

Summary
ATP binding and hydrolysis are critical for protein degradation by HslUV, a AAA+ machine containing one or two HslU6 ATPases and the HslV12 peptidase. Although each HslU homohexamer has six potential ATP-binding sites, we show that only three or four ATP molecules bind at saturation and present evidence for three functional subunit classes. These results imply that only a subset of HslU and HslUV crystal structures represent functional enzyme conformations. Our results support an asymmetric mechanism of ATP binding and hydrolysis and suggest that molecular contacts between HslU and HslV vary dynamically throughout the ATPase cycle. Nucleotide binding controls HslUV assembly and activity. Binding of a single ATP allows HslU to bind HslV, whereas additional ATPs must bind HslU to support substrate recognition and to activate ATP hydrolysis, which powers substrate unfolding and translocation. Thus, a simple thermodynamic hierarchy ensures that substrates bind to functional HslUV complexes, that ATP hydrolysis is efficiently coupled to protein degradation, and that working HslUV does not dissociate, allowing highly processive degradation.
Keywords: AAA+ protease, energy-dependent degradation, protein turnover, enzyme regulation, peptidase activation, HslVU
Introduction
ATP fuels the operation of most molecular machines, including energy-dependent intracellular proteases. ATP-dependent proteases typically consist of a barrel-shaped peptidase, with the active sites for peptide-bond cleavage sequestered in an aqueous internal chamber, and associated hexameric AAA+ ATPases that recognize specific protein substrates, unfold these molecules, and then translocate the denatured polypeptide into the peptidase chamber for degradation.1,2 AAA+ and related P-loop ATPases also operate in many other biological processes in which mechanical work is performed on macromolecules.3,4
HslUV is an ATP-dependent protease present in roughly 60% of eubacteria and many eukaryotic lineages.5 Functional HslUV enzymes are produced by the binding of one or two AAA+ HslU6 hexamers to HslV12, a double-ring dodecameric peptidase that shares an N-terminal active-site threonine and extensive structural homology with the β-subunits of the eukaryotic proteasome.6–8 Importantly, crystal structures of HslU6, HslV12, HslU6·HslV12, and HslU6·HslV12·HslU6 (Fig. 1A) have been solved.8–17 Moreover, many HslU structures differ in conformation, apparently as a consequence of differences in bound nucleotides, providing clues about potential nucleotide-dependent movements that may drive protein unfolding and translocation. Indeed, HslUV is the only ATP-dependent protease for which the entire structure is known. However, several issues have hampered further understanding of this enzyme. First, structures of HslU hexamers with six, four, or three bound nucleotides have been solved. Which of these structures are functionally relevant is unclear. Second, whether the nucleotide bound in different HslU crystal structures is ADP or ATP has been controversial.18 Third, little quantitative information is available about nucleotide binding to HslU in solution or about the functional consequences of this binding on HslU recognition of HslV and/or protein substrates.
Figure 1.
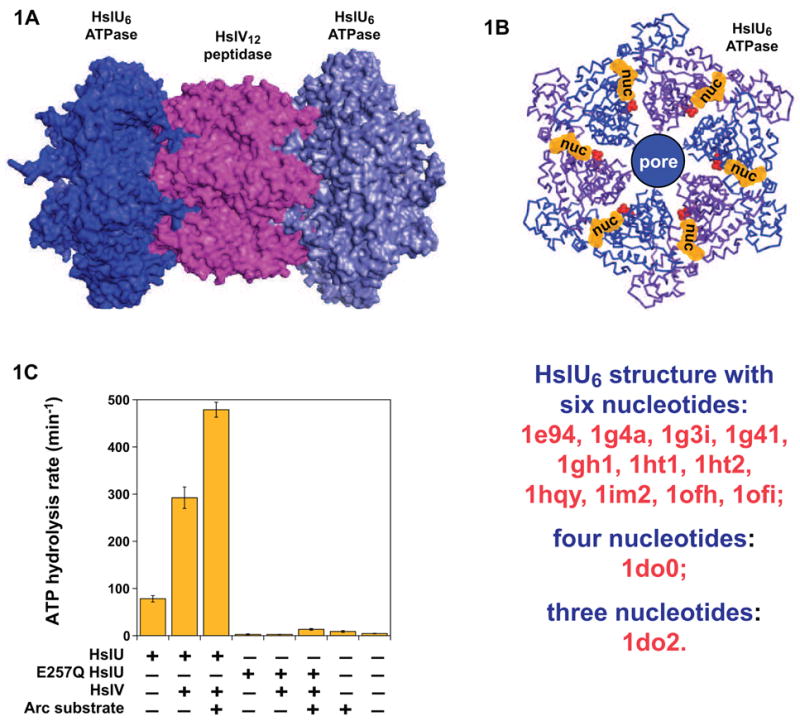
HslU. (A) Two HslU6 ATPases (blue) can assemble with the HslV12 peptidase (magenta). (B) HslU hexamers have six potential nucleotide binding sites, located at domain and subunit interfaces. From 3–6 nucleotides bind HslU6 in different crystal structures. (C) Hydrolysis of ATP (2.5 mM) was measured at 37 °C in PD buffer in the presence/absence of HslU or E257Q HslU (0.3 μM hexamer), HslV (0.8 μM dodecamer), and Arc-IA37-st11-titin-ssrA substrate27 (10 μM). Reactions components were preincubated with 10 μM ATPγS to promote HslU or HslUV association prior to addition of ATP.
ClpX is a hexameric ATPase that shares about 50% sequence homology and substantial structural homology with HslU.3,19 Under conditions of ATP saturation, hexamers of ClpX bind only three or four molecules of ATP.20 In addition, single-chain ClpX hexamers are functional with two subunits able to hydrolyze ATP, two hydrolytically inactive subunits capable of assuming an ATP-bound conformation, and two subunits that mimic ATP-free wild-type subunits.21 Studies of single-chain ClpX variants also suggest that hydrolysis is probabilistic in the sense that different subunits hydrolyze ATP in a sequential but not necessarily ordered fashion. If HslU and ClpX function in the same manner, then HslU hexamers would also operate sequentially, always containing a mixture of nucleotide-bound and nucleotide-free subunits. However, ClpX partners with ClpP, a peptidase that shares no structural homology with HslV,22 and thus it is not obvious that ClpX and HslU will operate by similar mechanisms. Indeed, structural studies of several other hexameric ATPases have been interpreted as evidence that these enzymes cycle between states that are fully ATP-bound or ADP-bound.23–25 Moreover, most HslU and HslUV crystal structures have six bound ATPs or ADPs (Fig. 1B), apparently supporting a symmetric or concerted mechanism of ATP hydrolysis.
Here, we demonstrate that only three or four molecules of ATP bind to the HslU homohexamer at saturation. Thus, some of the six potential binding sites in the hexamer remain empty. ATP binds the sites that can be filled with comparable affinities, although the kinetics of nucleotide dissociation reveals the existence of two classes of binding sites. Our results support an ATPase model in which HslU subunits function asymmetrically and highlight structures with three or four bound nucleotides as being most relevant for understanding HslU function. Importantly, a single bound ATP supports HslV binding by the HslU hexamer but does not allow ATP hydrolysis; multiple ATPs must be bound to allow protein-substrate binding and to activate hydrolysis. This fail-safe mechanism prevents hydrolysis of all bound ATP molecules, averting HslUV dissociation and facilitating processive degradation. We also show that interactions with HslV vary as subunits of the HslU hexamer transit through the ATPase cycle.
