffer, and samples were electrophoresed on 12% SDS-PAGE gels. Histone bands were visualized by Coomassie blue staining the gels, after which they were dried and apposed to a Kodak Biomax MS X-ray film. Band density was analyzed using Quantity One software (Biorad).
Large Scale Immunoisolation of Mutant and Wildtype Human SOD1
Large scale immunoisolation of mutant and wildtype hSOD1 was performed on brain lysate supernatants from G93A and wildtype hSOD1 transgenic samples using hSOD1 antibody (G-11) covalently coupled to protein G support using a Seize X protein G immunoprecipitation kit (Pierce, Rockford, IL). Eluted antigen was concentrated using microcon centrifugal filter devices (30kDa, Millipore, Bedford, MA). Protein concentrations of concentrated immunoislates were determined using a Qubit fluorometer (Invitrogen). Predetermined amounts of immunoisolate were added to 400μg of triceps surae lysate supernatants from non-transgenic samples. Following incubation for one hour at 4ºC, immunoprecipitation kinase activity assays were performed as described above.
Statistical Analysis
One-way and two-way ANOVA with least significant difference (LSD) post hoc test was performed using Statistica (1999 Edition for Windows©, StatSoft, Inc., Tulsa, OK). Unpaired Student’s t-test was performed using Excel (2003 edition for Windows©, Microsoft©, Redmond, WA).
Results
Reduced muscle cdk5 activity in very young presymptomatic G93A hSOD1 mice
Neuromuscular denervation is observed in G93A hSOD1 mice as early as 47 days of age [16]. Also, peripheral nerve injury in these mice accelerates muscle function deterioration [21]. It has been shown that muscle denervation resulting from nerve injury is accompanied by upregulation of p35 expression and muscle cdk5 activity [35]. p35 is a regulatory subunit of cdk5 [38]. To determine whether the early muscle denervation process in G93A hSOD1 mice is reflected in muscle cdk5 activity, we examined muscle cdk5 activity in G93A hSOD1 and non-transgenic mice at early at three pre-symptomatic time points: 27, 47, and 67 days of age.
Cdk5 immunoprecipitation kinase assay on triceps surae samples from non-transgenic mice showed higher cdk5 activity at 27- and 47-day time points compared to the 67-day time point (One-way ANOVA, F2,7=6.65, p=0.02; Multiple comparison post-hoc analysis (LSD test) 27- vs. 67-day [a vs c], p=0.05; 47- vs. 67-day [b vs. c], p=0.01), but such transient changes were not observed in the G93A-SOD1 mice samples (Fig. 1). Two-way ANOVA analysis of cdk5 activities observed in muscle samples from 27- and 47-day-old G93A hSOD1 and non-transgenic mice revealed a statistically significant genotype effect; wherein the muscle samples from G93A hSOD1 mice displayed significantly lower cdk5 activity compared to those from non-transgenic mice at these ages (Fig. 2A, F1,10=10.11, p=0.009). Multiple comparison post-hoc analysis showed a 23% and 20% decrease in muscle cdk5 activity for 27- and 47-day-old G93A hSOD1 mice, respectively (27-day-old mice: p=0.04; 47-day-old mice: p=0.04). Two-way ANOVA analysis on the levels of cdk5 and p35 in these muscle samples did not show any statistical significance (Fig. 2B and 2C) suggesting that diminished cdk5 activity in G93A hSOD1 mice at these ages are not due to altered levels of either cdk5 or p35.
Figure 1. Age-dependent changes in cdk5 activity in triceps surae is absent in G93A hSOD1 mice.
