

Abstract
The trp RNA-binding attenuation protein (TRAP) functions in many Bacilli to control the expression of the tryptophan biosynthesis genes. Transcription of the trp operon is controlled by TRAP through an attenuation mechanism, in which competition between two alternative secondary structural elements in the 5′ leader sequence of the nascent mRNA is influenced by tryptophan-dependent binding of TRAP to the RNA. Previously, NMR studies of the undecamer (11-mer) suggested that tryptophan-dependent control of RNA binding by TRAP is accomplished through ligand-induced changes in protein dynamics. We now present further insights into this ligand-coupled event from hydrogen/deuterium (H/D) exchange analysis, differential scanning calorimetry (DSC), and isothermal titration calorimetry (ITC). Scanning calorimetry showed tryptophan dissociation to be independent of global protein unfolding, while analysis of the temperature dependence of the binding enthalpy by ITC revealed a negative heat capacity change larger than expected from surface burial, a hallmark of binding-coupled processes. Analysis of this excess heat capacity change using parameters derived from protein folding studies, corresponds to the ordering of 17-24 residues per monomer of TRAP upon tryptophan binding. This result is in agreement with qualitative analysis of residue-specific broadening observed in TROSY NMR spectra of the 91 kDa oligomer. Implications for the mechanism of ligand-mediated TRAP activation through a shift in a preexisting conformational equilibrium and an induced fit conformational change are discussed.
Keywords: calorimetry, binding-coupled protein folding, allosteric regulation, oligomer, trp RNA-binding attenuation protein, induced fit, pre-existing conformational equilibrium
The undecameric (11-mer) trp RNA-binding attenuation protein (TRAP) is responsible for controlling the transcription (1-5), and in some cases the translation (6-11), of the genes responsible for tryptophan biosynthesis in many Bacilli. Transcriptional regulation of the trp operon in these Bacilli is achieved through attenuation, in which competing secondary-structural elements in the 5′ leader region of the nascent mRNA control the extent of transcriptional read-through of the structural genes (3-5). TRAP exercises transcriptional control by influencing the formation of these secondary-structural elements through tryptophan-dependent binding to the RNA. When tryptophan is limiting, TRAP is inactive and does not bind to the RNA. This allows a stable anti-terminator hairpin to form, promoting transcriptional read-through of the entire operon. However, when the intracellular tryptophan level is sufficiently high, TRAP binds to tryptophan and becomes activated to bind to eleven triplet repeats of (G/U)AG’s present in the 5′ leader region of the mRNA. When TRAP is bound, the anti-terminator RNA structure cannot form, allowing preferential formation of the terminator hairpin, thereby halting transcription (3-5).
The crystal structures of tryptophan-activated Bacillus stearothermophilus TRAP in binary complex with tryptophan (1QAW) (12) and in ternary complex with tryptophan and RNA (1C9S) (13, 14) reveal an oligomer of eleven identical subunits related by an eleven-fold rotational axis of symmetry. The structure is comprised of eleven anti-parallel beta-sheets, with each sheet consisting of three strands from one subunit and four strands from the neighboring subunit (Figure 1). The tryptophan ligand is completely buried between the beta-sheets at the interface between subunits in a cavity that is largely apolar with a few polar side-chain and backbone moieties making hydrogen bond contacts to the sidechain amide and backbone amino and carboxyl groups of the tryptophan (12, 14-16). The bound RNA wraps around the protein with conserved bases making specific contacts with TRAP residues (13, 16-19).
Figure 1.
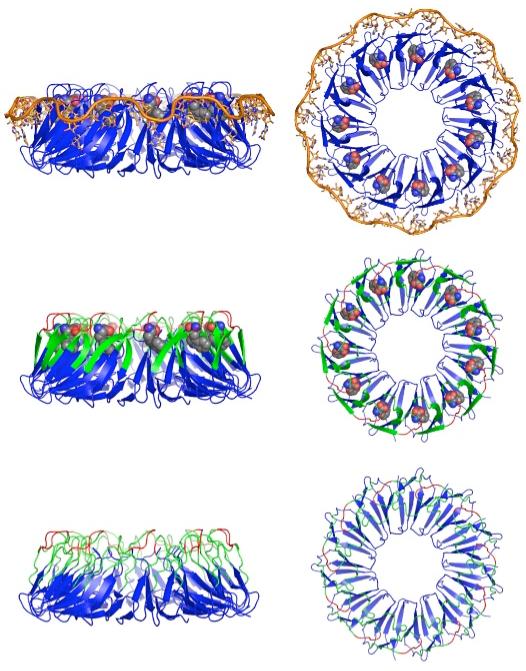
Structures and model of TRAP activation; left/right, side/top views. Crystal structures of B. stearothermophilus TRAP in ternary complex with tryptophan and RNA (Top; 1C9S (13)) and in binary complex with tryptophan (Middle; 1QAW (14)). (Top) The eleven subunits are displayed as blue cartoons, the tryptophan ligands are shown in CPK and the RNA in orange. The tryptophan ligands are buried in the interface between subunits and the RNA wraps around the perimeter of the TRAP oligomer making contacts to beta sheet residues. (Bottom) Model of the structure of TRAP in the absence of tryptophan illustrating dynamic disorder of residues in the Trp- and RNA-binding regions of the protein (21). (Middle, Bottom) Loop residues that exhibited severe exchange broadening in NMR spectra of apo-TRAP are green, while loop residues in red were exchange broadened in both apo- and holo-TRAP; in addition, residues 1-8 and 71-74 could not be assigned for either state (21).
Until recently, limited data were available on the structure of inactivate, apo-TRAP; therefore, little was known about the allosteric mechanism of tryptophan activation of TRAP for RNA-binding. The oligomeric state of TRAP (11-mer) is not altered by tryptophan, remaining oligomeric over a wide range of concentrations (10-6 - 10-2 M), ruling out assembly as an activation mechanism (20-22). TROSY NMR studies of the 91-kilodalton TRAP protein both free and bound to tryptophan revealed severe exchange broadening for the backbone amide resonances of 19 residues in the tryptophan and RNA binding regions of the protein (see Figure 1, Figure 2), while limited proteolysis revealed a ligand-dependent change in proteolytic accessibility for this region (21). Analytical ultra-centrifugation studies of apo- and holo-TRAP (22) also suggest that holo-TRAP is more tightly packed than apo-TRAP, consistent with the protein becoming more structured upon tryptophan binding. These findings are consistent with the proposal that tryptophan activates TRAP to bind to its RNA target through binding-coupled folding of the RNA binding surface of TRAP (21).
Figure 2.

Close-up view of tryptophan-mediated spectral perturbations. Two adjacent subunits in the undecamer are shown in grey and their tryptophan ligands are drawn as CPK. For the middle protomer the same color scheme is used as in Figure 1. Small spheres identify residues whose backbone amide hydrogens exhibited no measurable exchange with deuterium (H/D exchange) in either apo- or holo-TRAP, while the large spheres identify residues whose H/D exchange rate was reduced upon tryptophan binding.
Here we report results aimed at providing a thermodynamic description of the tryptophan binding-coupled transition that leads to TRAP activation. Hydrogen/deuterium (H/D) exchange experiments revealed increased protection of amide resonances upon binding tryptophan. A binding titration monitored by intrinsic tryptophan fluorescence indicated that the 11 tryptophan ligands bind to TRAP non-cooperatively. Scanning calorimetry showed that ligand dissociation involves a large thermal event separate from global protein unfolding. The binding-associated heat capacity change (ΔCp) measured by isothermal titration calorimetry (ITC) revealed a larger ΔCp than expected from burial of solvent accessible surface area (23-30). Interpretation of the excess ΔCp using protein folding models yields qualitative agreement with residue-specific conformational exchange broadening of residues in apo-TRAP. These results are consistent with a model in which TRAP activation proceeds through a mechanism involving both selection from a pre-existing conformational equilibrium, and an induced fit conformational change.