Results
HslU hydrolyzes ATP rapidly, complicating studies of ATP binding using the wild-type enzyme. To avoid this problem, we mutated Glu-257 in the Walker-B box of Escherichia coli HslU to Gln (E257Q). We anticipated that this mutant would be defective in ATP hydrolysis because the Glu side chain appears to be close to the γ phosphate of bound ATP, glutamate is invariant at this position in HslU orthologs, and the same mutation reduces ATP hydrolysis markedly in related AAA+ enzymes.4,9,11,20
HslU E257Q with an N-terminal His6 tag behaved similarly to His6-tagged wild-type HslU during purification steps involving anion-exchange chromatography and gel filtration, suggesting that both proteins had similar structures. However, unlike wild-type HslU, the ultraviolet spectrum of the E257Q protein following these steps indicated the presence of bound nucleotide, which was not added in any purification steps. This observation suggested that nucleotide from the cell extract remained bound to the mutant, perhaps because it binds nucleotide more tightly than wild-type HslU. Incubation with high salt, which dissociates the HslU hexamer,10 and activated charcoal, which binds nucleotide, resulted in a spectrum of the mutant protein that was similar to that of wild-type HslU. Moreover, in both cases, the spectra were those expected from the tyrosine and tryptophan content of the wild-type and mutant proteins. When wild-type HslU was treated with charcoal and high salt and then returned to a standard buffer, we detected no changes in spectrum, ATP-hydrolysis activity, stimulation of HslV peptidase activity, or ability to support HslV degradation of Arc repressor, a good substrate (data not shown).
Compared to wild-type HslU,26–27 the E257Q mutant had almost no ATPase activity by itself, or when HslV or Arc repressor and HslV were added (Fig. 1C). Indeed, ATP hydrolysis in reactions containing the E257Q mutant was similar to control reactions lacking this protein. Moreover, no degradation of Arc repressor was detected in the presence of HslV, E257Q HslU, and ATP (data not shown).
HslU hexamers bind three or four ATPs
Knowing how many ATPs bind to a HslU hexamer is important for understanding mechanism and for evaluating which crystal structures are likely to represent functional complexes. To determine stoichiometry, increasing amounts of ATP were added to a fixed concentration of the E257Q hexamer, and binding was assayed by isothermal titration calorimetry (ITC). As shown in Fig. 2A, saturation occurred at an ATP/hexamer ratio between three and four. Fitting of these data gave an average stoichiometry of 3.5 ATPs per hexamer (ΔH = –4.1 kcal/mol). In an independent experiment, the average binding stoichiometry was 3.3 ATPs per hexamer (ΔH = –4.1 kcal/mol). The Kd’s in both ITC experiments were poorly determined.
Figure 2.
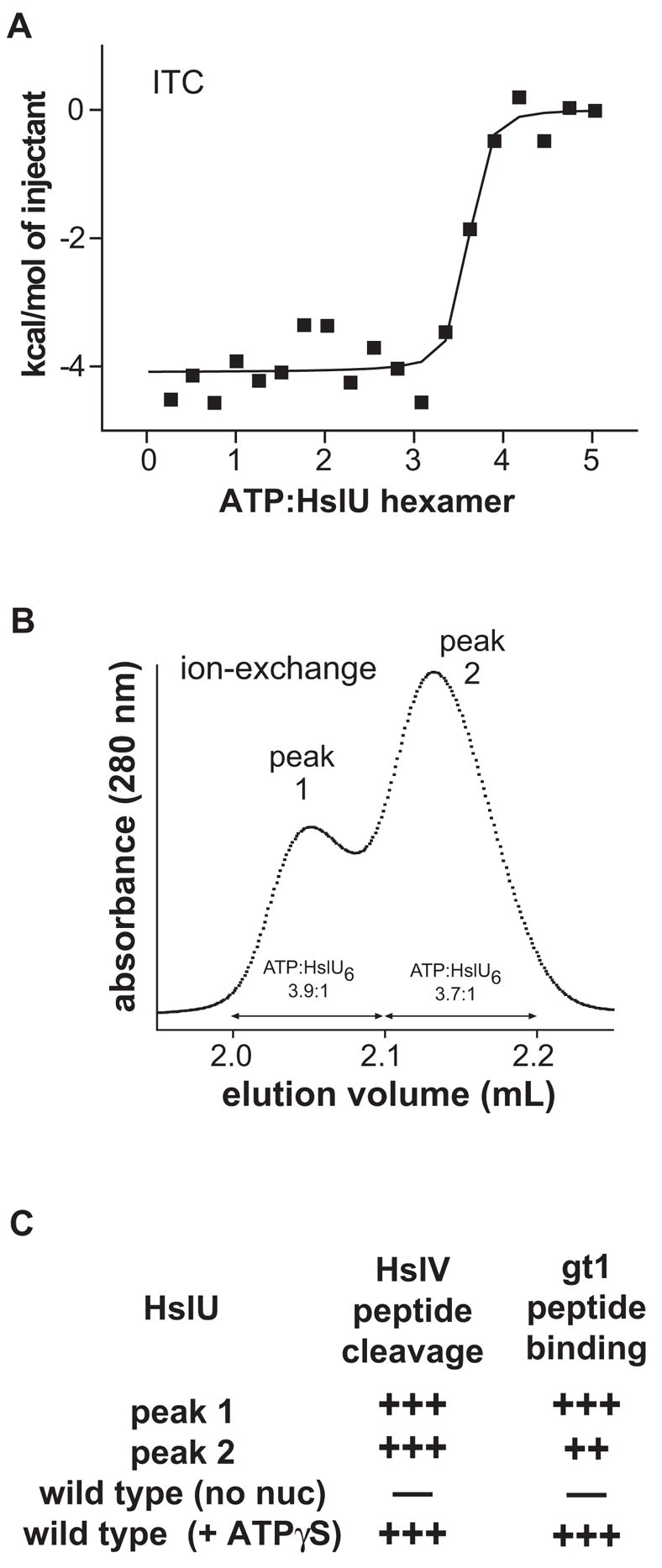
Binding stoichiometry. (A) ATP binding to the HslU E257Q mutant was assayed by ITC. 7.5 μL aliquots of ATP (131 μM) were injected into 1.5 mL of a solution of E257Q hexamer (7.2 μM) at 25 °C in PDN buffer. The solid line is a non-linear least squares fit of the data to a single set of sites model (n = 3.5 ± 0.05, ΔH −4.1 kcal/mol,= KD < 0.5 μM). (B) The HslU E257Q mutant (50 μL of a 12.5 μM solution of hexamer) was loaded onto a 0.1 mL Sepharose mono-Q column equilibrated in 50 mM Tris·HCl (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 10% glycerol, and 100 μM ATP Equilibration buffer was run for 1 mL and then a linear gradient (2.3 mL total) from equilibration buffer to 50 mM Tris·HCl (pH 7.5), 500 mM NaCl, and 10% glycerol was run. (C) HslU (1 μM hexamer with or without 1 mM ATPγS) or HslU E257Q from the pooled peak-1 or peak-2 samples in panel B were used in assays of HslV cleavage of Z-Gly-Gly-Leu-AMC or in binding of the fluorescein-labeled gt1 peptide (100 nM).
