

Summary
Phenytoin (DPH) is a clinically useful sodium (Na) channel blocker with efficacy against partial and generalized seizures. We have developed a novel hydantoin compound (HA) using comparative molecular field analysis (CoMFA) and evaluated its effects on hNav1.2 channels. Both DPH and HA demonstrated affinity for resting (Kr = 13.9 µM for HA, Kr = 464 µM for DPH) and slow inactivated channels (KI = 975 nM for HA, KI = 20.6 µM for DPH). However, HA also exhibited an affinity for fast inactivated channels (KI = 2.5 µM) and shifted the V1/2 for activation in the depolarizing direction. Furthermore, HA exhibited profound use dependent block at both 5 and 10 Hz stimulation frequencies.
In the 6 Hz seizure model (32 mA) HA had an ED50 of 47.1 mg/kg and a TD50 of 131 mg/kg (Protective Index (P.I.) = 2.8). In comparison, the ED50 for DPH was ~27.5 mg/kg with a TD50 of 35.6 mg/kg (P.I. ~ 1.3). These findings provide evidence for the utility of CoMFA in the design of novel anticonvulsant and support the hypothesis that selectivity plays an important role in achieving optimal protection with minimal side effects.
INTRODUCTION
Sodium (Na) channels are composed of a pore forming α subunit consisting of four homologous domains. Each domain contains six transmembrane segments. The alpha subunit associates with auxiliary β subunits that modulate channel gating properties (Catterall, 2000). To date, nine α isoforms (Goldin, 2001) and four β subunits (Isom et al., 1992;Isom et al., 1995;Morgan et al., 2000; Yu et al., 2003) have thus far been cloned. These channels are responsible for the generation and conduction of action potentials and are important targets for the control of neuronal excitability. Indeed, a number of antiepileptic drugs (AEDs) that target Na channels have been developed: carbamazepine, lamotrigine, valproate and phenytoin (Rogawski and Loscher, 2004).
Phenytoin, or Diphenylhydantoin (DPH), is a clinically useful and widely used first-line AED for the treatment of partial seizures and generalized tonic clonic seizures (Browne and Holmes, 2001). DPH induces a slight tonic or resting block of Na channels in their closed states. However, at more depolarized potentials, such as those observed during repetitive firing or seizure activity, the block is more pronounced. The voltage dependence of drug binding is well described by the modulated receptor hypothesis in which drugs have a much greater affinity for the inactivated states of the channel than for the resting states of the channel (Hille, 1977; Hondeghem and Katzung, 1977). This higher affinity for the inactivated states relative to the resting or closed states is thought to be the primary mechanism of many effective AEDs that target the Na channel (Macdonald and Kelly, 1994).
Despite recent advances in therapeutic options for the treatment of epilepsy, an estimated 30% of patients are refractory to currently available AEDs (Schmidt and Rogawski, 2002). In addition, for some patients adequate seizure control comes at the cost of significant toxic side effects. In view of this, there is a great need for the continued development of new AEDs with greater efficacy for pharmacoresistant seizures and/or improved side effect profiles. In this study we examined the effects on hNav1.2 of a novel compound previously developed using comparative molecular field analysis (CoMFA) (Brown et al., 1999). The CoMFA model was developed using a series of hydantoins to displace [3H]batrachotoxinin A 20-α -benzoate (BTX) from rat brain cerebral cortex synaptasomes (Brown et al., 1997). CoMFA samples the differences in steric and electrostatic fields surrounding a set of ligands to help define important 3-dimensional properties associated with the optimum binding of ligand to receptor. The CoMFA model revealed that replacing one of the phenyl rings in DPH with an n-alkyl substitution and an optimal length of 6–7 carbons was required for tight binding to the hydantoin site and that phenyl ring orientation is important for binding. This led to the discovery of the novel hydroxy amide structural class. However, since 3H-[BTX] displacement does not represent functional block of the Na channel, these compounds remained uncharacterized with respect to their ability to inhibit Na channel currents.
In this study, we have determined the effects of one compound, HA, on Na channel gating parameters and assessed its ability to block acute seizure activity induced by low frequency (6 Hz), long duration (3 sec) corneal stimulation in the 6 Hz partial psychomotor seizure test (Barton et al., 2001). HA’s performance was then compared to that of DPH in these in vitro and in vivo paradigms. The 6 Hz corneal stimulation test is considered a potential screen for compounds that may prove useful in therapy resistant epilepsy. Unlike other acute seizure models (ie, subcutaneous pentylenetetrazol (scPTZ) and maximal electroshock (MES)), the 6 Hz model is sensitive to novel compounds such as levetiracetam despite being relatively insensitive to phenytoin, lamotrigine, and topiramate (Barton et al., 2001). Our data suggests that in comparison to DPH, the novel analogue HA displayed profoundly greater inactivated channel affinity and frequency-dependent blockade of hNav1.2 channels. This modulation of neuronal Na channel gating could account for not only the anticonvulsant activity of HA observed, but perhaps more importantly, the markedly improved TD50 (131 mg/kg, i.p.) relative to that of DPH (35.6 mg/kg, i.p) observed in vivo. These studies suggest that CoMFA can be used to design novel sodium channel blockers with desired therapeutic characteristics.
METHODS
5.1. Compound Synthesis
Preparation of ketone from phenylnitrile: Mg+ turnings (1.2 equivalents (eq.)) were added to a flame dried round bottom flask equipped with a magnetic stir bar and a few crystals of I2 were added. The flask was gently heated until I2 vapor was produced. The flask was cooled to room temperature and 1-bromoheptane (1.1 eq.) in ether (10 mL) was slowly added. The reaction was stirred for 30 minutes at room temperature and then cooled to 0 °C in an ice bath and 3-chloro-bezonitrile in tetrahydrofuran (10 mL) was slowly added. 10 mg of CuBr was added and the reaction stirred for 3 hours. The reaction was quenched with 30 mL of 15% H2SO4 and stirred at room temperature overnight. The reaction was extracted with ethyl acetate (3 × 25 mL) and the combined organic extracts were dried with MgSO4 and evaporated in vacuo. Purification by flash column chromatography (10% Ethyl Acetate/90% Hexanes) gave the pure ketone 1-(3 -chlorophenyl)octan-1-one (Rf ~ 0.85) as a clear oil.