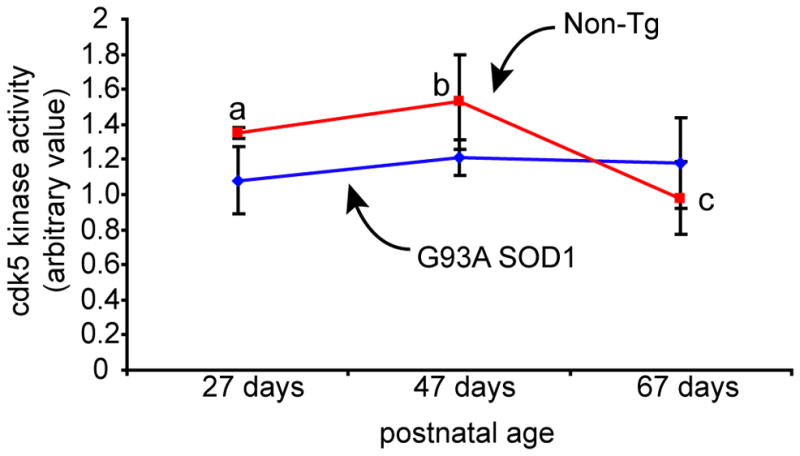
Cdk5 activity in triceps surae samples from G93A hSOD1 and Non-Tg littermates were examined at three different ages: 27 days, 47 days, and 67 days. A statistically significant age-dependent change in cdk5 activity was observed in the muscle samples from Non-Tg littermates (One-way ANOVA, F2,7=6.65, p=0.02). Multiple comparison post-hoc analysis showed higher cdk5 activity in 27-day-old and 47-day-old mice compared to 67-day-old mice (27 days vs. 67 days [a vs. c], p=0.03; 47 days vs. 67 days [b vs. c], p=0.009, least square difference test). Such transient change in cdk5 activity was not detected in the muscle samples from G93A hSOD1 at the ages tested (F2,7=0.454, p=0.65). The values in graphs are mean±SD.
Figure 2. Genotype-dependent changes in cdk5 activity are observed in the absence of changes in cdk5 and p35 protein levels.
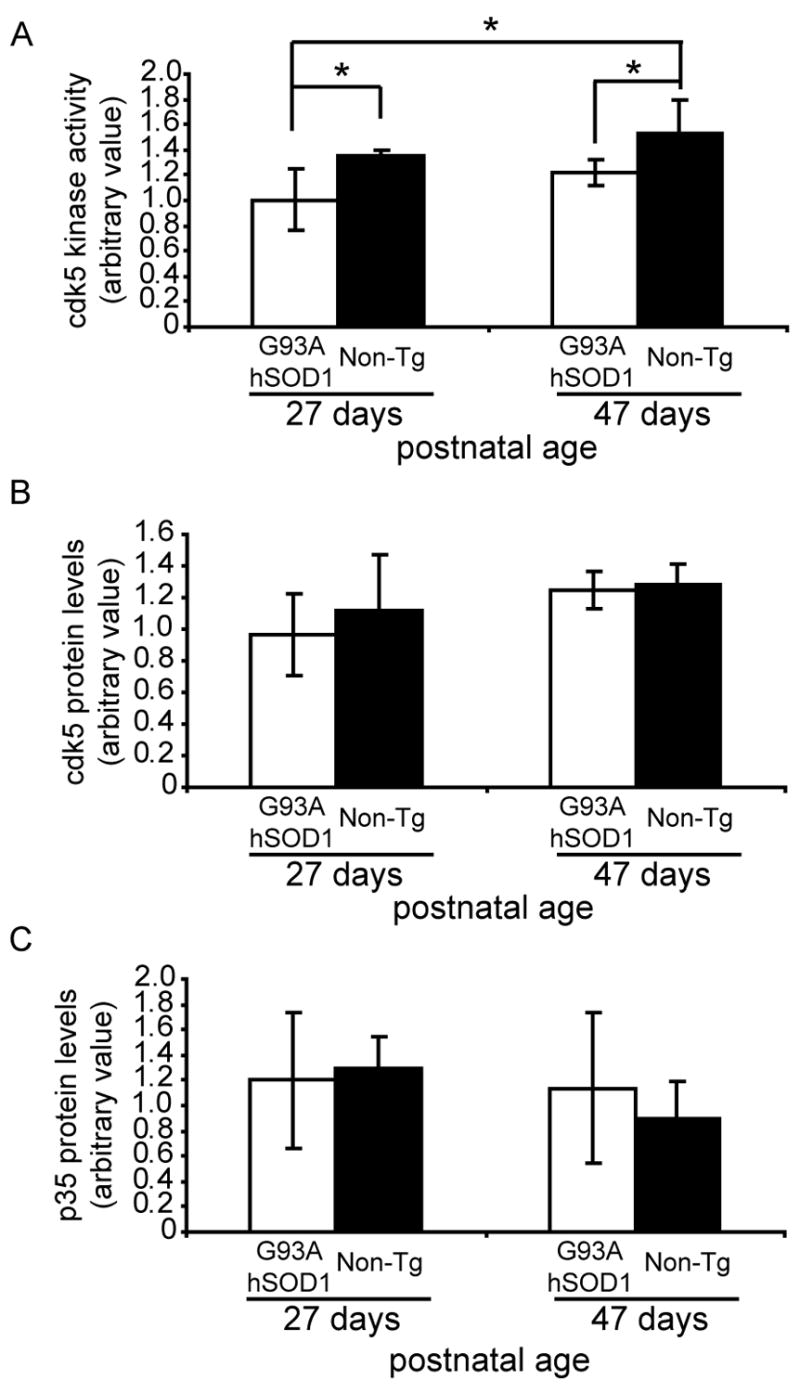
(A) Two-way ANOVA analysis of hindlimb muscle samples from 27- and 47-day-old G93A hSOD1 and Non-Tg mice showed significant reduction in cdk5 activity in samples from G93A hSOD1 mice compared to age-matched samples from Non-Tg mice (F1,10=10.11, p=0.009). Multiple comparison post hoc analysis showed statistically significant reduction in the cdk5 activity in samples from G93A hSOD1 mice compared to Non-Tg mice at both 27 and 47 days of age (27-day-old animal comparison: G93A hSOD1 vs. Non-Tg: p=0.04; 47-day-old animal comparison: G93A hSOD1 vs. Non-Tg; p=0.04; least-significant-difference test). Cdk5 activity in samples from 27-day-old G93A hSOD1 mice also showed significant decrease compared to samples from 47-day-old Non-Tg mice (p=0.006; least significant difference test). In contrast, no deficits were observed in cdk5 (B) and p35 (C) protein levels. The values in graphs are mean±SD.
Mutant G93A hSOD1 suppresses muscle cdk5 activity in vitro
The significant genotype effect on cdk5 activity at early ages suggests that mutant G93A hSOD1 has a role in the decreased muscle cdk5 activity in G93A hSOD1 mice. A recent study has shown that mutant huntingtin protein interacts with cdk5, resulting in the suppression of cdk5 activity [39]. Based on this finding, we surmised that the suppression of muscle cdk5 activity in G93A hSOD1 mice may arise from an abnormal interaction between cdk5 and mutant G93A hSOD1. However, we could not rule out the possibility that an aphysiological interaction between cdk5 and mutant G93A hSOD1 may be observed as a result of human protein overexpression. In order to distinguish between the mutant protein effect and protein overexpression effect, we used WT hSOD1 mice, which lack any pathological phenotype, as a control.