The binding of fewer than six nucleotides to each HslU homohexamer at saturation could be explained in two ways: (i) once three or four ATPs bind, conformational changes in the two unoccupied subunits preclude further binding (a type of negative cooperativity); or (ii) roughly two thirds of HslU hexamers bind six ATPs, whereas the remaining HslU hexamers fail to bind nucleotide because of conformational or chemical heterogeneity. In fact, ion-exchange chromatography performed in buffer containing 100 μM ATP revealed heterogeneity with two peaks of HslU E257Q (Fig. 2B). However, in assays that require ATP binding, HslU E257Q from both peaks activated peptide cleavage by HslV and bound a substrate-mimic peptide (Fig. 2C). Moreover, comparison of the absorbance spectra of column fractions containing ATP but no protein with fractions containing HslU E257Q revealed that both peaks contained about 3.8 ATPs per E257Q hexamer. When either peak-1 or peak-2 protein was rerun on the ion-exchange column, a mixture of the same two peaks was observed, establishing that both forms interconvert. Thus, HslU E257Q exists in discrete states with different chromatographic properties, but both states bind ATP and are functional in ATP-dependent HslU activities. Because we find no evidence for a form of HslU E257Q that cannot bind ATP, we conclude that three or four subunits of the hexamer bind ATP, whereas the remaining subunits do not.
Nucleotide affinity
Nitrocellulose-filter binding was initially used to assay the affinity of E257Q HslU for 32P-γ-ATP, titrating protein against a fixed concentration of ATP (Fig. 3A) or vice versa (Fig. 3B). Both binding curves were fit well by a model in which four subunits in the E257Q HslU hexamer each bind ATP with an equilibrium dissociation constant near 0.25 μM. The corresponding equilibrium constant for ATP association gives a binding free energy of approximately –9 kcal/mole. Using ΔH for ATP binding determined in the ITC experiment, TΔS for ATP binding is about 5 kcal/mol and thus the entropy of ATP association is favorable. Displaced solvent molecules or conformational rearrangements in HslU presumably account for this favorable binding entropy.
Figure 3.
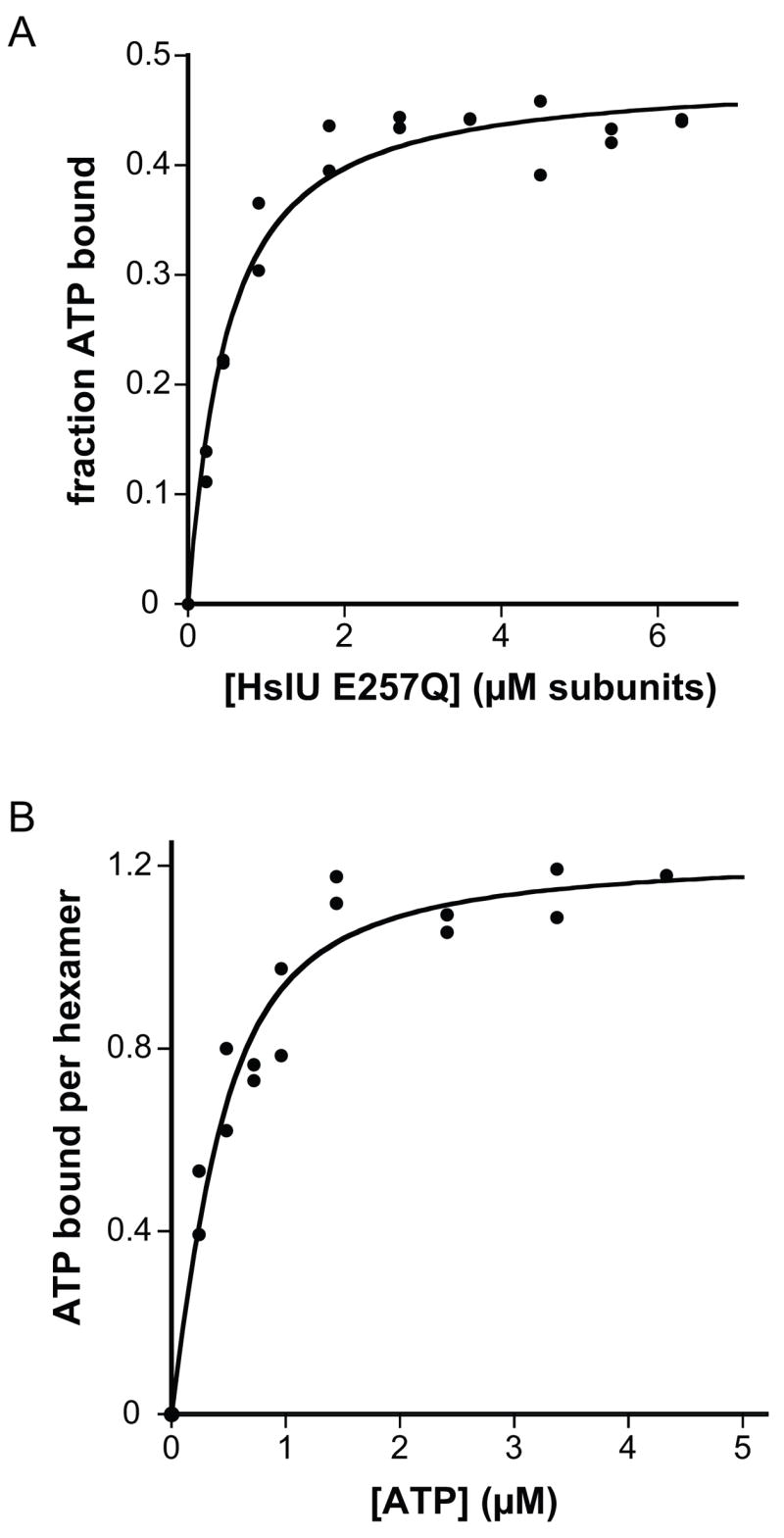
Nitrocellulose filter-binding assays. (A) Binding of increasing E257Q HslU to 0.1 μM 32P-γ-ATP. The solid line is a fit assuming that four of six subunits in the E257Q HslU hexamer bind ATP with an equilibrium dissociation constant of 0.27 ± 0.04 μM per site (B) Binding of increasing 32P-γ-ATP to 0.1 μM E257Q HslU hexamer. The solid line is a fit to a non-cooperative model assuming four identical ATP binding sites per HslU hexamer with an equilibrium dissociation constant of 0.21 ± 0.05 μM per site. Fitting of these data to the Hill equation gave an n-value of 1.1 ± 0.2. Binding assays were performed at room temperature in PD buffer. Some HslU·nucleotide complexes dissociated during the separation of bound and free ATP in these experiments, which may lead to an underestimate of affinity.
We also assayed binding of wild-type HslU or the E257Q mutant to N-methylanthraniloyl (mant) derivatives of ATP, ATPγS, and ADP by monitoring changes in the fluorescence center-of-mass of these nucleotides (Fig. 4A). E257Q HslU bound mant-ATP and mant-ATPγS with roughly similar affinities and bound mant-ADP about six-fold more tightly (Table 1). Competition of unmodified ATP, ATPγS, and ADP for mant-ATP binding to the E257Q protein showed that ADP competed more efficiently than ATP or ATPγS (data not shown), as expected from direct binding of the mant nucleotides. Wild-type HslU also bound mant-ATP and mant-ATPγS similarly, albeit less tightly than the E257Q variant, and bound mant-ADP about three-fold more strongly (Table 1). All of the Fig. 4A binding curves were fit well by a simple hyperbolic function, as expected if nucleotide binding to the three or four available sites in wild-type or E257Q HslU does not show strong positive or negative cooperativity.
Figure 4.

Nucleotide binding, dissociation, and hydrolysis. (A) Equilibrium binding of HslU or E257Q HslU (EQ) to 100 nM mant-ATP (circles), mant-ATPγS (triangles), or mant-ADP (squares). The fitted lines are for non-cooperative binding. (B) Dissociation kinetics. Following preincubation of E257Q HslU (2 μM hexamer) with mant-ATP (10 μM), excess ATP (5 mM) was added and dissociation was monitored by changes in fluorescence. The fitted line is a double exponential with time constants of 22 ± 1 s (amplitude 70%) and 193 ± 8 s (amplitude 30%) for the two phases. (C) The rate of ATP hydrolysis by HslU (0.3 μM hexamer) was measured as a function of ATP concentration in PD buffer at 25 °C. The solid fitted line is a non-linear least-squares fit to the Hill form of the Michaelis-Menten equation (half-maximal activity 9 ± 0.6 μM; Hill constant 2.0 ± 0.2. The dashed line shows the best fit to the non-cooperative Michaelis-Menten equation.