Materials and Methods
Preparation of apo-TRAP
B. stearothermophilus TRAP was expressed in E. coli and purified as described previously (21). Although tryptophan binding is relatively weak (∼1 μM), complete removal of the tryptophan from TRAP by dialysis is inefficient. Therefore, to remove the tryptophan that co-purifies with the protein, TRAP was purified by reversed phase HPLC on a 1 × 25 cm C4 column (Vydac model 214TP) at a flow rate of 3 ml/min using a gradient elution with 0.1% (v/v) trifluoroacetic acid (TFA) (Sigma) in water (buffer A) and 0.1% TFA in acetonitrile (buffer B). The gradient elution consisted of a 15 ml wash with 5% buffer B, a 30 ml gradient from 5-20% buffer B, a 30 ml wash with 20% buffer B (during which the tryptophan eluted), and a 90 ml gradient from 20-70% buffer B, where TRAP eluted between 40 and 60% acetonitrile. The fractions containing TRAP were then lyophilized and the powder (∼32 mg) was dissolved in 3 ml of 6 M guanidine hydrochloride (GdnHCl), 50 mM sodium phosphate at pH 8.0, and 100 mM sodium chloride (NaCl). The re-suspended TRAP was placed in a 3500 Da cutoff dialysis cassette (Pierce) and dialyzed against 0.5 L of 3 M GdnHCl, 50 mM sodium phosphate at pH 8.0, 100 mM NaCl for 12 hours at 55 °C. The GdnHCl was then diluted to 1.5 M with 50 mM sodium phosphate at pH 8.0 and 100 mM NaCl (buffer C) and the protein was dialyzed for an additional 12 hours at 55 °C. The protein was then dialyzed against 4 L of buffer C for 24 hours at 55 °C, followed by an additional dialysis against 4 L of buffer C for 48 hours at 4 °C. Previous studies have shown that TRAP can be denatured into unfolded monomers by GdnHCl and then renatured into fully functional 11-mers by simply removing the denaturant (20). The efficacy of the refolding procedure was confirmed by the ITC experiments, which yielded an n-value close to 11 (10.16-11.07, Table 1) for each oligomer. Distilled, deionized water (MilliQ, Millipore, Inc.) was used for all buffers.
Table 1.
Temperature dependent thermodynamics for the titration of tryptophan into TRAPa
| Temperature | n | ΔG | ΔH | TΔS | KA (/106) |
|---|---|---|---|---|---|
| 298 | 10.97 ± 0.03 | -9.55 ± 0.06 | -13.88 ± 0.09 | -4.33 ± 0.11 | 9.75 ± 0.16 |
| 303 | 11.07 ± 0.09 | -9.43 ± 0.01 | -15.60 ± 0.06 | -6.17 ± 0.07 | 6.28 ± 0.24 |
| 308 | 10.77 ± 0.29 | -9.35 ± 0.01 | -17.37 ± 0.07 | -8.02 ± 0.07 | 4.24 ± 0.05 |
| 313 | 10.40 ± 0.10 | -9.22 ± 0.32 | -19.13 ± 0.25 | -9.91 ± 0.41 | 2.72 ± 0.16 |
| 318 | 10.16 ± 0.23 | -9.13 ± 0.01 | -21.10 ± 0.43 | -11.97 ± 0.43 | 1.83 ± 0.04 |
| 323 | 10.53 ± 0.24 | -8.98 ± 0.02 | -23.03 ± 0.04 | -14.05 ± 0.05 | 1.17 ± 0.05 |
Reported errors are the standard deviation of three repeat data sets at each temperature. Values of ΔΗ, ΤΔS, and ΔG are in kcal mol-1, T is in Kelvin, KA is in M-1, and n is the number of tryptophan binding sites per TRAP oligomer.
H/D exchange of TRAP
Apo and holo-TRAP were exchanged into 90/10 (2H2O/1H2O) by a ten-fold dilution into buffer C in 99.9% 2H2O, adjusting the pH to account for the pH/pD difference (31). TRAP was then concentrated using an ultra-filtration centrifugal concentrator (Amicon). The first time point was taken approximately 1 hour after exchanging into 2H2O. Two-dimensional 1H-15N TROSY NMR spectra (32) were recorded on an 800 MHz NMR spectrometer at 55 °C, every hour for the first 24 hours, with additional time points taken after 48 hours and one week; samples were stored at 55 °C between spectra.
Tryptophan fluorescence
Protein concentration was estimated using the native extinction coefficient, 2798.6 M-1 cm-1 at 276 nm, determined using the theoretical extinction coefficient, 2900 M-1 cm-1 at 276 nm, and comparing the absorbance under denaturing (6 M GdnHCl and 100 mM sodium phosphate at pH 6.5) and native conditions (50 mM sodium phosphate at pH 8.0, 100 mM NaCl) (33). The tryptophan concentration was determined using the extinction coefficient 5579 M-1cm-1at 278 nm (34). For the concentrated solutions of protein or tryptophan, errors in the estimation of the concentrations were obtained from two different dilutions; for the dilute solutions, errors are the deviation of two measurements taken on the same solution. In order to ensure that the buffer conditions of the protein and ligand were identical, tryptophan (Sigma) was dissolved in the dialysate from the last dialysis of TRAP. All fluorescence experiments were performed on a Fluoromax-3 fluorometer with an excitation wavelength of 295 nm, an integration time of 0.1 s, 5 scan signal averaging, a slit width of 2 nm, and scans taken from 300-450 nm at 1 nm increments. The cell contained 2.8 ± 0.2 μM tryptophan and was titrated with 46.3 ± 0.3 μM apo-TRAP oligomer. Binding was monitored from the emission intensity at 332 nm after each addition of TRAP and the fraction of bound tryptophan was calculated from:
| (1) |
where ri is the fraction of bound tryptophan after the ith addition of TRAP, Ii is the signal intensity at 332 nm after the ith addition of TRAP, If is the intensity at 332 nm of free tryptophan, and Ib is the intensity at 332 nm of fully bound tryptophan. The resultant binding curves were fit to a standard binding quadratic:
| (2) |
where ri is the fraction of bound tryptophan, n is the number of binding sites, Kd is the dissociation constant, [Trp]T is the total tryptophan concentration, and [TRAP]T is the total TRAP oligomer concentration. The Hill coefficient was determined by taking the slope of the best fit line from the plot of the log (ri/(1-ri)) versus the log [TRAP]f (the free TRAP concentration).
ITC of TRAP
Protein and tryptophan concentrations were estimated as described above. All ITC data were obtained on a MicroCal VP-ITC with a 3 μl first injection followed by 5 μl injections with a spacing of 220 seconds between injections. The cell contained TRAP at a concentration of 5.1 ± 0.6 μM oligomer and the syringe contained 662.1 ± 12.0 μM tryptophan. The exothermic heat pulse detected after each injection was integrated, the heat of dilution subtracted from the integrated value, and the corrected heat value divided by the total moles of tryptophan injected. The resulting values were plotted as a function of the molar ratio of TRAP oligomer and fit to a one binding site model using a non-linear least squares method (Figure 5; Origin v. 7, MicroCal); ΔS values were obtained via Gibbs’ relation from the fitted KA and ΔH values. All experiments were performed in triplicate; reported errors are the standard deviation in the fitted values between the three data sets. Although the extremely low extinction coefficient of TRAP leads to ∼10% uncertainty in the protein concentration, the samples used for the ITC studies were prepared simultaneously from the same stock solution. Consequently, the small variation in the measured parameters reflects the exceptional signal-to-noise of the titration data, not the uncertainty in the protein concentration. In order to subtract the heat of dilution for both ligand and protein simultaneously, baseline runs were performed using the fully titrated TRAP (after removing the excess volume and refilling the syringe with ligand). The heat capacity change was obtained from a linear fit of the enthalpy data to:
| (3) |
where Th is the temperature at which the binding enthalpy is zero. The temperature at which the entropic contribution to binding is zero (Ts) was obtained from fitting the temperature dependence of the binding free energy to the modified Gibbs-Helmholtz equation:
| (4) |
Figure 5.