Preparation of HA from Ketone: Ketone (1 eq), KCN (2 eq), and (NH4)2CO3 (4 eq) were added to a stirring solution of 50% ethanol (25 ml). The solution was warmed to 50–65°C for 12 h. The precipitate was filtered, and the filtrate was acidified to pH 2 using concentrated HCl. The resulting solid was filtered, and the filtrate was made basic using 3% potassium hydroxide. This was concentrated to one-half volume and filtered again. The combined solids were recrystallized from hot ethanol to give pure hydantoins (Scheme 1). 5-(3-chlorophenyl)-5-heptyl-imidazolidine-2,4-dione (5) was obtained as a white solid (1.5 g, 83%). mp = 124–125°C; 1H NMR (300 MHz, d6-DMSO): δ 1.6 (t, 3H), 1.79–2.23 (m, 1H), 9.54 (s, 1H); 13C NMR: δ 14.83, 22.94, 24.14, 29.34, 29.53, 32.02, 39.35, 68.17, 125.07, 126.15, 128.66, 131.28, 134.14, 142.63, 157.30, 176.71.
Scheme 1.

Reagents: (a) 1-bromoheptane, Mg, I2, tetrahydrofuran; (b) 15% H2SO4, 24 hours; (c) (NH4)2CO3, KCN, 50% ethanol, 65°C, 3 days.
5.2. Molecular Modeling Studies
The x-ray coordinates for DPH were utilized in this study (Camerman and Camerman, 1970). The novel hydantoin was modified from the x-ray structure and was energy-minimized with the Tripos force field using conjugate gradient approach and 0.05 kCal/mol energy cutoff, without solvent, using default bond distances and angles and neglecting electrostatics. The minimization was completed by aggregating using the SYBYL/AGGREGATE module for only the x-ray structure atoms and allowing the modified portion to minimize. For internal consistency, we used the R-configuration for HA. We fit the C5, C4, N1, C6, C9 and C12 on the corresponding atoms of DPH (see figure 1). Figure 2C represents a hydantoin ring overlap of DPH (white) and HA (purple) in order to emphasize there structural differences.
Figure 1. Compound Structures.

Structure of DPH and the novel hydantoin analogue, HA. The major modifications to DPH were replacement of one of the phenyl rings with an n-heptyl group and substitution of chloride in the meta position the remaining phenyl ring.
Figure 2. Molecular Modeling.
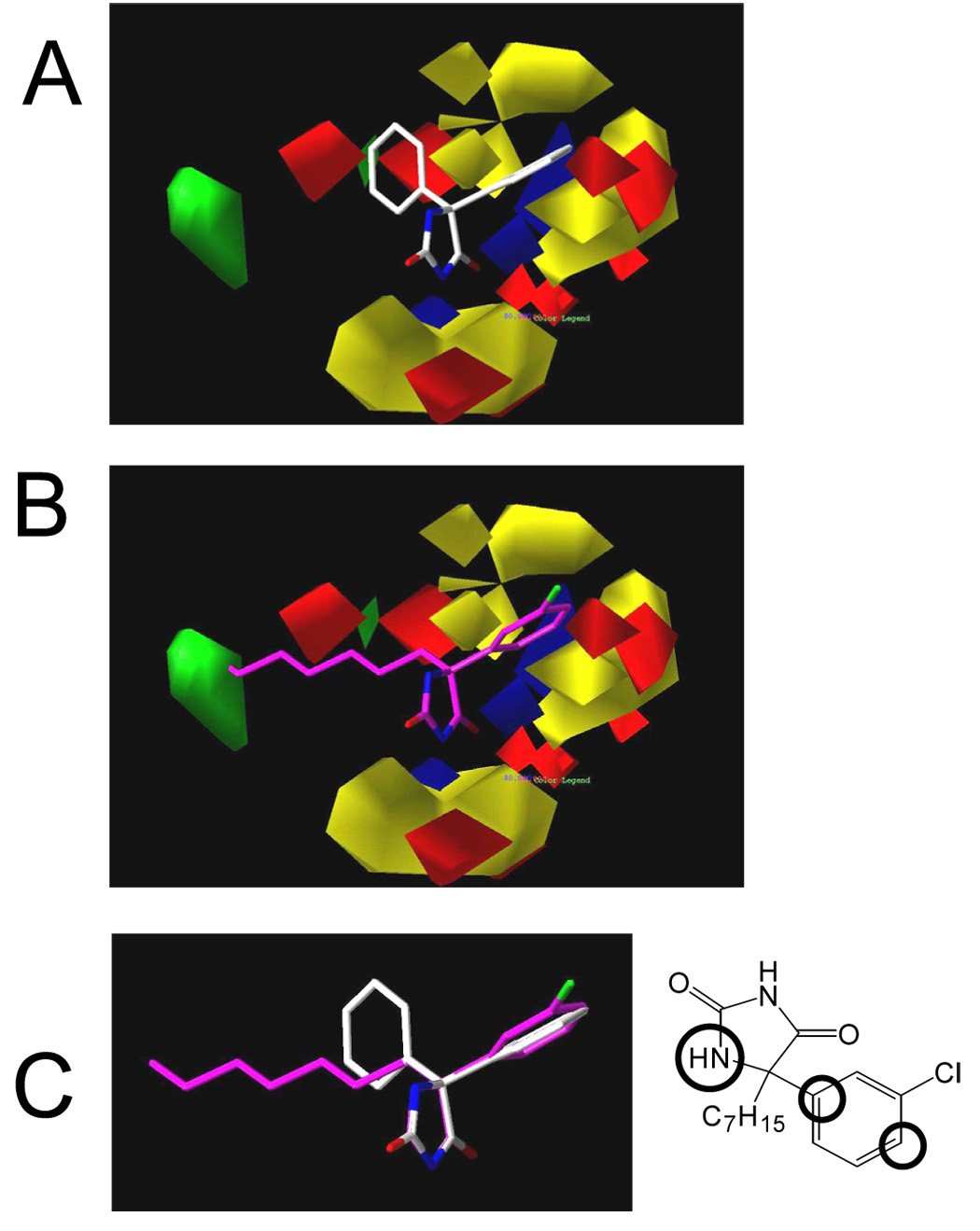
The x-ray coordinates for DPH were utilized in this study. The HA was modified from the x-ray structure and energy-minimized with the Tripos force field using default bond distances and angles and neglecting electrostatics. The minimization by the conjugate gradient approach was completed by aggregating atoms of the DPH structure common to HA using the SYBYL/AGGREGATE module. For internal consistency, we used the R-configuration HA. 2A, 3-D structures of DPH in the electrostatic and steric CoMFA fields. 2B, 3-D structures of HA in the electrostatic and steric CoMFA fields. 2C, Overlap of DPH and HA demonstrating the different relative position of the phenyl rings and the extension of the hydrophobic side chain of HA. Insert showing overlapping reference atoms for modeling (defined by circles).