Table 1.
Nucleotide affinities.
| ATP (μM) | ATPγS (μM) | ADP (μM) | mant-ATP (μM) | mant-ATPγS (μM) | mant-ADP (μM) | |
|---|---|---|---|---|---|---|
| HslU | 0.65 | 0.55 | 0.19 | 3.8 | 3.2 | 1.1 |
| E257Q | 0.24 | 0.21 | 0.04 | 1.4 | 1.2 | 0.2 |
Values are equilibrium dissociation constants of nucleotides for a single site in an HslU hexamer. Values for mant nucleotides are taken from the fits of the experiments shown in Fig. 4A. The value for ATP and E257Q HslU is an average of the values determined from the fits of the experiments shown in Fig. 3A and 3B. Values in italics were calculated by dividing the affinities for the corresponding mant-nucleotide by 5.8, the ratio of the affinities of E257Q HslU for mant-ATP versus ATP.
The mant-nucleotide binding experiments have implications for ATP hydrolysis by wild-type HslU. For example, wild-type HslU bound mant-ATP and mant-ATPγS with similar affinities but bound mant-ADP more tightly, suggesting that mant-ATP is not hydrolyzed to produce mant-ADP at a significant rate during the binding experiment. We were initially surprised by this result, as mant-ATP supported HslUV degradation of protein substrates (data not shown). As shown below, however, HslU hydrolyzes ATP very slowly at the low nucleotide concentrations (0.1 μM) used in these binding assays, and we assume that the same is true for hydrolysis of mant-ATP.
E257Q HslU bound mant-ATP, mant-ATPγS, and mant-ADP more tightly than the wild-type enzyme (Fig. 4A; Table 1). Stronger binding by the mutant is probably a consequence of replacing the negatively charged glutamate in the nucleotide-binding site with uncharged glutamine, thereby reducing electrostatic repulsion with the β and γ phosphates. This result is consistent with our finding that the mutant but not the wild-type protein purifies with bound nucleotide. Moreover, comparing the affinities of E257Q HslU for ATP and mant-ATP indicates that the mant group weakens binding about 6-fold. By applying a correction for the destabilizing effect of the mant group, we estimate that ATP and ATPγS bind accessible sites in wild-type HslU with an affinity of 0.6–0.7 μM, whereas ADP binds with an affinity of 0.2 μM (Table 1). This stronger binding of ADP relative to ATP helps explains why ADP is a potent inhibitor of HslU activity.28–29
Multiple classes of ATP binding sites
To determine the kinetics of nucleotide dissociation, we allowed complexes containing approximately three mant-ATP molecules per E257Q HslU hexamer to form, added excess unmodified nucleotide to block further mant-ATP binding, and assayed the rate of dissociation of the fluorescent nucleotide. As shown in Fig. 4B, when ATP was used as the competitor, the dissociation kinetics were biphasic. The same result was observed with competing ADP (data not shown). In each experiment, the fast phase had a time constant of roughly 20 s and the slow phase had a time constant of about 150 s. These dissociation experiments suggest a minimum of two classes of ATP-bound HslU subunits. Dissociation remained biphasic in the presence of HslV (fast phase 22 s; slow phase 107 s) or HslV and Arc substrate (fast phase 12 s; slow phase 88 s). Overall, these results support the existence of three classes of HslU subunits. One class fails to bind ATP. Of the two types that bind ATP, one class of subunits releases ATP more slowly than the other.
ATP hydrolysis
To probe nucleotide interactions with HslU under conditions where ATP hydrolysis provides an additional route for nucleotide escape, we measured changes in the steady-state rate of hydrolysis as a function of ATP concentration (Fig. 4C). Half-maximal hydrolysis activity was observed at a free ATP concentration of roughly 10 μM. Because this value is about 10-fold higher than the ATP concentration needed for half-maximal binding in the absence of hydrolysis, most bound ATP molecules must leave HslU via hydrolysis and subsequent release of ADP/Pi rather than by dissociation of ATP (see ref. 30). Importantly, the Fig. 4C data were fit poorly by the standard Michaelis-Menten equation (dashed line) but were fit well by the cooperative Hill form of this equation (solid line; rate=Vmax/(1+(KM/[ATP])n) with a Hill constant of 2.0 ± 0.2. Because we did not observe strong positive cooperativity in ATP binding to HslU, this result suggests that more than one ATP must bind to an HslU hexamer before the rate of hydrolysis becomes significant.
Linkage between ATP and peptide binding
We used binding of HslU and HslU E257Q to the substrate-mimic peptide gt1 (MRYFFKKKLRFY) to probe the relationship between recognition and ATP binding.27 In fluorescence-anisotropy assays performed in the presence of saturating ATP or ATPγS, HslU E257Q bound fluorescein-labeled gt1 peptide about 5-fold more weakly than did wild-type HslU (Fig. 5A). The presence of HslV in addition to HslU or the mutant in these binding reactions strengthened peptide binding modestly (Fig. 5A). The finding that the E257Q mutation reduces affinity for gt1 suggests that the Glu-257 side chain in the Walker-B motif of wild-type HslU plays a role in linking the energy of ATP binding to the conformational changes that allow binding of this substrate-mimic peptide.
Figure 5.

HslU binding to FL-gt1, a fluorescent substrate-mimic peptide. (A) Binding of FL-gt1 (100 nM) by wild-type HslU or E257Q HslU in the presence/absence of HslV12. Binding reactions contained 500 μM ATPγS (HslU) or 500 μM ATP (E257Q HslU) and were performed at 25 °C in PD buffer. The fitted lines are for non-cooperative binding with Kd’s of 0.76 μM (HslUV), 0.93 μM (HslU), 3.0 μM (E257Q HslUV), and 5.6 μM (E257Q). (B) Binding of E257Q HslU (1.5 μM hexamer) to FL-gt1 (0.1 μM) as a function of ATPγS concentration (top panel) or N6-methyl-ATP concentration (bottom panel). The data were fitted to the Hill equation. Half-maximal binding occurred at 0.8 μM ATPγS (n = 2.3 ± 0.23), 34 μM N6-methyl-ATP without HslV (n = 1.8 ± 0.15), and 2.3 μM N6-methyl-ATP with 3 μM HslV12 (n = 1.5 ± 0.12).
Next, we used a fixed concentration of E257Q HslU and assayed the dependence of gt1-peptide binding on ATPγS concentration (Fig. 5B; top panel). Although ATPγS binding was required for peptide binding, the total concentrations of ATPγS and HslU subunits were too close to calculate free concentrations in a model-independent fashion, and thus to determine if peptide and nucleotide binding were cooperatively linked. To obviate this problem, we used N6-methyl-ATP (Fig. 5B; lower panel), a derivative that binds more weakly to HslU. In this experiment, peptide binding occurred at N6-methyl-ATP concentrations in excess of HslU sites, and thus the free and total nucleotide concentrations were similar. The resulting binding curve showed clear cooperativity with a Hill constant of 1.8 ± 0.15, supporting the idea that the conformational changes needed to support substrate recognition require binding of at least two ATP molecules to HslU. When binding was assayed in the presence of HslV12, the concentration of N6-methyl-ATP required to support binding to the gt1 peptide was reduced more than 10-fold (Fig. 5B). Thus, the HslU·HslV complex must bind nucleotide more tightly than free HslU binds nucleotide.