Examination of the triceps surae samples from 27-day-old G93A and WT hSOD1 mice showed that more mutant G93A than wildtype hSOD1 co-immunoprecipitated with cdk5 (Fig. 3A; p=0.05). To determine whether this interaction is tissue specific, we examined spinal cord samples obtained from the same animals. When analyzed, there was no difference between the amounts of mutant G93A and wildtype hSOD1 co-immunoprecipitated with cdk5 in the spinal cord samples (Fig. 3A). The increased amount of mutant G93A hSOD1 pulled down with cdk5 in muscle samples is likely to be a result of a specific interaction with the mutant protein, as the overall expression levels of the human transgene was lower in the G93A hSOD1 compared to WT hSOD1 muscle samples (Fig. 3B).
Figure 3. Mutant G93A hSOD1 suppresses cdk5 activity in muscle lysates.

Cdk5 interaction with either mutant G93A or wildtype (WT) hSOD1 was examined in supernatants from muscle and spinal cord lysate samples from 27-day-old G93A and WT hSOD1 transgenic mice. (A) More G93A hSOD1, compared to WT hSOD1, co-immunoprecipitated with cdk5 in the muscle lysates (two-tailed Student’s t test; p=0.05). No increase in the interaction between mutant G93A hSOD1 and cdk5 was observed in spinal cord samples. (B) The greater interaction between mutant G93A hSOD1 and cdk5 is not a consequence of higher protein expression. In contrast, the hindlimb muscle lysates from WT hSOD1 mice showed higher hSOD1 levels than those from G93A hSOD1 mice, suggesting higher expression level for WT hSOD1. (C) Examination of cdk5 activity in the same samples corresponding to (A) showed decreased cdk5 activity in G93A hSOD1 muscle samples compared to WT hSOD1 muscle samples (two-tailed Student’s t-test; p=0.001). Again, no difference in the cdk5 activities was observed between the spinal cord samples from G93A hSOD1 and WT hSOD1 mice. (D) To determine whether the presence of mutant G93A hSOD1 is responsible for the reduced cdk5 activity, immunoprecipitates of either mutant G93A or wildtype hSOD1 was added to the supernatants of Non-Tg mouse muscle lysates prior to the immunopreciptation kinase assay. Immunoprecipitation kinase assay following the incubation with mutant G93A hSOD1 immunoisolated from brain resulted in a concentration dependent reduction of cdk5 activity (0 μg, n=6; 0.1 μg, n=1; 0.5 μg, n=2; 1 μg, n=2; 3 μg, n=3). Addition of 3 μg of WT hSOD1 (n=3) immunoisolated from brain to the supernatants of Non-Tg mouse muscle lysates did not result in a reduction of the cdk5 activity (One-way ANOVA, F5,11=6.06, p=0.006, post hoc multiple comparison test, * p<0.05). The values in graphs are mean±SD.