5.3. Sodium Channel Expression
Cell lines were prepared as previously described (Kirsch et al., 1994). Briefly, human embryonic kidney (HEK) cells stably expressing human Nav1.2 were grown in DMEM/F12 media (Invitrogen, CA, USA) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 µg/ml) and G418 (500 µg/ml; Sigma, MO, USA). Cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37 °C.
5.4. Electrophysiology Studies
Sodium currents were recorded using the whole-cell configuration of the patch clamp recording technique with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). All voltage protocols were applied using pCLAMP 9 software (Axon, USA) and a Digidata 1322A (Axon, USA). Currents were amplified and low pass filtered (2 kHz) and sampled at 33 kHz. Borosilicate glass pipettes were pulled using a Brown-Flaming puller (model P87, Sutter Instruments Co, Novato, CA) and heat polished to produce electrode resistances of 0.5–1.5 MΩ when filled with the following electrode solution (in mM); CsCl 130, MgCl2 1, MgATP 5, BAPTA 10, HEPES 5 (pH adjusted to 7.4 with CsOH). Cells were plated on glass coverslips and superfused with solution containing the following composition; (in mM) NaCl 130, KCl 4, CaCl2l, MgCl2 5, HEPES 5, and glucose 5 (pH adjusted to 7.4 with NaOH). Compounds were prepared as 100 mM stock solutions in Dimethly sulfoxide (DMSO) and diluted to desired concentration in perfusion solution. The maximum DMSO concentration used was 0.3% and had no effect on current amplitude. All experiments were performed at room temperature (20–22°C). After establishing whole-cell, a minimum series resistance compensation of 75% was applied and cells were held at −100 mV for 5 minutes to account for equilibrium gating shifts. Voltage error was calculated using the following equation:
where ΔV is the voltage error, Ip is the peak current, a is the series resistance compensation and R is the series resistance. Data from cells with a voltage error greater than 3mV were excluded. Solutions were applied by continuous perfusion at flow rates ranging from 1–5 mL/min. After control recordings, compound solutions were applied for five minutes to allow for bath equilibration. Tonic block was assessed by comparing peak sodium current in drug free conditions to peak current when drug was present. Dose response data were fitted using the Hill equation:
Where C is the drug concentration, IC50 is the concentration that blocks 50% of the current and H is the hill coefficient.
Construction of the voltage dependence of channel activation requires calculation of the ratio of channel conductance (g) from the peak sodium current (INa). This was accomplished using the equation g = INa/(V-ENa) where V is the test potential and ENa is the reversal potential. The voltage dependence of activation and steady state inactivation data were fitted by the equation:
where y is the normalized conductance (g/gmax) or the normalized current for activation and inactivation respectively, V1/2 is the voltage of half-maximal activation or inactivation and k is the slope factor. The difference between the V1/2 values in the presence and absence of compound is shown as ΔV1/2 (mV). Time constants for recovery from inactivation were obtained using a double exponential function:
A1 and A2 are the coefficients for the fast and slow exponentials, t is time (ms) and τ1 and τ2 are the fast and slow time constants respectively. The percentage of the current represented by the fast time constant was calculated from the equation 100% * A1/(A1+A2), where A1 and A2 are the amplitudes of the fast and slow gating modes respectively. Time constants for development of inactivation were also obtained using a double exponential function where C is the extent of development of inactivation.
5.5. 6Hz Psychomotor acute seizure test and rotorod toxicity tests
Anticonvulsant activity was assessed using the 6Hz seizure test. Adult, male CF-1 mice (Charles River Laboratories, Wilmington, MA) weighing between 20–30 g were used in all experiments. ‘Psychomotor’ seizures were induced via corneal stimulation (6 Hz, 0.2 ms rectangular pulse width, 3.0 s duration, 0.32 mA stimulation intensity) using a Grass S48 stimulator. At the time of test compound administration, a drop of 0.5% tetracaine was applied to the eyes of all animals. At the time of peak effect for each compound, a drop of 0.9% saline was placed on the eyes prior to corneal stimulation. Immediately following the stimulation, mice were observed for the presence or absence of minimal clonic seizure activity followed by stereotyped, automatistic behaviors consistent with the aura experienced by patients with partial seizures. The seizure activity observed is characterized by freezing, forelimb clonus, twitching of the vibrissae, and elevated (Straub) tail posture. Protection was defined as the absence of these partial seizure behaviors. All drugs were either dissolved or suspended in 0.5% (weight/volume) methylcellulose (MC) or 0.9% sodium chloride (SAL). All compounds were administered intraperitoneal (i.p.) in a volume of 0.01 ml/g body weight. To determine sedative and/or ataxic side effects of the test compounds, the rotorod toxicity test was performed (Dunham et al., 1957). Mice were observed by trained technicians on a spinning rotorod (6 rpm) for 1 minute to check for motor impairment, ataxia, or other signs of behavioral toxicity. Motor impairment was defined as the inability of a mouse to maintain equilibrium for one minute in three consecutive trials on the rotorod.