HslU binds HslV more weakly during ATP hydrolysis
HslU-HslV binding can be assayed by increases in the cleavage of a Z-Gly-Gly-Leu-AMC peptide by HslV31. When we titrated HslU6 against 15 nM HslV12 in the presence of saturating ATP, the resulting increase in peptidase activity fit well to a binding model with an apparent Kd of approximately 75 nM (Fig. 6A, upper panel). In the presence of saturating ATPγS, binding was substantially stronger (Kd ≤ 15 nM; Fig. 6A, upper panel). This result could mean that HslV is bound more strongly by HslU·ATPγS than by HslU·ATP. Alternatively, HslU may bind HslV less tightly when its subunits cycle through the different states of ATP hydrolysis, because a significant period of time is spent in lower-affinity states in which some ATP has been hydrolyzed. Importantly, however, ATP and ATPγS supported HslV binding by the hydrolysis-defective E257Q HslU6 mutant equally well (Fig. 6A, lower panel), supporting the idea that HslU states with lower HslV affinity are populated during the ATPase cycle (see Discussion).
Figure 6.

HslU·HslV binding. (A) Binding of HslU (top) or E257Q HslU (bottom) to HslV12 (15 nM) was assayed by changes in the rate of Z-Gly-Gly-AMC peptide cleavage. Reactions contained 1 mM ATPγS or 1 mM ATP and were performed in PD buffer at 25 °C. Data were fit to a quadratic form of a hyperbolic binding isotherm. Apparent KD values were 12 ± 2 nM (HslU; ATPγS), 78 ± 10 nM (HslU; ATP), 19 ± 5 nM (E257Q HslU; ATP) and 21 ± 2 nM (E257Q HslU; ATPγS). (B) The presence of a protein substrate27 (10 μM Arc-IA37-st11-titin-ssrA) strengthened binding of HslV to HslU (0.3 μM hexamer), as measured by changes in the rate of HslU ATP hydrolysis. HslUV·Arc complexes were preassembled by incubating for 5 min with 10 μM ATPγS at 37 °C. Reactions in PD buffer at 37 °C were initiated by addition of ATP and a regeneration system. (C) HslU activation of HslV12 (100 nM) cleavage of Z-Gly-Gly-Leu-AMC (200 μM) was assayed in the presence of 100, 200, 400 or 800 nM ATPγS. (D) Rates of Z-Gly-Gly-Leu-AMC (200 μM) cleavage were assayed in the presence of 1 μM HslU hexamer, 100 nM ATPγS, and increasing HslV12. (E) N6-methyl-ATP supports hyperbolic activation of HslU6 (50 nM) stimulation of HslV12 (200 nM) cleavage of Z-Gly-Gly-Leu-AMC (200 μM). The solid line is a fit (R = 0.997) to the equation activity = 100/(1+([0.85 μM][N6-methyl-ATP])).
Using surface-plasmon resonance (SPR) to measure HslU·HslV affinity in the presence of ATPγS, Azim et al. reported a Kd of approximately 1 μM,32 a value far weaker than we observe. It is possible that interactions of HslU and/or HslV with the SPR chip surface resulted in an underestimate of affinity. Alternatively, buffer differences may explain the weak binding observed in the SPR experiments, which used Ca++ rather than Mg++. Indeed, Ca++·ATP supports HslU activation of HslV peptidase activity against Z-GGL-AMC only about 1% as well as Mg++·ATP.33
We also titrated HslV against HslU and assayed binding by changes in the rate of ATP hydrolysis. These experiments were performed at 37 °C, because ATP hydrolysis was too slow to obtain reliable data at 25 °C, the temperature of other experiments. HslU·HslV affinity was about 140 nM when just HslU and HslV were present but became 10-fold stronger when an excess of protein substrate was added (Fig. 6B).
HslU6·ATP activates HslV
To probe the role of nucleotide in HslU activation of HslV, we titrated increasing HslU against 100 nM HslV12 in the presence of 100, 200, 400, and 800 nM ATPγS and measured Z-GGL-AMC cleavage (Fig. 6C). In this assay configuration, two HslU6 hexamers can bind each HslV12 dodecamer, and thus saturation should require binding of 200 nM HslU6, as Kwon et al. have shown that binding of an HslU hexamer activates only the cis but not the trans HslV ring.16 Notably, similar total concentrations of HslU6 resulted in half-maximal activation at each nucleotide concentration, indicating that binding of ATPγS, HslU6, and HslV12 are nearly stoichiometric under these conditions. Starting with 100 nM ATPγS, 100 nM HslV12, and 1 μM HslU6, we found that an additional 8-fold increase in HslV12 did not increase peptidase activity (Fig. 6D). Because nucleotide and HslV binding are linked, this result establishes that almost no free ATPγS or HslU6·ATPγSn are present under these conditions. Moreover, because we do not observe strong cooperativity in ATPγS binding to HslU, the dominant nucleotide-bound species in the 100 nM ATPγS curve in Fig. 6C should be HslU6·ATPγS·HslV12 (or HslU6·ATPγS·HslV12·HslU6·ATPγS). Thus, binding of a single ATPγS to an HslU hexamer appears to support HslV binding/activation. If this model is correct, then activation should show a hyperbolic dependence on nucleotide concentration. Indeed, E257Q HslU activation of HslV peptidase activity increased hyperbolically with N6-methyl-ATP concentration (Fig. 6E), as shown by the excellent fit of these data (R= 0.997) to a non-cooperative binding model.
By the arguments in the previous paragraph, 100 nM ATPγS should support HslU6 binding to half of the HslV rings in 100 nM HslV12, with nucleotide being the limiting factor. Consistently, increasing the ATPγS concentration increased the observed peptidase activity (Fig. 6C). By our model, however, most if not all of the HslV12 rings should have two HslU hexamers bound in the presence of 200 nM ATPγS, and yet peptidase activity continued to increase when ATPγS was increased to 400 and 800 nM ATPγS. There are two potential explanations. First, binding of more than one ATPγS to an HslU hexamer might increase the peptidase activity of the HslV ring to which it binds (see Discussion). Second, binding of the second HslU hexamer to HslV12 might be negatively cooperative, but the doubly bound species could have more than twice the peptidase activity of the singly bound species.
Discussion
Our results demonstrate that an HslU hexamer binds with substantial affinity to no more than four ATPs. Thus, two potential binding sites in the hexamer appear to be empty, even when nucleotide is saturating. Binding of ATP to the three or four available nucleotide sites occurs with an equilibrium constant of roughly 1 μM. However, two phases are observed in dissociation experiments, suggesting the existence of at least two classes of ATP-binding sites. These results suggest that HslU operates by an asymmetric mechanism, with different subunits in the hexamer serving different functional roles. Based on results with other AAA+ enzymes, it is likely that ATP hydrolysis occurs in one or two HslU subunits at a time, with the roles of individual subunits changing in a sequential fashion as repeated cycles of ATP binding, hydrolysis, and ADP/Pi release take place.21,34 An important implication of this model is that an all-ADP state or a fully nucleotide-free state of HslU is not part of the normal ATP cycle. Rather, the structural changes in HslU that power translocation and protein unfolding must occur as the enzyme cycles through a saturated ATP state (with 3 or 4 bound ATPs), a mixed ATP/ADP state, and a partially saturated ATP state.