We then examined the cdk5 activity in these samples to determine whether the amount of mutant hSOD1 interaction with cdk5 had an effect on the kinase activity. Interestingly, the amount of mutant G93A and wildtype hSOD1 pulled down by cdk5 was inversely correlated to the cdk5 activity in triceps surae. Muscle cdk5 activity in the G93A hSOD1 mice was 21% less than that of the WT hSOD1 mice (Fig. 3C; p=0.001). No such difference in the cdk5 activity between the two groups was detected in the spinal cord samples (Fig. 3C).
To directly test whether the decreased cdk5 activity in the muscle sample was indeed due to the presence of mutant hSOD1, we introduced either the mutant G93A or WT hSOD1 to lysates of triceps surae samples from non-transgenic animals. Mutant G93A and WT hSOD1 were immunoisolated from brains using a monoclonal hSOD1 antibody (G-11). Muscle tissue was not used for immunoisolation as a precaution against potential muscle-specific co-factors that may co-immunoprecipitate with immunoisolates and potentially confound the results.
The addition of mutant G93A hSOD1 immunoisolate to non-transgenic triceps surae lysate resulted in a concentration-dependent reduction in the cdk5 activity (Fig. 3D, one-way ANOVA, F5,11=6.8, p=0.006). We observed 30% decrease in cdk5 activity with the addition of 3μg of mutant G93A hSOD1 immunoisolate (Fig. 3D, p=0.05, post hoc LSD multiple comparison test). In contrast, the addition of 3μg of wildtype hSOD1 immunoisolate to the lysate did not result in suppression of cdk5 activity (Fig. 3D). These results suggest that the presence of mutant G93A, but not WT, hSOD1 leads to the suppression of endogenous cdk5 activity in triceps surae.
MyoD and cyclin D1 levels are reduced in G93A hSOD1 mouse triceps surae
Although much of what we understand regarding cdk5 function is derived from its role in the central nervous system, it has also been shown to be associated with various muscle functions. In Xenopus, it is involved in the regulation of muscle development [34]. In rats, cdk5 activity is upregulated in the hindlimb muscle following nerve injury [35]. In C2 myoblasts, cdk5 activity is associated with, and positively modulates, myogenic differentiation [33].
In order to determine whether the diminished cdk5 activity in triceps surae of G93A hSOD1 mice reflects a potential myogenic dysfunction in these animals, we examined the levels of MyoD. MyoD is a well-defined regulatory factor involved in myogenic determination and differentiation [40, 41]. A comparison of muscle samples from 27-day-old G93A hSOD1 and Non-Tg mice using Western immunoblot assay revealed 61% less MyoD in G93A hSOD1 samples (Fig. 4A, p=0.001, two-tailed t-test). The decreased MyoD levels in G93A hSOD1 mice seem to suggest altered myogenic function in 27-day-old G93A hSOD1 mice.
Figure 4. Myogenic and G1 phase cell cycle markers are altered in the muscle of G93A hSOD1 animals.

Protein levels of a myogenic marker and various cell cycle regulators were examined in the muscle samples of 27-day-old G93A hSOD1 and non-transgenic (Non-Tg) mice. Protein levels for (A) MyoD and (B) cyclin D1, a G1 phase cell-cycle regulator, were significantly lower in the muscle samples from G93A hSOD1 compared to those from Non-Tg mice (two-tailed Student’s t-test, p=0.001 and p=0.01, respectively). We did not detect any differences in the protein levels for (C) cdk4, another G1 phase cell-cycle marker, (D) PCNA, an S phase cell-cycle regulator and (E) cyclin B1, a G2/M phase cell-cycle regulator, in the muscle samples between the G93A hSOD1 and Non-Tg mice. The values in graphs are mean±SD.