For each compound, the time of peak effect (TPE) was determined and then used to calculate an ED50 and TD50. The protective index (PI) value was determined based on these values. The PI value is calculated as the TD50/ED50 ratio. At the conclusion of each experiment, animals were sacrificed in accordance with the guidelines established by the National Institutes of Health guide for the care and use of laboratory animals, the Institute of Laboratory Resources and the University of Utah’s policy on the humane care and use of laboratory animals.
5.6. Data Analysis
Data analysis was performed using Clampfit software (v8, Axon Instruments, CA, USA) and Origin (v6, Microcal Software, MA, USA). Statistical analyses were performed using the standard one way ANOVA followed by Tukey’s post hoc test, or the Rank Sum test for non-normalized data (SigmaStat, Jandel). Averaged data are presented as means ± standard error of the mean (S.E.M). Statistical significance was set at P < 0.05.
RESULTS
6.1. Development of novel ligand
Figure 1 shows the structures of DPH and the novel hydantoin analogue (HA). The novel hydantoin was developed in order to satisfy all of the constraints of the DPH pharmacophore previously developed by our group (Brown et al., 1999). Figure 2A and B show the fit of HA into the steric and electrostatic fields of the CoMFA model. Regions in green represent areas where increased steric interactions are favored. Yellow regions are areas where steric interactions are unfavored. The blue regions represent areas where increased positive charge is favored and red areas represent areas where more negative charges are required. Compound HA was designed to meet the requirements of this CoMFA model. Figure 2A shows DPH in the CoMFA field. This clearly shows that a longer chain is needed to reach into the lipophilic region (green area). HA has a heptyl side chain that partitions into this region (Figure 2B). The addition of the chlorine on the phenyl ring places steric and electrostatic density in a favorable position in the aromatic region. This resulting structural analogue was predicted to have a much lower IC50 than DPH, 3 versus 40 µM, when optimally fit using our CoMFA model.
6.2. Comparison of resting state affinity
Sodium currents were evoked by a depolarizing step from a holding potential of −120 mV to +10 mV for 25 msec. Currents were recorded under control conditions and after five minutes perfusion with test compound. Figure 3A shows the dose response curves for HA and DPH. The IC50 value and Hill slope for HA were calculated using the Hill equation and were found to be 13.9 ± 0.9 µM and 1.8 ± 0.6 respectively (n = 10). At the highest concentration tested (200 µM) DPH decreased current amplitude by only 26.7 ± 4.7 % (n = 5). Higher concentrations were not attempted due to the limited solubility of DPH, thus a full dose response curve could not be developed; however, assuming a hill slope of 1 yielded an estimated IC50 of ≈ 500 µM for DPH. Representative current tracings showing the effects of HA and DPH are given in figure 2 B and C respectively.
Figure 3. Tonic block.

Currents were elicited by a step depolarization from −120 mV to +10mV for 25 ms. A, dose response relationship for HA and DPH. Data represent mean ± S.E.M. Smooth lines represent the least squares fit when data were fitted with the Hill Equation. B and C show representative current traces for the application of increasing concentrations of HA and DPH respectively
6.3. Modulation of channel activation
We examined the effects of HA and DPH on channel activation. The current-voltage relationship was determined by applying a 25 ms voltage step ranging from −80 to +60 mV in steps of 5 mV from a holding potential of −120 mV at 1 second intervals. In order to assess effects on channel activation, the voltage dependence of whole cell conductance was derived from the current-voltage relationship. DPH and HA were tested at 100 µM and 10 µM respectively. The half activation (V1/2) and slope (k) values for control data were −16.8 ± 0.9 mV and −6.3 ± 0.2 mV (n = 6) respectively and were not significantly changed in the presence of DPH. In contrast, HA induced a significant (p<0.05) change in both the half activation potential and slope value, shifting the V1/2 value to −9.4 ± 0.8 mV and increasing the k value to −7.6 ± 0.5 mV (figure 4, n = 6).
Figure 4. Channel Conductance.