Our results highlight two crystal structures of the HslU hexamer that show partial occupancy of nucleotide-binding sites.9 In the 1do0 dimer-of-trimers structure, adjacent subunits in each trimer are nucleotide-free, ATP-bound, and ATP·Mg++-bound, respectively. This structure contains three distinct types of subunits and is thus most compatible with our results. Why is the ATP in this structure not hydrolyzed? One possibility, raised by Wang and colleagues, is that the four ATPs were misidentified in the crystallographic analysis and are actually ADPs.18 If just the Mg++-free nucleotides were ADP, then this structure might also be inactive if more than two bound ATPs are needed to support hydrolysis. Our results suggest that ATP hydrolysis requires at least two HslU-bound ATPs, but this is a minimum estimate. Another HslU structure (1do2) is trimer-of-dimers hexamer, with alternating nucleotide-free and AMPPNP-bound subunits.
In apparent contrast to our results, six nucleotides are bound to the HslU hexamer in 11 crystal structures at resolutions of 3.5 Å or better.9–11,13,15,16 How can we reconcile our finding that HslU binds a maximum of four ATPs in solution with structures showing six bound nucleotides? One possibility is that conformational changes required for HslU to bind six nucleotides occur slowly during the long periods required for crystallization but are not accessible on the much shorter time scales used for solution experiments. Another possibility is that that six nucleotides molecules can bind HslU if Mg++ is absent, as most structures with six bound nucleotides do not have Mg++ in the nucleotide-binding site. In either case, based on our studies, it is unlikely that HslU structures with six bound nucleotides represent functional species in the normal ATPase cycle. Although six nucleotides can obviously bind HslU under some circumstances, this enzyme binds HslV and protein substrates, hydrolyzes ATP, and supports active proteolysis at ATP concentrations where no more than four sites in the hexamer are occupied.
Huang and Goldberg found that low micromolar ATP concentrations activated peptide cleavage by HslUV, whereas 100-fold higher ATP concentrations were required for degradation of casein, and proposed that that ATP binding to a high-affinity site in HslU reduced affinity for other sites.33 We find no evidence to support this proposal. Indeed, the three or four HslU subunits that bind nucleotide do so with similar affinities. Rather, we propose that binding of one ATP molecule to HslU supports a conformational change that allows HslV peptidase activation but not robust ATP hydrolysis or the binding of protein substrates. The binding of additional ATPs to the HslU hexamer then drives a second set of conformational changes that activate hydrolysis and substrate binding. When ATP is hydrolyzed, however, apparent “affinity” becomes weaker because dissociation of ADP/Pi rather than ATP becomes the main route of nucleotide release (i.e., if ATP hydrolysis is substantially faster than dissociation, then KM will be substantially greater than Kd). Our model also explains the results of Yoo et al., who concluded that HslU affinity for ATPγS was roughly 20-fold stronger than for ATP.35 By contrast, we find that the actual affinity of HslU for these nucleotides is almost identical. Again, ATP appears to bind HslU more weakly than ATPγS because bound ATP dissociates rapidly from the enzyme via hydrolysis, whereas bound ATPγS does not.
Several of our results support a model in which HslU conformational changes required for HslV activation can occur independently from the structural changes needed for other activities. First, ATP hydrolysis and substrate-mimic binding by HslU require binding of two or more ATPs, whereas HslV activation is supported by a single bound ATP. Second, compared to wild type, the E257Q mutation reduces HslU affinity for the gt1 substrate-mimic peptide but does not reduce affinity for HslV. Thus, different structural changes appear to be involved in these distinct activities.
ATP or ATPγS binding to HslU is generally required to activate HslV peptidase activity.28,31,35 Nevertheless, an isolated C-terminal peptide of HslU can activate HslV in the absence of nucleotide.36,37 In some HslUV crystal structures, this C-terminal peptide assumes an extended conformation that packs between HslV subunits, thereby stabilizing the active conformation of the catalytic site for peptide-bond cleavage.11,13,16 In other HslU or HslUV structures, this peptide folds back against the rest of HslU and is not positioned to interact with HslV (Bochtler et al, 2000; Song et al, 2000; Wang et al, 2001a). Because HslU·ADP·HslV complexes are observed in crystal structures,9–10,15 ATP is clearly not required for HslU binding to HslV. However, crosslinking studies show that HslU·ATPn·HslV complexes are more stable than HslU·ADPn·HslV complexes.35 We find that the affinity of HslU for ATP increases when HslV is present. By thermodynamic linkage, ATP-binding must also stabilize HslU·HslV binding. This finding is expected if ATP-dependent conformational changes in HslU release its C-terminal peptides to interact with HslV, thereby stabilizing and activating the proteolytic complex.11,18 An apparent challenge to this model is the finding that the C-terminal tails of an HslU variant lacking the intermediate domain interact with HslV in an ADP-bound form.16 However, each nucleotide site in this structure (1ofh) contains both ADP and Pi, which may mimic ATP.
Our results show that affinity for HslV is stronger when ATP/ATPγS is bound to HslU but is not hydrolyzed. As discussed above, HslU must cycle through states with three or four bound ATPs, with a mixture of ATP/ADP, and with sub-saturating ATP during hydrolysis. Because affinity for HslV deceases when ATP is hydrolyzed, HslU appears to spend the majority of cycle time in one of the latter states. Indeed, because ADP is a potent inhibitor of HslU-catalyzed ATP hydrolysis,29 and binds HslU more tightly than ATP, it would be surprising if ADP release were not the rate-limiting step in ATP hydrolysis. By this model, the mixed ATP/ADP state would be most highly populated because it precedes the rate-determining step. If the C-terminal tails or other regions of ADP-bound HslU subunits in hexamers containing ATP/ADP mixtures did not interact with HslV, then this would provide a straightforward structural basis for the decrease in HslV affinity during ATP turnover. In the related ClpXP system, contacts between the ATPase and peptidase also appear to change dynamically throughout the ATPase cycle.38 Moreover, if HslU hexamers are asymmetric during the ATPase cycle, then interactions with HslV may also be asymmetric. The number or strength of molecular contacts with HslV could also vary depending on the number of ATPs bound to an HslU hexamer, explaining the graded activation of HslUV peptidase activity that we observe.
Several aspects of this system favor assembly of productive HslUV·substrate complexes over assembly of HslU·substrate complexes and minimize wasteful ATP hydrolysis. First, binding of HslU to HslV occurs with nanomolar affinity and requires only a single bound ATP. By contrast, the binding of protein substrates to HslU or HslUV requires several bound ATPs and typically occurs with micromolar affinity. Thus, if ATP were scarce, intact protease complexes would assemble, but neither these complexes nor free HslU would bind substrate. Moreover, because several bound ATPs are also required to activate ATP hydrolysis, little energy would be wasted via non-productive hydrolysis under these conditions. At higher ATP concentrations, both substrate binding and ATP hydrolysis can occur. Because HslUV complexes bind ATP more tightly than HslU6 alone, however, the intact HslUV protease would out compete free HslU6 for binding available ATP and substrate. This feature of the system should diminish the probability of free HslU hexamers binding substrates, unfolding them at a significant cost in terms of ATP hydrolysis, and translocating them back into solution to refold and/or rebind. Thus, a simple thermodynamic hierarchy ensures that HslU binds one ATP, then binds HslV, and then binds additional ATP and protein substrate prior to hydrolysis-driven unfolding and translocation of the substrate into HslV for degradation.