Since myogenesis in vivo involves proliferation of satellite cells, leading to their fusion onto mature myofibers [42, 43], we also examined the levels of various cell proliferation markers. The level of cyclin D1, a marker for the G1 phase of the cell cycle, was 38% lower in the G93A hSOD1 muscle (Fig. 4B; p=0.01, two-tailed t-test). Despite the significantly reduced level of cyclin D1, the levels of cdk4 (Fig. 4C), PCNA (Fig. 4D), and cyclin B1 (Fig. 4E) in G93A hSOD1 mice were not statistically different from the levels observed in non-transgenic mice. Additionally, we did not detect any phospho-Rb in either the G93A hSOD1 or non-transgenic muscle samples (data not shown). The lack of changes in the levels for other protein markers of G1, S, and G2/M phases seems to suggest that the alteration of the cyclin D1 levels in the muscle of G93A hSOD1 mice may reflect a cell-cycle independent event.
To better evaluate the association of cdk5 and mutant hSOD1 in situ, and in the context of seemingly altered myogenic function, we performed double immunofluorescence staining for cdk5 and mutant hSOD1 in muscle sections from G93A hSOD1 mice. A distinct nuclear localization of cdk5 was detected via immunofluorescence in muscle fibers (Fig. 5A). Mutant hSOD1 also showed nuclear localization, but at a much reduced frequency relative to cdk5 (Fig. 5A). However, some of the mutant hSOD1 staining showed clear co-localization with cdk5 in nuclei (Fig. 5A; white arrows). We also noted significant differences in the distribution of DAPI stained nuclei between G93A hSOD1 and non-transgenic muscle fibers. Although a highly organized patterning of nuclei was observed in muscle sections from non-transgenic mice (Fig. 5B), the G93A hSOD1 mice muscle sections were characterized by a distribution of nuclei that was clustered and disorganized (Fig. 5B).
Figure 5. Nuclear co-localization of cdk5 and mutant G93A hSOD1 in muscle sections.

In vitro analysis of muscle samples revealed a mutant hSOD1 interaction-mediated reduction in cdk5 activity. Cdk5 and mutant hSOD1 localization were examined in muscle sections to determine whether the observed interactions are detectable in situ. (A) Nuclear staining of cdk5 is more widespread than that of mutant hSOD1. Only a subpopulation of cdk5 positive nuclei showed mutant hSOD1 immunostaining. Triple immunofluorescence shows distinct co-localization of cdk5 and mutant hSOD1 in the myonuclei. (B) Muscle fibers in G93A hSOD1 mice show aberrant nuclear patterning compared to those from Non-Tg mice. Nuclear patterning in the G93A hSOD1 mice muscle was clustered and disorganized in appearance compared to non-transgenic mice as shown by DAPI staining. Scale bar = 50 μm.
Discussion
In this report, we have highlighted biochemical changes in the hindlimb muscle of 27-day-old G93A hSOD1 mice in comparison to similar muscle samples from age matched WT hSOD1 and Non-Tg mice. Our results show reduced cdk5 activity in hindlimb muscle of 27- and 47-day-old G93A hSOD1 mice compared to their Non-Tg littermates. Comparison of hindlimb muscle from 27-day-old G93A and WT hSOD1 mice using a co-immunoprecipitation assay revealed a greater protein-protein interaction between cdk5 and mutant G93A hSOD1. In vitro, a concentration-dependent decrease in cdk5 activity mediated by mutant hSOD1 was observed. Furthermore, MyoD and cyclin D1 levels were lower in hindlimbs of 27-day-old G93A hSOD1 mice compared to age-matched Non-Tg mice.
Past studies have shown evidence of early neuromuscular changes in G93A hSOD1 mice that precede motor neuron loss. Muscle denervation has been observed in these animals as early as 47 days of age [15, 16, 18]. Electrophysiological examination has revealed decreases in muscle contractile force and functional motor units as early as 40 days of age [18, 44]. Although axonal sprouting has been reported in these mice [16, 18], contractile force decline in coordination with the decline in motor unit numbers in G93A hSOD1 mice suggests no functional compensation by sprouting [44]. Additionally, altered muscle function in G93A hSOD1 mice is suggested by a study showing accelerated muscle function deficit following peripheral nerve injury [21].