ITC of TRAP. (a) Thermogram from a titration of tryptophan into TRAP (25 °C) showing the change in power required to maintain a constant temperature difference between the sample and reference cells upon injections of tryptophan. (b) Time-integrated heat data normalized per mole of injectant (squares) with nonlinear fit to identical and independent sites model (line). Best fit parameters are shown in Table 1.
Differential scanning calorimetry
Protein and tryptophan concentrations were estimated as described above. All DSC data were acquired on a MicroCal VP-DSC with a scan rate of 60 °C/hour and scans taken from 25 to 130 °C at a starting pressure of 29 psi and cells were refilled at 27 °C. The protein concentration for apo-TRAP was 51.1 μM TRAP (562.3 μM monomer) and for holo-TRAP was 50.0 μM (550.5 μM monomer). 26.6 mM tryptophan was added to a final concentration of 560 μM in the holo-TRAP sample. Data were normalized to the protein concentration and the holo-TRAP data were y-axis translated to make the initial baselines for the apo and holo-TRAP data coincident using Origin v. 7 (MicroCal).
Solvent accessible surface area, , and ΔSHE
Solvent accessible surface areas (ASA) of both the tryptophan ligand and the tryptophan-binding cavities of TRAP were determined using STC v. 5.0 (35) with extended atom radii and a probe radius of 1.4 Å. To measure the change in solvent accessible surface area upon tryptophan binding, separate PDB files were constructed containing one protomer of TRAP and one tryptophan ligand from the crystal structure of the binary complex of tryptophan with Bacillus stearothermophilus TRAP (1QAW) (14). In all, 23 files were generated, two for each protomer (one with each of the two tryptophan ligands that are contacted by that protomer). The average polar and non-polar solvent accessible surface areas of the tryptophan ligands were obtained from the STC calculated values for the free tryptophans. The average change in polar and non-polar solvent accessible surface area of the tryptophan binding pockets was calculated by adding the change in solvent accessible surface area for the two protomers that contact the same tryptophan. The overall change in polar and non-polar solvent accessible surface is the sum of the solvent accessible surface area of the tryptophan and the solvent accessible surface area of the binding pocket. The heat capacity change expected from the structural data () was calculated from the change in ASA using (28):
| (5) |
where ΔAp is the change in polar solvent accessible surface area, ΔAnp is the change in non-polar solvent accessible surface area, and ΔCp is the heat capacity change. The change in entropy due to the hydrophobic effect (ΔSHE) in the absence of a change in conformation or folding was calculated from (28):
| (6) |
where Ts is the temperature at which the entropy of association is zero.
Proton and ion linkage effects
To determine whether proton or ion linkage contributes to the heat capacity of tryptophan binding to TRAP (30, 36-38), apo-TRAP was prepared as described above, but with one additional dialysis step. After refolding, the resultant protein was split into seven aliquots, which were placed into separate 3500 Da cutoff dialysis cassettes and dialyzed into the appropriate buffers. Buffers used to test for ion linkage (38) were 50 mM sodium phosphate at pH 8.0 with no NaCl, 100 mM NaCl, 250 mM NaCl, and 500 mM NaCl. The buffers used for analysis of proton-linkage contributions were 50 mM sodium phosphate at pH 8.0, 100 mM NaCl; 20 mM Tris at pH 8.0, 100 mM NaCl; 20 mM HEPES at pH 8.0, 100 mM NaCl; ionization enthalpies used were as reported (37).
ΔCp and binding-coupled folding
As proposed by Spolar and Record (28), the experimentally measured heat capacity change was used to estimate the loss in protein conformational entropy upon binding tryptophan. Once the contributions to ΔCp from other sources are determined (30, 36-38), the remaining (excess) heat capacity was used to calculate the contribution to association from the hydrophobic effect (ΔSHE):
| (7) |
where the factor 1.35 assumes a ΔAp/ΔAnp = 0.59 (which is approximately the case in the folding of globular proteins)(28), and 386 K is the temperature at which the entropy contribution of the hydrophobic effect vanishes (23). The entropy of association (ΔSassoc) can be further dissected at the characteristic temperature (Ts) where the overall association entropy change is zero:
| (8) |
where ΔSrt is the change in entropy due to loss of rotational and translational freedom and ΔSother is the change in entropy due to other events attributed predominantly to the loss of conformational mobility upon protein folding (28). Since at the Ts the change in entropy of molecular association is zero, the magnitude of ΔSHE must equal the sum of ΔSrt and ΔSother. For molecular associations in which no conformational rearrangements of the ligand or receptor occur (rigid-body association), ΔSother should be negligible and ΔSHE should be entirely balanced by ΔSrt. Analysis of a number of proteins for which structural information suggests that there are no binding-coupled conformational rearrangements or folding yields an empirical estimate for ΔSrt of -50 ± 10 cal K-1 mol-1 (28). Thus, from the remainder (ΔSother) one obtains an estimate of the change in protein conformation entropy upon ligand binding.
Empirical studies of the thermodynamics of protein folding suggest that the unfavorable entropic cost of protein folding is relatively constant when expressed per residue, ΔSℜ, although reported values are somewhat dependent on the dataset used to derive it (28, 39, 40), ranging from -4.1 to -5.6 cal K-1 (mol res)-1. This value can be used to estimate the number of residues that become folded upon binding (ℜ) from:
| (9) |
Estimation of ΔCp for binding-coupled folding from structural data
The 19 residues in the tryptophan and RNA binding sites that become exchange broadened upon removal of ligand (21-25, 31-35, 45-46, and 48-54), were modeled in an extended conformation using MOLMOL (41) and ASA calculations were performed using STC v. 5.0 (35) with both the extended structures and the tryptophan treated as the “ligand” for the calculations. Although the use of fully extended conformations for these loops will result in an overestimate of the ASA change upon folding these residues, in the absence of accurate models of the loops in apo-TRAP, use of the extended conformation provides an upper limit for the ASA change. The ASA buried by the linkage between the structured and unstructured regions was subtracted from the total ASA and the resulting value was used to calculate the ΔCp for binding-coupled folding using equation 3. Inclusion of the six loop residues that remain exchange-broadened in Trp-bound TRAP (26-30 and 47, shown in red, in Figure 2) would increase the estimated ASA change roughly proportionally (∼30%).