When ATP is plentiful under normal cellular conditions, HslUV complexes would be stable and have, on average, three to four bound ATPs. However, species with fewer ATPs would be populated transiently as a consequence of hydrolysis and ADP/Pi dissociation. Under these conditions, the need for multiple ATPs for hydrolysis but just one ATP for binding HslV provides a fail-safe mechanism, which may be especially important for an enzyme that binds ADP more tightly than ATP. If, for example, just one ATP remained bound to HslUV during substrate degradation, then hydrolysis/degradation would cease and the complex would remain intact but catalytically inert until additional ATPs could bind. Both this mechanism and the fact that ATP-dependent binding of protein substrate increases HslU·HslV affinity would combine to make degradation highly processive, as is observed.39
Materials and Methods
Buffers and materials
Buffer D contains 20 mM Tris-Cl (pH 7.5), 1 mM EDTA, and 10% (v/v) glycerol. Nickel-wash buffer contains 50 mM NaHPO4 (pH 8), 300 mM NaCl, and 10 mM imidazole. Nickel-elution buffer contains 50 mM NaHPO4 (pH 8), 300 mM NaCl, and 250 mM imidazole. PD buffer contains 25 mM HEPES-KOH (pH 7.6), 5 mM KCl, 5 mM MgCl2, 0.032% (v/v) Igepal CA-630 (NP-40), and 10% (v/v) glycerol; PDN buffer lacks NP-40 but is otherwise identical. Lysis buffer contains 50 mM Tris-Cl (pH 7.5), 300 mM NaCl, 1 mM EDTA, and 10% (v/v) glycerol. Immediately prior to use, charcoal buffer was prepared by adding 4 M NaCl, 50 mM Tris-Cl (pH 7.5) to 10 g of activated charcoal (Sigma C-2764) to a final volume of 50 mL. The mixture was shaken for 30 min at room temperature, and the liquid phase was decanted for use.
Sodium salts of ATP and ADP were purchased from Sigma (St. Louis, Missouri); concentrations were determined by absorbance at 259 nm using an extinction coefficient of 15400 M−1cm−1. The lithium salt of ATPγS was purchased from Roche Diagnostics (Indianapolis, Indiana). Triethylammonium salts of mant-ATP, mant-ATPγS, and ADP were purchased from Jena Biosciences GmbH (Jena, Germany); concentrations were determined by absorbance at 355 nm using an extinction coefficient of 5,800 M−1cm−1. N6-methyl ATP was purchased from TriLink BioTechnologies (San Diego, CA); concentration was determined by absorbance at 265 nm using an extinction coefficient of 15,567 M−1cm−1. Z-Gly-Gly-Leu-AMC was purchased from Bachem.
Proteins
Arc substrates were gifts from Eyal Gur (MIT) and Randy Burton (MIT). HslV-H6 was purified as previously described.27 Cells over-expressing H6-HslU were lysed as described,27 substituting nickel-wash buffer and nickel-elution buffer for buffers A and B. Following elution from Ni2+-NTA resin (Qiagen), fractions containing protein were identified by the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, California), pooled, and loaded onto a Sephacryl-300 HR26/60 gel-filtration column (Amersham) equilibrated with buffer D containing 0.1 M NaCl. Fractions containing HslU were pooled, loaded onto a HR16/10 Q-Sepharose column equilibrated in buffer D containing 0.1 M NaCl, and eluted with a linear gradient between buffer D containing 0.1 M NaCl and 1M NaCl. Fractions containing HslU were pooled, concentrated, exchanged into buffer D containing 0.25 M NaCl by PD-10 column chromatography (Amersham), and frozen at −80 °C. H6-HslU E257Q was purified like the wild-type protein with one exception. Following the nickel column, the eluate containing H6-HslU E257Q was mixed 1:1 with charcoal buffer for 30 min at 4 °C, and then passed through a 0.2 μm Acrodisc syringe filter (PN4612, Pall Life Sciences, Ann Arbor, Michigan) to remove charcoal particles before continuing the purification. Protein concentrations were determined by UV absorbance at 280 nm using extinction coefficients in units of M−1cm−1as follows: HslU6, HslU E257Q, and HslU lacking 7 residues from its C-terminus, 140,820; HslV12, 129,720; Arc-IA37-st11-titin-ssrA, 15,220.
Assays
Unless noted, all assays were performed at 25 °C in PDN buffer. Isothermal titration calorimetry was performed and analyzed as described,20 using a Microcal VP-ITC instrument (Amherst, Massachusetts). All components were degassed, and 159 μM ATP was loaded into a 300-μL syringe and injected in 15 μl aliquots at 10 min intervals into a 1.4-ml cell containing E257Q H6-HslU (7 μM hexamer). Binding of E257Q H6-HslU to γ-labeled 32P-ATP (GE Healthcare) was assayed by nitrocellulose-filter binding as described (Hersch et al, 2005). Fluorescence was measured using a PTI QM-2000-4SE spectrofluorimeter (Lawrenceville, New Jersey). Binding of mant nucleotides to HslU was assayed by changes in the fluorescence center-of-mass (excitation 340 nm; emission 400–500 nm). To assay dissociation kinetics, 2 μM E257Q HslU hexamer was preincubated with 10 μM mant-ATP. At time zero, non-fluorescent competitor (5 mM ATP or ADP) was added to prevent rebinding of dissociated mant-ATP and the time course of dissociation was monitored by changes in fluorescence (excitation 340 nm; emission 445 nm). In some dissociation experiments, HslV (2 μM) or Arc-IA37-st11-titin-ssrA (10 μM) were also present in the preincubation. For equilibrium-competition experiments, 3 μM E257Q HslU was added to 100 nM mant-ATP and different concentrations of unmodified ATP, ATPγS, or ADP were added for 5 min before determining the degree of competition by the reduction in fluorescence.
Ion-exchange chromatography on a MonoQ PC 1.6/5 column (GE Healthcare) was conducted using a SMART system (Pharmacia). The column was equilibrated in 50 mM Tris-Cl (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 10% glycerol, and 100 μM ATP. 50 μL of 15 μM HslU (hexamer) was loaded and eluted using a linear gradient between equilibration buffer and equilibration buffer with a final concentration of 500 mM NaCl.
ATP hydrolysis was assayed using a coupled system at 25 °C or 37 °C in PD buffer.30 Binding of fluorescein-labeled gt1 peptide to HslU was assayed by changes in fluorescence anisotropy (excitation: 467 nm; emission: 520 nm) using motorized Glan Thompson polarizers.27 Data were collected over 120 s and averaged. When titrating protein against fixed nucleotide, anisotropy values were corrected for scattering. Cleavage of Z-Gly-Gly-Leu-AMC by HslV in the presence or absence of HslU was assayed by changes in fluorescence (excitation: 380 nm; emission: 455 nm) as described.33
Non-linear least-squares fitting of binding and kinetic data was performed using algorithms implements in KaleidaGraph 3.6.2 (Synergy Software).