Nerve injury in normal muscle results in the activation and proliferation of satellite cells. Denervation of normal muscle results in the upregulation of MyoD in muscle fiber nuclei and satellite cells [45–47]. In transgenic mice displaying localized IGF-1 overexpression in skeletal muscle, nerve injury results in an enhancement of satellite cell activation accompanied by MyoD and cyclin D1 upregulation, and enhanced muscle reinnervation [25]. Interestingly, muscle targeted IGF-1 expression in G93A hSOD1 mice significantly delayed disease onset and progression [22, 24]. On the other hand, myostatin inhibition in G93A hSOD1 slowed muscle atrophy but did not delay disease progression or rescue motor neurons [27]. Myostatin is known to induce cyclin D1 degradation [48] and myostatin inhibition leads to upregulation in MyoD [49] in muscle. Thus, the evidence suggests that diminished MyoD and cyclin D1 levels in muscle are unlikely to play a primary role in ALS, but may have an impact on the deterioration of muscle function associated with the disease.
Another change that was observed in our study is the diminished muscle cdk5 activity in G93A hSOD1 mice. Although the functional role of cdk5 in the muscle is limited in knowledge compared to its role in the central nervous system [28, 29], cdk5 activity has been shown to be upregulated during myofiber formation in C2 murine cells [33]. Accordingly, dominant negative cdk5 expression in these cells inhibits myoblast differentiation [33]. In Xenopus embryos, dominant negative cdk5 expression results in aberrant somitic muscle patterning, which is accompanied by reduced MyoD expression [34]. Cdk5 activity has also been shown to be upregulated in muscle following nerve injury [35]. Thus, currently available research evidence of cdk5’s role in muscle suggests that it is involved in muscle regeneration and repair mechanisms; diminished cdk5 activity at a very young, presymptomatic age may result in an inappropriate muscle response to progressive muscle denervation taking place in these mice. On the other hand, p35 null/G93A hSOD1 double mutant mice did not present with an enhanced disease phenotype [50]. It may be that in the muscle of G93A hSOD1 mice, the additional reduction in cdk5 activity mediated by p35 knock-out does not contribute further to the dysfunction. Normally, cdk5 activity is much higher in spinal cord compared to muscle. In our study we observed decreased cdk5 activity in the muscle but not in the spinal cord of G93A hSOD1 mice. In the study by Takahashi and Kulkarni, only the cdk5 activity in the spinal cord samples was measured [50] so we can only speculate about the additional reduction of muscle cdk5 activity in these double mutant mice. Perhaps the lack of effect in p35 null/G93A hSOD1 double mutant mice may be due to a floor effect of cdk5 activity in muscle. Additional p35 knock-out-mediated reduction of already lower cdk5 activity in muscle of G93A mice may not hasten disease progression.
Our in vitro analysis showed that the presence of mutant hSOD1 was sufficient to reduce the cdk5 activity in the muscle lysate in a concentration-dependent manner. However, a recent study by Miller and colleagues showed that neither siRNA-mediated knock down nor Cre recombinase-mediated knockout of mutant hSOD1 had any beneficial effect on the disease progression in these animals [12]. The lack of an effect in their study could be due the fact that the partial knock-down of mutant hSOD1 is insufficient in reducing the deleterious actions of the mutant protein in the muscle. Also, Cre recombinase-mediated knockout in their study targeted only a subset of cells in the muscle leaving open the possibility that the remaining cells in the muscle may mediate some the mutant hSOD1 toxicity. Even if the presence of mutant hSOD1 in the muscle does not directly contribute to the disease progression, we speculate that postnatal accumulation of mutant hSOD1 protein in muscle [51] may inihibit cdk5 activity, which may in turn contribute to the acceleration of muscle function deterioration in these mice following peripheral nerve injury [21].
To our knowledge these are the earliest observed biochemical changes in the hindlimb muscle of G93A hSOD1 mice. Diminished levels of MyoD, cyclin D1 and cdk5 activity in the muscle suggest that the muscle regenerative mechanisms may be defective in these animals. Although the recent evidence suggests that the muscle does not play a primary role in mediating mutant hSOD1 toxicity, the deterioration of muscle function in response to denervation in these animals may be enhanced by the early alteration of this intrinsic muscle property. Through a deeper understanding of the mechanisms underlying these biochemical changes and their impact on the disease, it may be possible to delay the deterioration of muscle function in the affected individuals and contribute positively to their quality of life.