Results and Discussion
Tryptophan binding to B. stearothermophilus TRAP is non-cooperative
Prior to detailed analysis of the mechanism of TRAP activation we examined whether tryptophan binding to B. stearothermophilus TRAP is cooperative. We monitored binding of TRAP to tryptophan at 25 °C using the change in intrinsic fluorescence of the latter upon binding (Figure 3). The wavelength for the emission maximum of tryptophan fluorescence shows the typical blue shift (from 357 nm to 332 nm) and increase in intensity (∼6-fold) that is expected upon transfer of tryptophan from a polar to nonpolar environment (42). The data were well described by a single-site binding quadratic (Figure 3, bottom) and was fit with an equilibrium binding constant, KA, of 6.32 ± 0.3 × 106 M-1. Analysis of the binding data using a Hill plot (Figure 3, inset) yielded highly linear data with a Hill coefficient near unity. Titrations repeated over a range of temperatures (25-70) yielded similar Hill coefficients (not shown). Thus, although there are 11 tryptophan-binding sites in each TRAP oligomer, this analysis indicates that tryptophan binding to B. stearothermophilus TRAP can be reasonably treated as involving 11 identical and independent sites, instead of requiring the use of more complicated partition functions (43).
Figure 3.

Non-cooperative binding of tryptophan to TRAP. (a) Titration of tryptophan with TRAP monitored by tryptophan fluorescence (25 °C). (b) A typical binding isotherm for TRAP binding tryptophan (squares) with a non-linear fit to equation 2 (line); at 25 °C the dissociation constant (Kd) was 0.16 ± 0.01 μM, with a best-fit stoichiometry of 10.6 ± 0.9 tryptophans per TRAP 11-mer. Inset, Hill plot demonstrating lack of cooperativity in tryptophan binding to TRAP; Hill coefficient is 1.02 ± 0.01 (slope of the best fit line).
Structural changes upon tryptophan binding: H/D exchange
On the basis of site-specific NMR resonance linebroadening we previously proposed that ligand binding to TRAP involved a ligand-mediated change in protein dynamics - coupled folding (21). The structure of holo-TRAP reveals an all beta sheet protein (12, 14); therefore, if tryptophan binding leads to stabilization of secondary structural elements, one might expect a concomitant increase in beta-sheet signature in the circular dichroism (CD) spectrum. However, the relative inaccuracy of quantitating beta sheet structure (44, 45), coupled with interference from the tryptophan ligand (46, 47), rendered CD studies uninformative of ligand-induced structural changes (data not shown). On the other hand, ligand-induced stabilization of the protein structure could be ascertained from hydrogen/deuterium exchange experiments.
If folding were coupled to ligand binding, upon tryptophan addition to TRAP the amide hydrogens of residues involved in the folding transition should show increased protection from exchange with solvent. Unfortunately, the majority of the TRAP residues of most interest (i.e., those in the tryptophan and RNA-binding regions; colored red and green in Figure 1 and Figure 2) are broadened beyond detection in the apo-protein (21), preventing NMR-based measurement of the hydrogen/deuterium (H/D) exchange rates of these residues. Nevertheless, H/D exchange experiments (Figure 4) provided important insight into the ligand induced structural changes in TRAP. Many of the amides show no measurable exchange even after a week, revealing the presence of an extremely stable structured core in both apo- and holo-TRAP. Thus, we conclude that the amides with measurable rates exchange via local, not global fluctuations in the protein structure. On the other hand, increased protection from exchange in holo-TRAP is evident for several residues, including Ala12, Val19, Ile20, Leu36, Asp37, Glu40 and Ile61 (large spheres in Figure 2), reflecting local ligand-induced stabilization of the protein structure. The increased protection of amides from Asp37 and Glu40 (Figure 2, Figure 4) is particularly telling, since these residues are involved in RNA base recognition (13), reflecting the mechanism by which ligand binding is transmitted to the RNA binding residues. These data are consistent with the hypothesis (21) that apo-TRAP is unable to bind RNA due to conformational disorder of the RNA-binding residues, and that tryptophan binding likely proceeds through preferential binding to one conformation of a pre-existing conformational equilibrium (48).
Figure 4.

Hydrogen/deuterium exchange in apo- and holo-TRAP. An expanded region of 1H-15N correlation spectra of apo- (bottom) and holo- (top) TRAP recorded before and at a series of time intervals (left to right: 0, 1, 5, 9 and 15 hours) after exchange into 90/10 2H2O/1H2O.
Thermodynamics of tryptophan binding to TRAP
Difficulties in characterizing the ligand-induced structural/dynamic changes in TRAP using spectroscopic techniques led us to use isothermal titration calorimetry (ITC) to measure the change in thermodynamic heat capacity (ΔCp) as a means of characterizing binding-coupled changes in protein structure (23-28, 30). Excellent quality ITC data were obtained (Figure 5), with high signal-to-noise over a range of temperatures, enabling accurate determination of association constants (KA), enthalpy changes (ΔH), and the stoichiometry of binding (n) (Table 1). Consistent with the fluorescence titration, the ITC data fit well to a one-site model allowing the sites to be treated as identical and independent, simplifying further analysis.
ITC data were recorded at five-degree intervals between 25 and 50 °C. Strong enthalpy/entropy compensation was observed, making the Gibbs free energy of binding (ΔG) nearly independent of temperature with a change of only 22 cal mol-1 K-1 over the temperature range (Figure 6). Tryptophan binding was found to be slightly temperature dependent as noted previously (21), with KA values ranging from 9.75 × 106 M-1 at 25 °C to 1.17 × 106 M-1 at 50 °C.† The temperature dependence of the binding enthalpy was found to be linear in the measured temperature range, and a large negative change in heat capacity (ΔCp) of -370 cal mol-1 K-1 was obtained from the temperature dependence of the binding enthalpy, δΔH/δT (Figure 6). Analysis of the temperature dependence of the binding entropy, TδΔS/δT, yielded a value of 287.0 K for the characteristic temperature (Ts) at which the entropic contribution to binding (ΔSassoc) is zero. Notably, the linearity of the ΔH versus T plot indicates that there is no measurable shift in the pre-existing equilibrium between binding-competent and binding-incompetent conformational states of apo-TRAP, in the temperature range studied (36, 49, 50).
Figure 6.

Temperature dependence of tryptophan binding to TRAP. The error bars represent the standard deviation in three repeat measurements at each temperature. Lines represent the best fit of the Gibbs-Helmholtz equation to the data. The data show the temperature dependence of the binding enthalpy (ΔHassoc, circles), entropy (TΔSassoc, triangles), and free energy change (ΔGassoc, squares). A linear fit of the temperature dependence of the enthalpy yields a heat capacity change (ΔCp) of -370 cal mol-1 K-1, while fitting the temperature dependence of the free energy to the Gibbs-Helmholtz equation yields 287.0 K as the temperature (Ts) at which the association entropy change is zero.
Thermal (DSC) scans of apo- and holo-TRAP (Figure 7) showed the partial specific heat capacity to be linear in the pre-transition temperature range where ITC experiments were carried out. DSC analysis of apo- and holo-TRAP also showed the transition for ligand dissociation to be separate from that of global unfolding, occurring between 70 and 100 °C, with a midpoint temperature (Tm) of 85 °C. As a bimolecular association, the Tm for ligand dissociation would be expected to be concentration dependent; this was confirmed by a DSC scan at five-fold lower concentration which showed the Tm to be slightly lower (83 °C) but still well separated from global unfolding. Assignment of the low temperature transition to ligand dissociation was confirmed by monitoring the temperature dependent dissociation of tryptophan by fluorescence (not shown). Interestingly, the observed independence of ligand dissociation and global unfolding in holo-TRAP is reminiscent of the thermal unfolding behavior of proteins with independent domains (51). Further, the high thermal stability for global unfolding of the oligomeric protein (> 100 °C) is evident from the data. Non-reversibility of the high-temperature transition and absence of a post-transition baseline precluded detailed deconvolution analysis of the traces. The fact that there are two thermal transitions in addition to that associated with ligand binding merits more careful investigation, but may indicate that thermal disassembly and denaturation of the oligomeric complex are not tightly coupled.