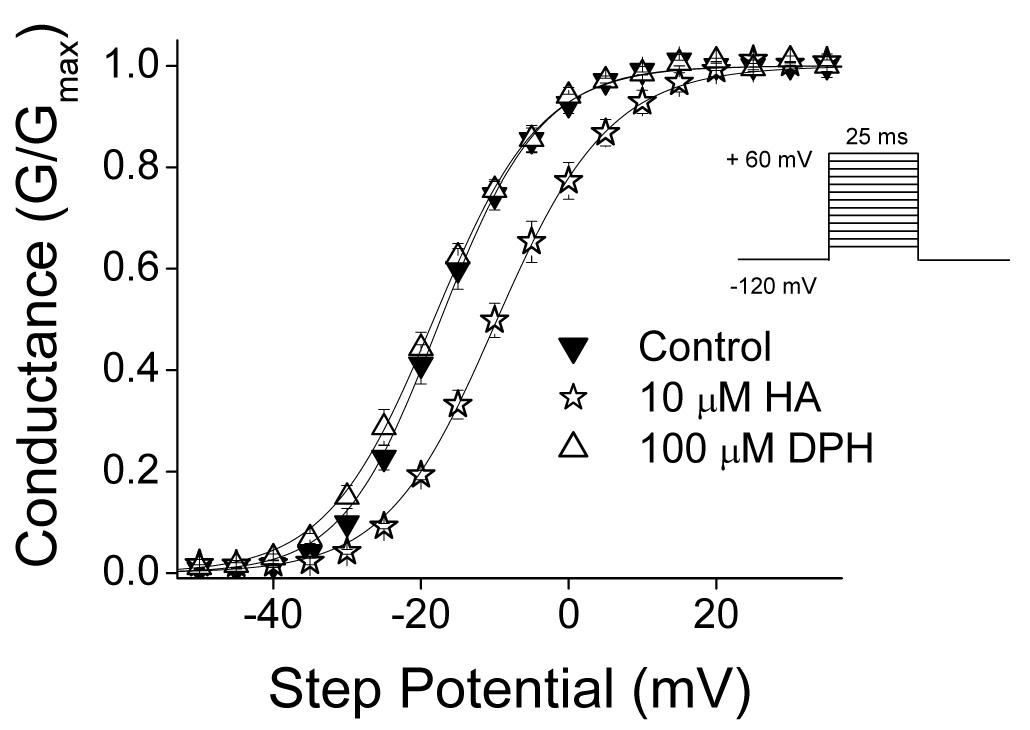
The voltage dependence of channel conductance was derived from the current-voltage relationship as described in the methods. DPH and HA were tested at 100 µM and 10 µM respectively. Data represent mean ± S.E.M. Smooth lines correspond to the average of the least squares fits when data were fitted by the Boltzman equation as described in the methods.
6.4. Modulation of steady state inactivation; affinity for inactivated sodium channels
To determine the voltage dependence of drug action, we examined the effects of HA and DPH on steady state inactivation. Steady state inactivation was examined using either a short 10 msec prepulse, or a longer 1 sec prepulse as previously described by others (Xie et al., 2001). Cells were held at −100 mV and a prepulse ranging from −120 mV to +30 mV was applied for the duration of the test prepulse followed by a step to +10 mV for 6 msec to determine channel availability. Current amplitudes were normalized to the first pulse in order to eliminate effects of tonic inhibition and data were fitted using a Boltzmann function. In Figure 5A the effects of 10 µM HA and 100 µM DPH on steady state inactivation using a short 10 msec prepulse are shown. Under control conditions the V1/2 and k values were −38.0 ± 0.8 mV and 8.3 ± 0.3 mV (n = 6) respectively and were not significantly changed in the presence of 100 µM DPH. In contrast, 10 µM HA caused a significant (p< 0.05) hyperpolarizing shift in V1/2 (V1/2: −47.8 ± 1.7 mV) and increased the slope value (k: 10.4 ± 0.2 mV; n = 6). When a longer 1 sec prepulse duration was applied, an effect of DPH on steady-state inactivation was observed. Under these conditions modulation by DPH (100 µM) and HA (10 µM) were indistinguishable (figure 5B). Under control drug free conditions the V1/2 and k values were −51.2 ± 1.2 mV and 5.4 ± 0.2 mV (n = 5) respectively. In the presence of 10 µM HA the recorded V1/2 was −62.9 ± 1.9 mV (p<0.001, n = 7) whilst in the presence of 100 µM DPH, the V1/2 shifted to −61.9 ± 1.8 mV (p<0.01, n = 6). Slope values remained unchanged from control values.
Figure 5. Channel Availability.

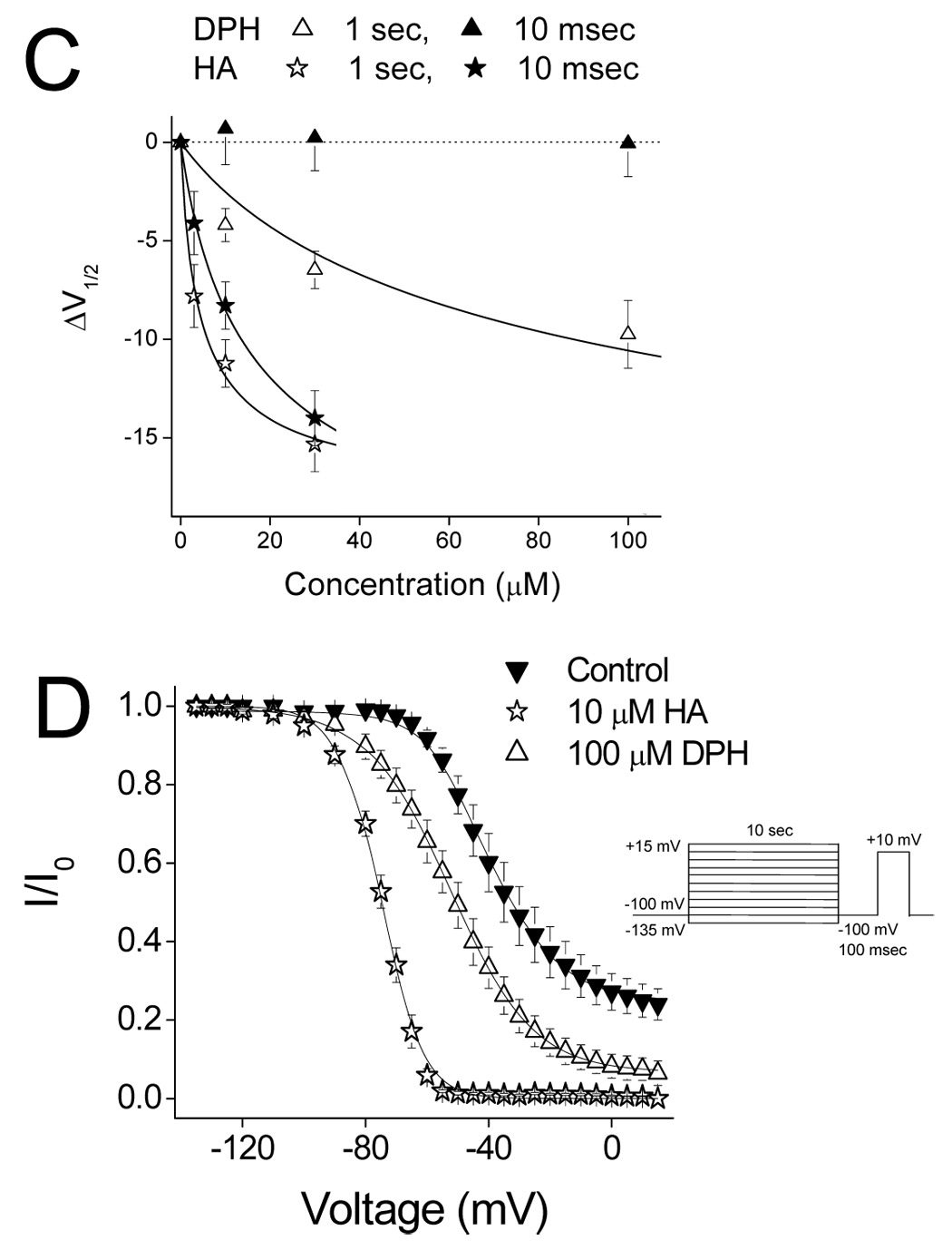
Steady state inactivation was determined using either a conditioning pulse of 1 sec (A), 10 msec (B) or 10 sec (D). Data was recorded under control drug free conditions, in the presence of DPH (100µM) or HA (10µM). Data represent Mean ± S.E.M. Smooth lines correspond to the average of the least squares fits when data were fitted by the Boltzmann equation as described in the methods. C) Concentration dependence of the shift of the steady state inactivation curve for DPH and HA. At each concentration the shift of the steady state inactivation curve was determined using either the 1 sec (open symbols) or 10 msec (closed symbols) prepulse protocol. Dotted line represents baseline (ΔV1/2 = 0). Data represent mean ± S.E.M. Smooth lines correspond to the average of the least squares fits when data were fitted by the equation ΔV1/2 = k log [1 + (D/KI)]/[1+(D/KR)] to calculate the affinity for the inactivated state (KI).
The affinities of DPH and HA for the inactivated states of the channel were determined by assessing the concentration dependent shifts in the steady state availability curves (Bean et al., 1983;Kuo and Bean, 1994). This method employs use of the following equation:
where KI and KR are the affinities for channels in the inactivated and resting state respectively, D is the drug concentration, k is the slope of the steady state inactivation curve and ΔV1/2 is the shift of the steady state inactivation curve in the presence of drug compared to control. By plotting the ΔV1/2 values against drug concentration, and fitting the curve using the above equation, values for KI and KR were determined (Figure 5C). Using the 1 second prepulse protocol values of KI and KR for DPH were 20.6 µM and 464.1 µM respectively and were consistent with previously published results (Ragsdale et al., 1996;Xie et al., 2001). In comparison HA demonstrated a nearly twenty fold higher affinity with a KI value of 975 nM. Furthermore, in contrast to DPH, HA also demonstrated an appreciable affinity for inactivated sodium channels when using the shorter 10 msec prepulse, yielding a calculated KI of 2.5 µM (Figure 5A and 5C).
These results suggest a greater affinity for presumably slow inactivated channels since during a longer depolarizing prepulse a greater number of Na channels would be expected to enter the slow inactivated state. To further examine this hypothesis a longer prepulse duration of 10 sec was used to allow a greater number of Na channels to enter the slow inactivated state. This was immediately followed by a 100 msec pulse to −100 mV to recover fast inactivated channels (Figure 5D). Under these conditions V1/2 and k values recorded were −38.0 ± 2.8 mV and 9.9 ± 0.8 mV (n = 18) respectively and the extent of inactivation reached 74 ± 4%. In the presence of DPH (100 µM), the extent of inactivation was increased to 93 ± 3% and was coupled with a significant hyperpolarizing shift in the steady state inactivation curve and also an increase in the slope value to −52.5 ± 2.5 mV and 13.1 ± 0.5 mV respectively (ΔV1/2 = −17.5 mV: p<0.05; n = 6). HA (10 µM) caused complete inactivation and a larger hyperpolarizing shift in the steady state inactivation curve (V1/2 = −74.7 ± 1.2 mV: p<0.05). Slope values, although increased, did not reach significance 6.3 ± 0.4 mV (n = 6). Since slow inactivation is thought to contribute to overall membrane excitability, spike-frequency adaptation and termination of action potential bursts occurring during repetitive neuronal depolarization, this modulation could be essential to achieve seizure suppression (Rogawski and Loscher, 2004).
6.5. Use Dependent Block at 5 and 10 Hz
A greater affinity for inactivated channels should lead to accumulation of inactivated drug-bound channels during repetitive, high frequency stimulation, a process known as use dependent block. Use dependent block was assessed using a depolarizing pulse from a holding potential of −120 mV to +10 mV for 25 msec at two pulse frequencies: 5 and 10 Hz (figure 6). Peak current amplitude was normalized to the first pulse in each experiment in order to account for tonic block. At 5 Hz stimulation frequency, DPH (100 µM) had no effect on current amplitude. In direct contrast, HA caused profound use dependent block reducing current amplitude by 78.3 ± 3.2 % (10 µM; n = 4) at steady state (Figure 6A). Representative current traces are shown in figure 6B and C for HA and DPH respectively. This lack of use dependent block exhibited by DPH may be due to the relatively slow stimulation frequency. For this reason we also examined use dependent blockade at a 10 Hz pulse frequency (Figure 6D). Under control condition, there was an accumulation of inactivated channels supported by a reduction in peak current amplitude by 16.0 ± 2.1 % (n = 4) as a result of the protocol. At this frequency, DPH (100 µM) exhibited use dependent block reducing the current amplitude by 38.9 ± 5.7 % (n=8). Not surprisingly, use dependent block by HA was accentuated with current amplitudes reduced by 96.0 ± 4.8 % (n=3) at the end of the protocol. In order to investigate open channel block by HA, a 3 msec depolarizing pulse was applied at 5 Hz stimulation frequency (Figure 6A; n=4). This short depolarizing pulse protocol did not induce a significant use dependent reduction in current amplitude, suggesting a lack of open channel block.