Acknowledgments
We thank R. Biasell (University of Washington) and B. Cai (University of British Columbia) for maintaining the mouse colony and Dr. Blair Leavitt, Scott Neal and Judy Park for helpful comments on the manuscript. This work was supported by grants from NIH (AG12721:I.Vincent., and T32 00057: K.H.J. Park, P. Rabinovitch, PI) and the Muscular Dystrophy Association (I.Vincent).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 2.Mulder DW, Kruland LT, Offord KP, Beard CM. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology. 1986;36 doi: 10.1212/wnl.36.4.511. [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4.Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- 5.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide disumtase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 7.Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their non-neuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 8.Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 9.Boillée S, Yamanaka K, Lobsinger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 10.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nature neuroscience. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao Q, Zhao W, Beers DR, Yen AA, Xie W, Henkel JS, Appel SH. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem. 2007;102:2008–2019. doi: 10.1111/j.1471-4159.2007.04677.x. [DOI] [PubMed] [Google Scholar]
- 12.Miller TM, Kim SH, Yamanaka K, Hester M, Umapathi P, Arnson H, Rizo L, Mendell JR, Gage FH, Cleveland DW, Kaspar BK. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19546–19551. doi: 10.1073/pnas.0609411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Musarò A, Dobrowolny G, Rosenthal N. The neuroprotective effects of a locally acting IGF-1 isoform. Exp Gerontol. 2007;42:76–80. doi: 10.1016/j.exger.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- 15.Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nature neuroscience. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- 16.Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Experimental Neurology. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: an electromyographic study. Neuroreport. 1996;7:1427–1431. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- 19.Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. The Journal of comparative neurology. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- 20.Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp PS, Dick JR, Greensmith L. The effect of peripheral nerve injury on disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neuroscience. 2005;130:897–910. doi: 10.1016/j.neuroscience.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 22.Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musarò A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. JCB. 2005;168:193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Brakefield D, Pan Y, Hunter D, Myckatyn TM, Parsadanian A. Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Experimental Neurology. 2007;203:457–471. doi: 10.1016/j.expneurol.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 24.Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 25.Rabinovsky ED, Gelir E, Gelir S, Lui H, Kattash M, DeMayo FJ, Shenaq SM, Schwartz RJ. Targeted expression of IGF-1 transgene to skeletal muscle accelerates muscle and motor neuron regeneration. FASEB J. 2003;17:53–55. doi: 10.1096/fj.02-0183fje. [DOI] [PubMed] [Google Scholar]
- 26.Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27:195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 27.Holzbaur EL, Howland DS, Weber N, Wallace K, She Y, Kwak S, Tchistiakova LA, Murphy E, Hinson J, Karim R, Tan XY, Kelley P, McGill KC, Williams G, Hobbs C, Doherty P, Zaleska MM, Pangalos MN, Walsh FS. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiology of disease. 2006;23:697–707. doi: 10.1016/j.nbd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Xie Z, Samuels BA, Tsai LH. Cyclin-dependent kinase 5 permits efficient cytoskeletal remodeling -- a hypothesis on neuronal migration. Cereb Cortex. 2006;16:i64–i68. doi: 10.1093/cercor/bhj170. [DOI] [PubMed] [Google Scholar]
- 29.Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 30.Fu AK, Fu WY, Cheung J, Tsim KW, Ip FC, Wang JH, Ip NY. Cdk5 is involved in neuregulin-induced AChR expression at the neuromuscular junction. Nature neuroscience. 2001;4:374–381. doi: 10.1038/86019. [DOI] [PubMed] [Google Scholar]