Figure 7.

DSC of apo- and holo-TRAP. Scanning thermograms of apo (solid line) and holo-TRAP (dashed line) reveal (1) the hyperthermal stability of the oligomeric protein, (2) the thermal separation between tryptophan dissociation and global unfolding of the protein, and (3) the linearity of the partial specific heat capacity in the temperature range studied by ITC (25-50 °C). Quantitative deconvolution analyses of these traces is not possible however, due to the absence of a post-transition baseline and by lack of reversibility of the high temperature global unfolding (as illustrated by the re-scan of holo-TRAP, dotted line).
Solvent accessible surface area and the expected ΔCp
The heat capacity change upon protein folding or protein-ligand binding is often well described by solvation effects arising from changes in solvent accessible surface area (ASA) (23-30). Coupling of conformational changes (30, 36), including local protein folding (28, 52), to ligand binding leads to a greater heat capacity change than that expected from surface burial (ΔASA) upon binding. The heat capacity change expected from the burial of polar and nonpolar surface area () has been parametrized (29, 53-55) and can be calculated from the three-dimensional structure of the complex. The change in polar and non-polar ASA due to burial of the tryptophan in the tryptophan-binding cavity was determined from the crystal structure of B. stearothermophilus TRAP in complex with tryptophan (1QAW) (14) using the program STC (35) (Table 2). From the change in ASA, and the parameters of Spolar & Record (28) we calculated an expected change in heat capacity upon association () in the absence of protein folding of -88 cal M-1 K-1; similar values are obtained with alternate parametrizations (29, 53-55), including when aromatic and aliphatic carbons are treated differently (29). The difference between the calculated value and that measured by ITC (-370 cal M-1 K-1) demonstrates that the large negative ΔCp is poorly accounted for by surface burial, indicating a large contribution from other binding-coupled events.
Table 2.
Thermodynamics of the binding-coupled folding of TRAP
| Change in Solvent Accessible Surface Area (ΔASA): | |
| Absence of folding a | |
| Polar | 239 Å2 |
| Nonpolar | 378 Å2 |
| Presence of folding b | |
| Polar | 1328 Å2 |
| Nonpolar | 1916 Å2 |
| Change in Heat Capacity (ΔCp): | |
| Expected from tryptophan burial c | -88 cal mol-1 K-1 |
| Experimental | -370 cal mol-1 K-1 |
| Upper limit for folding d | -427 cal mol-1 K-1 |
| Binding-Coupled Folding (ℜ): | |
| ITC | 17-24 residues/monomer |
| NMR e | 19 residues/monomer |
ΔASA for the burial of tryptophan measured from the structure 1QAW (14); see methods.
ΔASA for the burial of tryptophan and the 19 residues that exhibit ligand-dependent line broadening, measured from the structure 1QAW (14); see methods.
Expected heat capacity change in the absence of folding calculated from ΔASA a using equation 1.
Upper limit for the heat capacity change in the presence of folding if all the residues go from an extended structure to a folded structure calculated from ΔASA b using equation 1.
Number of residues for which the NMR resonances are exchange-broadened beyond detection in apo-TRAP but are present in holo-TRAP and are located in the tryptophan or RNA-binding sites.
Contributions to ΔCp
Binding-associated changes in ΔCp can have a number of origins (23, 30, 36): a change in solvent accessible surface area (solvation effects), a change in the protonation state of either binding partner (proton linkage) (37), release of ions due to salt bridge formation during binding (ion linkage) (38), burial of structural water molecules (56, 57), loss of translational and rotational degrees of freedom, changes in conformational entropy (28), and induced fit conformational changes (23-28, 36). In order to understand the nature of the binding event all possible contributors to ΔCp should be considered.
The intermolecular contacts between TRAP and bound tryptophan involve mainly hydrophobic contacts and polar interactions between groups that ionize outside of physiological pH ranges (14). This suggested that proton or ion linkage were unlikely to contribute to the heat capacity change; nonetheless, potential contributions from these sources were explored by performing titrations of the binding partners in buffers with different enthalpies of ionization (37) and salt concentrations (38, 58). The change in enthalpy (ΔH) of tryptophan binding to TRAP at 25 °C was measured in buffers with ionization enthalpies spanning 10 kcal mol-1 (Figure 8). The enthalpy of tryptophan binding was found to be nearly independent of the heat of ionization of the buffer. The binding affinity of TRAP for tryptophan was similarly found to be largely independent of salt concentration (Figure 8). Therefore, the contributions to the observed ΔCp from proton and ion linkage are very small.
Figure 8.

Proton and ion linkage. (a) There is very little change in the binding enthalpy in titrations of tryptophan into TRAP in the presence of buffers with different enthalpies of ionization (a slope of -0.08 compared to a slope of ∼1 in the case of a single proton uptake (37, 71)), demonstrating that changes in protonation state upon binding do not contribute significantly to the observed heat capacity change. (b) Changes in salt concentration show little effect on the association constant (a slope of -0.17), demonstrating that uptake or release of ions also does not contribute significantly to the observed heat capacity change (38, 58).
Burial of structural water molecules in molecular interfaces has been implicated in contributing to the measured ΔCp (56, 57), so possible contribution from bound water molecules was considered. Examination of the tryptophan-binding sites in B. stearothermophilus holo-TRAP alone (1QAW; 2.5 Å resolution)(14) and bound to RNA (1C9S; 1.9 Å solution)(13) reveals a surface-exposed water molecule bound between the carboxylate group of the tryptophan ligand and the sidechain of His49 in the majority of the binding pockets. Estimates for the contribution of burying a water molecule in an interface (i.e., transfer of a water molecule from solution to the protein) have been obtained from sorption isotherms (59), the standard enthalpies of anhydrous and hydrated inorganic salts (60), and from high-resolution structural data (57). These analyses suggest an upper limit of -12 cal mol-1 K-1 for the contribution to ΔCp of burying a water molecule in an interface. Use of these estimates suggests that the contribution to the observed ΔCp of crystallographically-observed water molecules in the binding cavity is small compared to the observed ΔCp (-370 cal K-1 mole-1). It should be noted, however, that the nature of these bound water-mediated contributions to ΔCp are not yet well understood and it has been proposed they can be insignificant (60) or even significantly larger (56, 61, 62).
Binding-coupled folding of TRAP
Changes in structure and conformational mobility that accompany induced-fit binding (i.e., coupled folding) strongly impact measured heat capacity changes (23, 28, 30). We followed the approach of Spolar and Record (28), which provides an empirical estimate of the number of residues involved in coupled folding (ℜ) from the experimentally measured ΔCp (equation 7, methods). Using this approach, neglecting contributions from trapped interfacial water, and using a value for the per-residue entropic loss (ΔSℜ) of -5.6 cal K-1 (mol res)-1 (28), we find that the excess heat capacity change corresponds to the folding of 18 residues per monomer upon tryptophan binding to TRAP (equation 7, methods; Table 2). If we assume that the crystallographically observed water molecules contribute up to -12 cal mol-1 K-1 (57, 59, 60) to the measured heat capacity, this lowers the estimate of ℜ to 17 residues per monomer. Use of -4.1 cal K-1 (mol res)-1 for ΔSℜ (40) increases the estimate to 23 and 24 residues with and without a contribution from water burial, respectively. (We note, however, that there remains considerable disagreement in the literature over the magnitude of the contribution of ΔSrt to the overall entropy of association and has been proposed to be much smaller, on the order of 5 ± 4 cal mol-1 K-1 (63); use of this smaller value would increase ℜ by ∼9 residues.) Thus, following this empirical approach we find that the measured ΔCp corresponds to ligand-coupled loss of conformational mobility (i.e., folding) of 17-24 residues (ℜ) per monomer of TRAP (Table 2); these numbers corresponds to roughly one quarter to one third of the 74 residues in each protomer.