Figure 6.


Effects of DPH and HA on use dependent block. Use dependent block was assessed using a depolarizing pulse from −120 mV to +10 mV for either 25 msec (open symbols) or 3 msec (closed symbol) at a 5 Hz pulse frequency (A) or a 10 Hz frequency (D). Representative current traces for 5 Hz stimulation frequency are shown for 10 µM HA (B) and 100 µM DPH (C). Peak current amplitude was normalized to the first pulse in each experiment in order to account for tonic block.
6.6. Recovery from drug block at −100 mV
The stark difference in use dependent blockade could result from a differential ability to inhibit channel recovery from the inactivated state. In order to explore this idea, we examined the effects of DPH and HA on recovery of channel availability using a two pulse protocol. Cells were depolarized to +10 mV for 1 sec from a holding potential of −100 mV. Repriming rates at −100 mV were determined between 1 msec and 60 seconds. Data were normalized to the peak current amplitude in control conditions and fitted using a double exponential function as described in the methods sections (figure 7A). Repriming parameters are summarized in table 1. DPH and HA both delayed recovery of channel availability. DPH significantly delayed τ1 (P<0.05) and τ2 (p<0.05) but did not significantly affect the proportion of channels repriming with the fast time constant. Data for HA were best fitted with a single exponential function and the single time constant for HA was significantly slower than the slow time constants under control conditions and in the presence of DPH (P<0.05).
Figure 7.
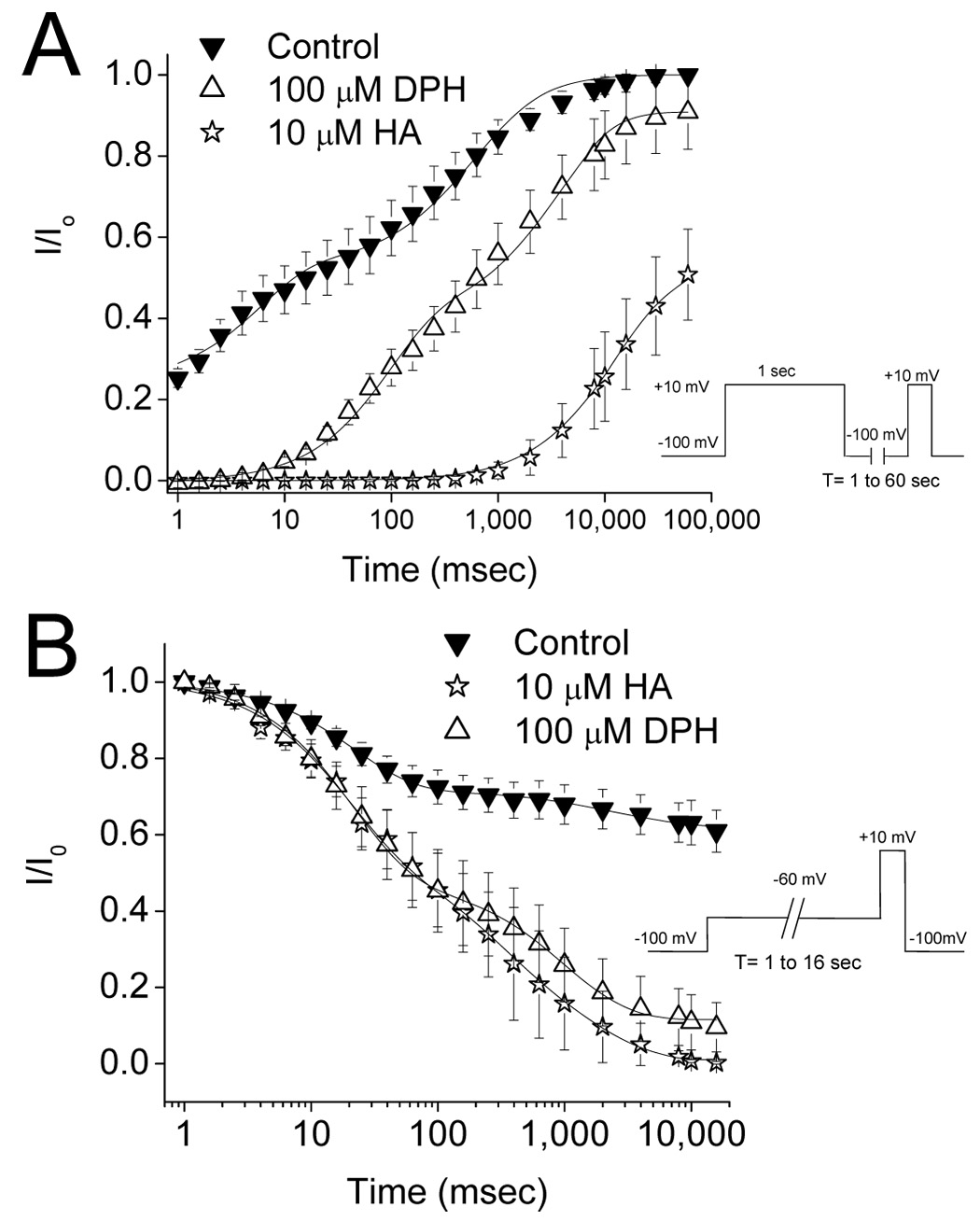
Effects of DPH and HA on recovery from inactivation and development of inactivation. DPH was examined at 100 µM and HA at 10 µM. A. Recovery from inactivation was assessed using a two-pulse protocol. A pre-pulse from −100 mV to +10 mV for 1 sec was applied to inactivate all channels. Cells were then held at −100 mV for a variable period (1–60 sec) to allow channels to recovery and then the proportion of recovered channels was assessed with a voltage step to +20mV. B. Development of inactivation was assessed using a two pulse protocol. From a holding potential of −100 mV a prepulse to −60 mV was applied ranging from 1 msec to 16 secs followed immediately by a step to +10 mV for 20 msec to elicit sodium current. Data points represent the mean ± S.E.M. Smooth lines correspond to the average of the least squares fits when data were fitted by a double exponential function as described in the methods section.