- 31.Fu AK, Ip FC, Fu WY, Cheung J, Wang JH, Yung WH, Ip NY. Aberrant motor axon projection, acetylcholine receptor clustering, and neurotransmission in cyclin-dependent kinase 5 null mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15224–15229. doi: 10.1073/pnas.0507678102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin W, Dominguez B, Yang J, Aryal P, Brandon EP, Gage FH, Lee KF. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron. 2005;46:569–579. doi: 10.1016/j.neuron.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 33.Lazaro JB, Kitzmann M, Poul MA, Vandromme M, Lamb NJC, Fernandez A. Cyclin dependent kinase 5, cdk5, is a positive regulator of myogenesis in mouse C2 cells. J Cell Sci. 1997;110:1251–1260. doi: 10.1242/jcs.110.10.1251. [DOI] [PubMed] [Google Scholar]
- 34.Philpott A, Porro EB, Kirschner MW, Tsai LH. The role of cyclin-dependent kinase 5 and a novel regulatory subunit in regulating muscle differentiation and patterning. Genes Dev. 1997;11:1409–1421. doi: 10.1101/gad.11.11.1409. [DOI] [PubMed] [Google Scholar]
- 35.Fu WY, Fu AK, Lok KC, Ip FC, Ip NY. Induction of Cdk5 activity in rat skeletal muscle after nerve injury. Neuroreport. 2002;13:243–247. doi: 10.1097/00001756-200202110-00014. [DOI] [PubMed] [Google Scholar]
- 36.Hallows JL, Chen K, DePinho RA, Vincent I. Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci. 2003;23:10633–10644. doi: 10.1523/JNEUROSCI.23-33-10633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci. 2002;22:6515–6525. doi: 10.1523/JNEUROSCI.22-15-06515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 39.Luo S, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implication for mutant huntingtin toxicity. JCB. 2005;169:647–656. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Megeney L, Rudnicki MA. Determination versus differentation and the MyoD family of transcription factors. Biochem Cell Biol. 1995;73:723–732. doi: 10.1139/o95-080. [DOI] [PubMed] [Google Scholar]
- 41.Perry RL, Rudnicki MA. Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci. 2000;5:D570–D767. doi: 10.2741/perry. [DOI] [PubMed] [Google Scholar]
- 42.Yablonka-Reuveni Z, Rudnicki MA, Rivera AJ, Primig M, Anderson JE, Natanson P. The transition from proliferation to differentiation is delayed in satellite cells from mice lacking MyoD. Dev Biol. 1999;210:440–455. doi: 10.1006/dbio.1999.9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996;10:1173–1183. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- 44.Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiology of disease. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Koishi K, Zhang M, McLennan IS, Harris AJ. MyoD protein accumulates in satellite cells and is neurally regulated in regenerating myotubes and skeletal muscle fibers. Dev Dyn. 1995;202:244–254. doi: 10.1002/aja.1002020304. [DOI] [PubMed] [Google Scholar]
- 46.Ishido M, Kami K, Masuhara M. In vivo expression patterns of MyoD, p21, and Rb proteins in myonuclei and satellite cells of denervated rat skeletal muscle. Am J Physiol Cell Physiol. 2004;287:C484–493. doi: 10.1152/ajpcell.00080.2004. [DOI] [PubMed] [Google Scholar]
- 47.Weis J. Jun, Fos, MyoD1, and myogenin proteins are increased in skeletal muscle fiber nuclei after denervation. Acta neuropathologica. 1994;87:63–70. doi: 10.1007/BF00386255. [DOI] [PubMed] [Google Scholar]
- 48.Yang W, Zhang Y, Li Y, Wu Z, Zhu D. Myostatin induces cyclin D1 degradation to cause cell cycle arrest through a phosphatidylinositol 3-kinase/AKT/GSK-3b pathway and is antagonized by insuling-like growth factor 1. J Biol Chem. 2007;282:3799–3808. doi: 10.1074/jbc.M610185200. [DOI] [PubMed] [Google Scholar]
- 49.Siriett V, Salerno MS, Berry C, Nicholas G, Bower R, Kambadur R, Sharma M. Antagonism of myostatin enhances muscle regeneration during sarcopenia. Mol Ther. 2007;15:1463–1470. doi: 10.1038/sj.mt.6300182. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi S, Kulkarni AB. Mutant superoxide dismutase 1 causes motor neuron degeneration independent of cyclin-dependent kinase 5 activation by p35 or p25. J Neurochem. 2004;88:1295–1304. doi: 10.1046/j.1471-4159.2003.02256.x. [DOI] [PubMed] [Google Scholar]
- 51.Turner BJ, Lopes EC, Cheema SS. Neuromuscular accumulation of mutant superoxide dismutase 1 aggregates in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2003;350:132–136. doi: 10.1016/s0304-3940(03)00893-0. [DOI] [PubMed] [Google Scholar]