The results of these calorimetric estimations (17-24 residues folded) are in reasonable agreement with NMR studies which showed that 19 residues in the tryptophan and RNA binding regions of TRAP were observed in the presence of tryptophan but were severely exchange broadened in the absence (21) (shown in green in Figure 1 and Figure 2). As a check on the underlying approximations, we tested whether folding of these 19 residues would generate a ΔCp comparable to that measured experimentally. We computed the ΔASA for binding-coupled folding of the 19 residues (see Methods) and obtained a ΔCp of -427 cal mol-1K-1 for a transition between fully extended and exposed conformation, and the fully folded conformation (Table 2). Because the unfolded residues are in loops between regions that remain structured even in apo-TRAP, this number would be expected to over-estimate the possible ASA change, but is of similar magnitude to the experimentally measured value of -370 cal mol-1 K-1.
The agreement between these quantitative and qualitative analyses leads us to conclude that most of the ΔCp measured by ITC arises from the same ligand-coupled changes in protein structure and dynamics (i.e., folding), that lead to the observed differences in the spectra between the apo/inactive and holo/activated states of TRAP. Those structural and dynamic changes are localized principally to the RNA- and tryptophan-binding regions of TRAP (Figure 2). On the other hand, it is clear from the observation that ligand binding affects the H/D exchange rates of residues outside of those 19 broadened by exchange (Figure 2, Figure 4), that the thermodynamic consequences of ligand binding are not entirely limited to those 19 residues and affect the structure, dynamics and thermodynamics of (at least those) additional residues.
Binding-coupled folding and a model for allosteric control of TRAP
The data presented lend support for a model for allosteric control of TRAP in which tryptophan-dependent coupled folding of the tryptophan and RNA-binding residues control RNA-binding (Figure 1) (21). In this model, residues in the tryptophan and RNA-binding region of TRAP are dynamically disordered in the apo protein (i.e., a pre-existing conformational equilibrium (48)), preventing RNA binding and allowing entry of the tryptophan to its binding-site. Initial tryptophan binding to one state of the protein induces ordering of the tryptophan- and RNA-binding residues, thereby shifting the equilibrium, forming a stable RNA-binding interface, and activating TRAP to bind to its RNA target. We propose that the loss of conformational mobility (28) (reduction in “soft vibrational modes”(23)) in this tryptophan and RNA bindingregion upon ligand binding is responsible for much of the large negative heat capacity detected in the calorimetric experiments.
However, it is worth noting that because the tryptophan ligand is completely buried in holo-TRAP (14), the binding-competent conformation of apo-TRAP must be distinct from that of holo-TRAP. Consequently, it seems necessary that upon Trp binding, TRAP must also undergo an induced-fit conformational change (64) from a binding-competent state to the ligand-buried holo- state. Indeed, the absence of a significant population of a holo-like state in apo-TRAP is essential from a regulatory standpoint: because RNA binds very tightly to the activated form of TRAP, if that state were significantly populated in the absence of ligand, RNA binding would drive the equilibrium towards the activated form, thereby degrading the effectiveness of regulation. The equilibrium thermodynamic parameters measured in this study would capture, without distinction, both the initial binding event, and the ensuing conformational change.
Since the tryptophan is completely buried in tryptophan-activated TRAP, it seems reasonable that some of the residues in the tryptophan-binding site must remain dynamic even in the RNA-bound state in order to allow TRAP to remain sensitive to the levels of intracellular tryptophan. This prediction is in agreement with the observation that NMR resonances from the tryptophan-binding region remain exchange-broadened in tryptophan-activated TRAP (regions in red, Figure 1, Figure 2)(21). This observation is also consistent with the observed lack of cooperativity in tryptophan binding (Figure 3, Figure 5), as persistent dynamics would result in, at best, weak coupling between adjacent binding sites. If, on the contrary, ligand binding were to lead to complete rigidification of one interprotomer interface, conventional allostery models (43, 65-68) would predict strong coupling and lead to undesirably high affinity tryptophan binding and loss of regulation.
The apparent independence of thermally induced ligand dissociation and protein denaturation (Figure 7) merits a final point of discussion. Tight coupling between ligand binding and global unfolding of proteins has been widely observed, and deconvolution of thermograms from such systems has been used to obtain the relevant thermodynamic parameters (69, 70). In contrast, the observed independence of local and global unfolding in holo-TRAP is reminiscent of the thermal unfolding behavior of proteins with independent domains (51). The fascinating and complex mystery of how TRAP regulates transcription is hereby further accentuated by the observation that TRAP activation seems to involve the discrete stabilization of an RNA binding “domain” that is thermodynamically distinct from the underlying oligomeric protein scaffold that supports it.
Supplementary Material
{kind=link}
Acknowledgement
The authors thank J. Cowan (OSU) for access to instrumentation, and V. Fresca (Microcal), J. Cowan, S. Walsh (OSU), members of the Foster laboratory for helpful discussions, and Stephen Edmondson (UAH) and John Ladbury (UCL) for critical reading of the manuscript. This work was supported by a Structural Biology supplement from the National Institutes of Health award number GM62750-01 to PG and is based upon work supported by the National Science Foundation under the award number MCB-0092962 to MPF. CAM was supported in part by NIH training grant T32GM008512.
Footnotes
In these studies the temperature dependence of binding was found to be in the opposite direction from the previous studies. Although the origin of this discrepancy is unknown, it is small, and may be a consequence of the differing conditions compared to the previous NMR-based experiment where the protein concentration (between 11-12 mM) was much higher than the Kd (∼1 μM at 50 °C, Table 1).