Acknowledgments
We thank G. Hersch, R. Burton, and E. Gur for advice and materials. Supported by NIH grant AI-15706. The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319 and NIH GM68762) is also gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 2.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ESC, Siddiqui SM, Wah DA, Baker TA. Sculpting the proteome with AAA+ proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 4.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz-Gonzalez MX, Marin I. Proteasome-related HslU and HslV genes typical of eubacteria are widespread in eukaryotes. J Mol Evol. 2006;63:504–512. doi: 10.1007/s00239-005-0282-1. [DOI] [PubMed] [Google Scholar]
- 6.Rohrwild M, Pfeifer G, Santarius U, Muller SA, Huang HC, Engel A, Baumeister W, Goldberg AL. The ATP-dependent HslVU protease from Escherichia coli is a four-ring structure resembling the proteasome. Nat Struct Biol. 1997;4:133–139. doi: 10.1038/nsb0297-133. [DOI] [PubMed] [Google Scholar]
- 7.Kessel M, Wu W, Gottesman S, Kocsis E, Steven AC, Maurizi MR. Six-fold rotational symmetry of ClpQ, the E. coli homolog of the 20S proteasome, and its ATP-dependent activator, ClpY. FEBS Lett. 1996;398:274–278. doi: 10.1016/s0014-5793(96)01261-6. [DOI] [PubMed] [Google Scholar]
- 8.Bochtler M, Ditzel L, Groll M, Huber R. Crystal structure of heat shock locus V (HslV) from Escherichia coli. Proc Natl Acad Sci USA. 1997;94:6070–6074. doi: 10.1073/pnas.94.12.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 10.Song HK, Hartmann C, Ramachandran R, Bochtler M, Behrendt R, Moroder L, Huber R. Mutational studies on HslU and its docking mode with HslV. Proc Natl Acad Sci USA. 2000;97:14103–14108. doi: 10.1073/pnas.250491797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sousa MC, Trame CB, Tsuruta H, Wilbanks SM, Reddy VS, McKay DB. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103:633–643. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 12.Sousa MC, McKay DB. Structure of Haemophilus influenzae HslV protein at 1.9 Å resolution, revealing a cation-binding site near the catalytic site. Acta Crystallogr D Biol Crystallogr. 2001;57:1950–1954. doi: 10.1107/s090744490101575x. [DOI] [PubMed] [Google Scholar]
- 13.Sousa MC, Kessler BM, Overkleeft HS, McKay DB. Crystal structure of HslUV complexed with a vinyl sulfone inhibitor: corroboration of a proposed mechanism of allosteric activation of HslV by HslU. J Mol Biol. 2002;318:779–785. doi: 10.1016/S0022-2836(02)00145-6. [DOI] [PubMed] [Google Scholar]
- 14.Trame CB, McKay DB. Structure of Haemophilus influenzae HslU protein in crystals with one-dimensional disorder twinning. Acta Crystallogr D Biol Crystallogr. 2001;57:1079–1090. doi: 10.1107/s0907444901007673. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Song JJ, Franklin MC, Kamtekar S, Im YJ, Rho SH, Seong IS, Lee CS, Chung CH, Eom SH. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure (Camb) 2001;9:177–184. doi: 10.1016/s0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 16.Kwon AR, Kessler BM, Overkleeft HS, McKay DB. Structure and reactivity of an asymmetric complex between HslV and I-domain deleted HslU, a prokaryotic homolog of the eukaryotic proteasome. J Mol Biol. 2003;330:185–195. doi: 10.1016/s0022-2836(03)00580-1. [DOI] [PubMed] [Google Scholar]
- 17.Song HK, Bochtler M, Azim MK, Hartmann C, Huber R, Ramachandran R. Isolation and characterization of the prokaryotic proteasome homolog HslVU (ClpQY) from Thermotoga maritima and the crystal structure of HslV. Biophys Chem. 2003;100:437–452. doi: 10.1016/s0301-4622(02)00297-1. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Song JJ, Seong IS, Franklin MC, Kamtekar S, Eom SH, Chung CH. Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure. 2001;9:1107–1116. doi: 10.1016/s0969-2126(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 19.Kim DY, Kim KK. Crystal structure of ClpX molecular chaperone from Helicobacter pylori. J Biol Chem. 2003;278:50664–50670. doi: 10.1074/jbc.M305882200. [DOI] [PubMed] [Google Scholar]
- 20.Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase, allosteric control of a protein machine. Cell. 2005;121:1017–1027. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 21.Martin A, Baker TA, Sauer RT. Rebuilt AAA+ motors reveal operating principles for ATP-fueled machines. Nature. 2005;437:1115–1120. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 23.Gai D, Zhao R, Li D, Finkielstein CV, Chen XS. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell. 2004;119:47–60. doi: 10.1016/j.cell.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Chen B, Doucleff M, Wemmer DE, De Carlo S, Huang HH, Nogales E, Hoover TR, Kondrashkina E, Guo L, Nixon BT. ATP ground- and transition states of bacterial enhancer binding AAA+ ATPases support complex formation with their target protein, sigma54. Structure. 2007;15:429–440. doi: 10.1016/j.str.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S, Choi JM, Tsai FT. Visualizing the ATPase cycle in a protein disaggregating machine, structural basis for substrate binding by ClpB. Mol Cell. 2007;25:261–271. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon AR, Trame CB, McKay DB. Kinetics of protein substrate degradation by HslUV. J Struct Biol. 2004;146:141–147. doi: 10.1016/j.jsb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Burton RE, Baker TA, Sauer RT. Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat Struct Mol Biol. 2005;12:245–251. doi: 10.1038/nsmb898. [DOI] [PubMed] [Google Scholar]
- 28.Rohrwild M, Coux O, Huang HC, Moerschell RP, Yoo SJ, Seol JH, Chung CH, Goldberg AL. HslV-HslU, A novel ATP-dependent protease complex in Escherichia coli related to the eukaryotic proteasome. Proc Natl Acad Sci USA. 1996;93:5808–5813. doi: 10.1073/pnas.93.12.5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seol JH, Yoo SJ, Shin DH, Shim YK, Kang MS, Goldberg AL, Chung CH. The heat-shock protein HslVU from Escherichia coli is a protein-activated ATPase as well as an ATP-dependent proteinase. Eur J Biochem. 1997;247:1143–1150. doi: 10.1111/j.1432-1033.1997.01143.x. [DOI] [PubMed] [Google Scholar]
- 30.Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT. Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J. 2001;20:3092–3100. doi: 10.1093/emboj/20.12.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoo SJ, Seol JH, Shin DH, Rohrwild M, Kang MS, Tanaka K, Goldberg AL, Chung CH. Purification and characterization of the heat shock proteins HslV and HslU that form a new ATP-dependent protease in Escherichia coli. J Biol Chem. 1996;271:14035–14040. doi: 10.1074/jbc.271.24.14035. [DOI] [PubMed] [Google Scholar]
- 32.Azim MK, Goehring W, Song HK, Ramachandran R, Bochtler M, Goettig P. Characterization of the HslU chaperone affinity for HslV protease. Protein Sci. 2005;14:1357–13562. doi: 10.1110/ps.04970405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H, Goldberg AL. Proteolytic activity of the ATP-dependent protease HslVU can be uncoupled from ATP hydrolysis. J Biol Chem. 1997;272:21364–21372. doi: 10.1074/jbc.272.34.21364. [DOI] [PubMed] [Google Scholar]
- 34.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 35.Yoo SJ, Seol JH, Seong IS, Kang MS, Chung CH. ATP binding, but not its hydrolysis, is required for assembly and proteolytic activity of the HslVU protease in Escherichia coli. Biochem Biophys Res Commun. 1997;238:581–585. doi: 10.1006/bbrc.1997.7341. [DOI] [PubMed] [Google Scholar]
- 36.Ramachandran R, Hartmann C, Song HK, Huber R, Bochtler M. Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY) Proc Natl Acad Sci USA. 2002;99:7396–73401. doi: 10.1073/pnas.102188799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seong IS, Kang MS, Choi MK, Lee JW, Koh OJ, Wang J, Eom SH, Chung CH. The C-terminal tails of HslU ATPase act as a molecular switch for activation of HslV peptidase. J Biol Chem. 2002;277:25976–25982. doi: 10.1074/jbc.M202793200. [DOI] [PubMed] [Google Scholar]
- 38.Martin A, Baker TA, Sauer RT. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell. 2007;27:41–54. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishii W, Takahashi K. Determination of the cleavage sites in SulA, a cell division inhibitor, by the ATP-dependent HslVU protease from Escherichia coli. FEBS Lett. 2003;553:351–354. doi: 10.1016/s0014-5793(03)01044-5. [DOI] [PubMed] [Google Scholar]