REFERENCES
- (1).Babitzke P, Yanofsky C. Reconstitution of Bacillus subtilis trp attenuation in vitro with TRAP, the trp RNA-binding attenuation protein. Proc Natl Acad Sci U S A. 1993;90:133–7. doi: 10.1073/pnas.90.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gollnick P. Regulation of the Bacillus subtilis trp operon by an RNA-binding protein. Mol Microbiol. 1994;11:991–7. doi: 10.1111/j.1365-2958.1994.tb00377.x. [DOI] [PubMed] [Google Scholar]
- (3).Gollnick P, Babitzke P. Transcription attenuation. Biochim Biophys Acta. 2002;1577:240–50. doi: 10.1016/s0167-4781(02)00455-4. [DOI] [PubMed] [Google Scholar]
- (4).Babitzke P. Regulation of tryptophan biosynthesis: Trp-ing the TRAP or how Bacillus subtilis reinvented the wheel. Mol Microbiol. 1997;26:1–9. doi: 10.1046/j.1365-2958.1997.5541915.x. [DOI] [PubMed] [Google Scholar]
- (5).Babitzke P. Regulation of transcription attenuation and translation initiation by allosteric control of an RNA-binding protein: the Bacillus subtilis TRAP protein. Curr Opin Microbiol. 2004;7:132–9. doi: 10.1016/j.mib.2004.02.003. [DOI] [PubMed] [Google Scholar]
- (6).Du H, Babitzke P. trp RNA-binding attenuation protein-mediated long distance RNA refolding regulates translation of trpE in Bacillus subtilis. J Biol Chem. 1998;273:20494–503. doi: 10.1074/jbc.273.32.20494. [DOI] [PubMed] [Google Scholar]
- (7).Merino E, Babitzke P, Yanofsky C. trp RNA-binding attenuation protein (TRAP)-trp leader RNA interactions mediate translational as well as transcriptional regulation of the Bacillus subtilis trp operon. J Bacteriol. 1995;177:6362–70. doi: 10.1128/jb.177.22.6362-6370.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sarsero JP, Merino E, Yanofsky C. A Bacillus subtilis gene of previously unknown function, yhaG, is translationally regulated by tryptophan-activated TRAP and appears to be involved in tryptophan transport. J Bacteriol. 2000;182:2329–31. doi: 10.1128/jb.182.8.2329-2331.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yakhnin H, Babiarz JE, Yakhnin AV, Babitzke P. Expression of the Bacillus subtilis trpEDCFBA operon is influenced by translational coupling and Rho termination factor. J Bacteriol. 2001;183:5918–26. doi: 10.1128/JB.183.20.5918-5926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yakhnin H, Zhang H, Yakhnin AV, Babitzke P. The trp RNA-binding attenuation protein of Bacillus subtilis regulates translation of the tryptophan transport gene trpP (yhaG) by blocking ribosome binding. J Bacteriol. 2004;186:278–86. doi: 10.1128/JB.186.2.278-286.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yang M, de Saizieu A, van Loon AP, Gollnick P. Translation of trpG in Bacillus subtilis is regulated by the trp RNA- binding attenuation protein (TRAP) J Bacteriol. 1995;177:4272–8. doi: 10.1128/jb.177.15.4272-4278.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Antson AA, Otridge J, Brzozowski AM, Dodson EJ, Dodson GG, Wilson KS, Smith TM, Yang M, Kurecki T, Gollnick P. The structure of trp RNA-binding attenuation protein. Nature. 1995;374:693–700. doi: 10.1038/374693a0. [DOI] [PubMed] [Google Scholar]
- (13).Antson AA, Dodson EJ, Dodson G, Greaves RB, Chen X, Gollnick P. Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature. 1999;401:235–42. doi: 10.1038/45730. [DOI] [PubMed] [Google Scholar]
- (14).Chen X, Antson AA, Yang M, Li P, Baumann C, Dodson EJ, Dodson GG, Gollnick P. Regulatory features of the trp operon and the crystal structure of the trp RNA-binding attenuation protein from Bacillus stearothermophilus. J Mol Biol. 1999;289:1003–16. doi: 10.1006/jmbi.1999.2834. [DOI] [PubMed] [Google Scholar]
- (15).Babitzke P, Yanofsky C. Structural features of L-tryptophan required for activation of TRAP, the trp RNA-binding attenuation protein of Bacillus subtilis. J Biol Chem. 1995;270:12452–6. doi: 10.1074/jbc.270.21.12452. [DOI] [PubMed] [Google Scholar]
- (16).Yang M, Chen X, Militello K, Hoffman R, Fernandez B, Baumann C, Gollnick P. Alanine-scanning mutagenesis of Bacillus subtilis trp RNA-binding attenuation protein (TRAP) reveals residues involved in tryptophan binding and RNA binding. J Mol Biol. 1997;270:696–710. doi: 10.1006/jmbi.1997.1149. [DOI] [PubMed] [Google Scholar]
- (17).Elliott MB, Gottlieb PA, Gollnick P. Probing the TRAP-RNA interaction with nucleoside analogs. Rna. 1999;5:1277–89. doi: 10.1017/s1355838299991057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Elliott M, Gottlieb P, Gollnick P. Using nucleotide analogs to probe protein-RNA interactions. Methods. 2001;23:255–63. doi: 10.1006/meth.2000.1136. [DOI] [PubMed] [Google Scholar]
- (19).Elliott MB, Gottlieb PA, Gollnick P. The mechanism of RNA binding to TRAP: initiation and cooperative interactions. Rna. 2001;7:85–93. doi: 10.1017/s135583820100173x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li PT, Scott D, Gollnick P. Creating hetero-11-mers composed of wild-type and mutant subunits to study RNA binding to TRAP. J Biol Chem. 2002;227:11838–44. doi: 10.1074/jbc.M110860200. [DOI] [PubMed] [Google Scholar]
- (21).McElroy C, Manfredo A, Wendt A, Gollnick P, Foster M. TROSY-NMR studies of the 91kDa TRAP protein reveal allosteric control of a gene regulatory protein by ligand-altered flexibility. J Mol Biol. 2002;323:463–73. doi: 10.1016/s0022-2836(02)00940-3. [DOI] [PubMed] [Google Scholar]
- (22).Snyder D, Lary J, Chen Y, Gollnick P, Cole JL. Interaction of the trp RNA-binding attenuation protein (TRAP) with anti-TRAP. J Mol Biol. 2004;338:669–82. doi: 10.1016/j.jmb.2004.03.030. [DOI] [PubMed] [Google Scholar]
- (23).Sturtevant JM. Heat capacity and entropy changes in processes involving proteins. Proc Natl Acad Sci U S A. 1977;74:2236–40. doi: 10.1073/pnas.74.6.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Baldwin RL. Temperature dependence of the hydrophobic interaction in protein folding. Proc Natl Acad Sci U S A. 1986;83:8069–72. doi: 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Privalov PL, Gill SJ. Stability of protein structure and hydrophobic interaction. Adv Protein Chem. 1988;39:191–234. doi: 10.1016/s0065-3233(08)60377-0. [DOI] [PubMed] [Google Scholar]
- (26).Privalov PL, Khechinashvili NN. A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J Mol Biol. 1974;86:665–84. doi: 10.1016/0022-2836(74)90188-0. [DOI] [PubMed] [Google Scholar]
- (27).Spolar RS, Ha JH, Record MT., Jr. Hydrophobic effect in protein folding and other noncovalent processes involving proteins. Proc Natl Acad Sci U S A. 1989;86:8382–5. doi: 10.1073/pnas.86.21.8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Spolar RS, Record MT., Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–84. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- (29).Makhatadze GI, Privalov PL. Energetics of protein structure. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/s0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- (30).Prabhu NV, Sharp KA. Heat capacity in proteins. Annu Rev Phys Chem. 2005;56:521–48. doi: 10.1146/annurev.physchem.56.092503.141202. [DOI] [PubMed] [Google Scholar]
- (31).Glasoe PK, Long FA. Use of Glass Electrodes to Measure Acidities in Deuterium Oxide. J. Phys. Chem. 1960;64:188–190. [Google Scholar]
- (32).Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–71. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–26. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- (34).Fasman GD. Handbook of biochemistry and molecular biology. 3d ed. CRC Press; Cleveland: 1975. [Google Scholar]
- (35).Lavigne P, Bagu JR, Boyko R, Willard L, Holmes CF, Sykes BD. Structure-based thermodynamic analysis of the dissociation of protein phosphatase-1 catalytic subunit and microcystin-LR docked complexes. Protein Sci. 2000;9:252–64. doi: 10.1110/ps.9.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Eftink MR, Anusiem AC, Biltonen RL. Enthalpy-entropy compensation and heat capacity changes for protein-ligand interactions: general thermodynamic models and data for the binding of nucleotides to ribonuclease A. Biochemistry. 1983;22:3884–96. doi: 10.1021/bi00285a025. [DOI] [PubMed] [Google Scholar]
- (37).Baker BM, Murphy KP. Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys J. 1996;71:2049–55. doi: 10.1016/S0006-3495(96)79403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lohman TM, Overman LB, Ferrari ME, Kozlov AG. A highly salt-dependent enthalpy change for Escherichia coli SSB protein-nucleic acid binding due to ion-protein interactions. Biochemistry. 1996;35:5272–9. doi: 10.1021/bi9527606. [DOI] [PubMed] [Google Scholar]
- (39).Murphy KP, Privalov PL, Gill SJ. Common features of protein unfolding and dissolution of hydrophobic compounds. Science. 1990;247:559–61. doi: 10.1126/science.2300815. [DOI] [PubMed] [Google Scholar]
- (40).Robertson AD, Murphy KP. Protein Structure and the Energetics of Protein Stability. Chem Rev. 1997;97:1251–1268. doi: 10.1021/cr960383c. [DOI] [PubMed] [Google Scholar]
- (41).Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- (42).Creed D. The Photophysics and Photochemistry of the Near-UV Absorbing Amino Acids - I. Tryptophan and Its Simple Derivitives. Photochem Photobiol. 1984;39:537–562. [Google Scholar]
- (43).Saroff HA, Kiefer JE. Analysis of the binding of ligands to large numbers of sites: the binding of tryptophan to the 11 sites of the trp RNA-binding attenuation protein. Anal Biochem. 1997;247:138–42. doi: 10.1006/abio.1997.2039. [DOI] [PubMed] [Google Scholar]
- (44).Deleage G, Geourjon C. An interactive graphic program for calculating the secondary structure content of proteins from circular dichroism spectrum. Comput Appl Biosci. 1993;9:197–9. doi: 10.1093/bioinformatics/9.2.197. [DOI] [PubMed] [Google Scholar]
- (45).Sreerama N, Venyaminov SY, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal Biochem. 2000;287:243–51. doi: 10.1006/abio.2000.4879. [DOI] [PubMed] [Google Scholar]
- (46).Freskgard PO, Martensson LG, Jonasson P, Jonsson BH, Carlsson U. Assignment of the contribution of the tryptophan residues to the circular dichroism spectrum of human carbonic anhydrase II. Biochemistry. 1994;33:14281–8. doi: 10.1021/bi00251a041. [DOI] [PubMed] [Google Scholar]
- (47).Grishina IB, Woody RW. Contributions of tryptophan side chains to the circular dichroism of globular proteins: exciton couplets and coupled oscillators. Faraday Discuss. 1994:245–62. doi: 10.1039/fd9949900245. [DOI] [PubMed] [Google Scholar]
- (48).Tsai CJ, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–90. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bruzzese FJ, Connelly PR. Allosteric properties of inosine monophosphate dehydrogenase revealed through the thermodynamics of binding of inosine 5′-monophosphate and mycophenolic acid. Temperature dependent heat capacity of binding as a signature of ligand-coupled conformational equilibria. Biochemistry. 1997;36:10428–38. doi: 10.1021/bi9708040. [DOI] [PubMed] [Google Scholar]
- (50).Privalov PL, Jelesarov I, Read CM, Dragan AI, Crane-Robinson C. The energetics of HMG box interactions with DNA: thermodynamics of the DNA binding of the HMG box from mouse sox-5. J Mol Biol. 1999;294:997–1013. doi: 10.1006/jmbi.1999.3285. [DOI] [PubMed] [Google Scholar]
- (51).Privalov PL. Stability of proteins. Proteins which do not present a single cooperative system. Adv Protein Chem. 1982;35:1–104. [PubMed] [Google Scholar]
- (52).Ha JH, Spolar RS, Record MT., Jr. Role of the hydrophobic effect in stability of site-specific protein-DNA complexes. J Mol Biol. 1989;209:801–16. doi: 10.1016/0022-2836(89)90608-6. [DOI] [PubMed] [Google Scholar]
- (53).Spolar RS, Livingstone JR, Record MT., Jr. Use of liquid hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry. 1992;31:3947–55. doi: 10.1021/bi00131a009. [DOI] [PubMed] [Google Scholar]
- (54).Murphy KP, Freire E. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv Protein Chem. 1992;43:313–61. doi: 10.1016/s0065-3233(08)60556-2. [DOI] [PubMed] [Google Scholar]
- (55).Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–48. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bergqvist S, Williams MA, O’Brien R, Ladbury JE. Heat capacity effects of water molecules and ions at a protein-DNA interface. J Mol Biol. 2004;336:829–42. doi: 10.1016/j.jmb.2003.12.061. [DOI] [PubMed] [Google Scholar]
- (57).Morton CJ, Ladbury JE. Water-mediated protein-DNA interactions: the relationship of thermodynamics to structural detail. Protein Sci. 1996;5:2115–8. doi: 10.1002/pro.5560051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Holbrook JA, Tsodikov OV, Saecker RM, Record MT., Jr. Specific and non-specific interactions of integration host factor with DNA: thermodynamic evidence for disruption of multiple IHF surface salt-bridges coupled to DNA binding. J Mol Biol. 2001;310:379–401. doi: 10.1006/jmbi.2001.4768. [DOI] [PubMed] [Google Scholar]
- (59).Bryan WP. The thermodynamics of water-protein interactions. J Theor Biol. 1980;87:639–61. doi: 10.1016/0022-5193(80)90109-5. [DOI] [PubMed] [Google Scholar]
- (60).Dunitz J. The Entropic Cost of Bound Water in Crystals and Biomolecules. Science. 1994;264:670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- (61).Holdgate GA, Tunnicliffe A, Ward WH, Weston SA, Rosenbrock G, Barth PT, Taylor IW, Pauptit RA, Timms D. The entropic penalty of ordered water accounts for weaker binding of the antibiotic novobiocin to a resistant mutant of DNA gyrase: a thermodynamic and crystallographic study. Biochemistry. 1997;36:9663–73. doi: 10.1021/bi970294+. [DOI] [PubMed] [Google Scholar]
- (62).Cooper A. Heat capacity effects in protein folding and ligand binding: a re-evaluation of the role of water in biomolecular thermodynamics. Biophys Chem. 2005;115:89–97. doi: 10.1016/j.bpc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- (63).Tamura A, Privalov PL. The entropy cost of protein association. J Mol Biol. 1997;273:1048–60. doi: 10.1006/jmbi.1997.1368. [DOI] [PubMed] [Google Scholar]
- (64).Koshland J,DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Changeux JP, Gerhart JC, Schachman HK. Allosteric interactions in aspartate transcarbamylase. I. Binding of specific ligands to the native enzyme and its isolated subunits. Biochemistry. 1968;7:531–8. doi: 10.1021/bi00842a007. [DOI] [PubMed] [Google Scholar]
- (66).Cooper A, Dryden DT. Allostery without conformational change. A plausible model. Eur Biophys J. 1984;11:103–9. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- (67).Jardetzky O. Protein dynamics and conformational transitions in allosteric proteins. Prog Biophys Mol Biol. 1996;65:171–219. doi: 10.1016/s0079-6107(96)00010-7. [DOI] [PubMed] [Google Scholar]
- (68).Perutz MF. Mechanisms of cooperativity and allosteric regulation in proteins. Q Rev Biophys. 1989;22:139–237. doi: 10.1017/s0033583500003826. [DOI] [PubMed] [Google Scholar]
- (69).Brandts JF, Lin LN. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry. 1990;29:6927–40. doi: 10.1021/bi00481a024. [DOI] [PubMed] [Google Scholar]
- (70).Jelesarov I, Bosshard HR. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J Mol Recognit. 1999;12:3–18. doi: 10.1002/(SICI)1099-1352(199901/02)12:1<3::AID-JMR441>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- (71).Jelesarov I, Bosshard HR. Thermodynamics of ferredoxin binding to ferredoxin:NADP+ reductase and the role of water at the complex interface. Biochemistry. 1994;33:13321–8. doi: 10.1021/bi00249a019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.