Table 1.
Recovery from inactivation parameters
| Control (n = 7) | DPH (100 µM, n = 4) | HA (10 µM, n = 4) | |
|---|---|---|---|
| % Fast | 47.6 ± 6.3 | 45.6 ± 3.8 | 0 |
| τ1 (msec) | 6.1 ± 0.4 | 99.8 ± 11* | NA |
| τ2 (sec) | 0.81 ± 0.11 | 4.4 ± 0.9* | 18.8 ± 3.5* |
P <0.05. Data represent the Mean ± S.E.M.
6.7. Development of drug block at −60 mV
To test if DPH and HA affected the time course at which channels become unavailable to open, a two pulse development of inactivation protocol was used. Cells were held at −100 mV and the development of inactivation was assessed using a −60 mV prepulse for durations of 1 msec to 16 seconds. The degree of inactivation was determined using a depolarizing step to +10 mV for 20 msec. Data were normalized to the peak current amplitude and fitted using a double exponential function. Under control conditions, fast (τ1) and slow (τ2) time constants for development were 23.3 ± 1.9 msec and 2.8 ± 0.5 sec respectively (figure 7B) and were not significantly different in the presence of 100 µM DPH (τ1 = 23.2 ± 2.9 msec, τ2 = 1.1 ± 0.2 sec, n = 4) or 10 µM HA (τ1 = 20.3 ± 5.4 msec, τ2 = 1.0 ± 0.7 sec, n = 4). However, both DPH and HA did significantly increase the extent of development of inactivation compared to control (p < 0.05). At steady state, current amplitude was reduced by 38.9 ± 4.8% under control conditions. In the presence of DPH or HA, steady state currents were similarly reduced by 88.4 ± 7.1% and 99.0 ± 0.5% respectively.
6.7. Anticonvulsant testing in mice using the 6 Hz psychomotor seizure test
In view of these electrophysiology studies, protection by HA against partial seizure activity evoked in the 6 Hz seizure test was assessed and compared with DPH (Table 2). To determine the time of peak effect (TPE) in the 6 Hz test at the 32 mA stimulus intensity, compounds were assessed at 0.25, 0.5, 1.0 and 2.0 hours following i.p. administration. The TPE for HA was determined to be 0.5 hours. Phenytoin was tested at the predetermined TPE of 1.0 hour. Dose-dependent protection was subsequently assessed at 10–200 mg/kg, i.p. (n=8 per dose) HA and 10–100 mg/kg, i.p. (n=8 per dose) DPH at the TPE for each compound. At the 0.5 hour TPE, the ED50 value for HA was calculated to be 47.1 mg/kg following i.p. administration (95% confidence interval (C.I.) = 26.5 – 68.9 mg/kg; slope ± SEM = 2.29 ± 0.5). The TD50 for HA was calculated to be 131 mg/kg (95% C.I. = 117 – 148 mg/kg; slope ± SEM = 15.3 ± 4.9). From these values, the P.I. value for HA was determined to be approximately 2.8. Unlike HA which afforded clear protection at doses well below the onset of toxicity in most animals, an accurate determination of an ED50 for DPH was problematic given the severe toxicity observed at doses of DPH ≥ 50 mg/kg. These animals were sedated and ataxic following i.p. administration of 50 mg/kg with 7/8 mice failing the rotorod test. At 100 mg/kg, all mice were severely sedated/ataxic with 8/8 mice demonstrating an inability to grasp the rotorod and 3/8 mice demonstrating loss of righting reflex. The ED50 value for DPH was therefore calculated as ~ 27.5 mg/kg (95% C.I. = 16.3 – 45.5 mg/kg; slope ± SEM = 3.68 ± 1.27) using the results obtained following i.p. administration of 10–50 mg/kg DPH. The TD50 value for DPH was calculated as 35.6 mg/kg (95% C.I. = 25.6 – 48.9 mg/kg; slope ± SEM = 7.64 ± 2.66). The P.I. value for DPH was therefore estimated as 1.3 based on these values. As suggested by the overlap in the 95% confidence interval ranges shown above for DPH, there was no significant difference found between the ED50 and TD50 values. Interestingly, despite the relatively similar ED50 values calculated for HA and DPH, there was a 3.7 fold difference between the TD50 values which resulted in a greater calculated PI for HA compared with DPH. Taken together, these in vivo studies demonstrate that HA not only exhibits anticonvulsant activity in the 6 Hz seizure model but perhaps more importantly provides protection with reduced toxicity and motor impairment compared with DPH.
Table 2.
Anticonvulsant activity in 6 Hz psychomotor seizure test in mice. Values shown in brackets represent 95% confidence intervals
| ED50 (mg/kg) | TD50 (mg/kg) | T.I. | T.P.E. (hours) | |
|---|---|---|---|---|
| DPH | 27.5 (16.3 – 45.5)† | 35.6 (25.6 – 48.9)† | 1.3† | 1 |
| HA | 47.1 (26.5 – 68.9) | 131 (117 – 148) | 2.8 | 0.5 |
The significant toxicity observed at doses of ≥ 50 mg/kg, i.p. precluded accurate determination of an ED50 value for phenytoin in this model. Based on the results up to 50mg/kg, an estimated ED50 value and protective index (PI) have been calculated for phenytoin (DPH) using Probit software.
DISCUSSION
Despite the availability of many AED drugs a significant number of patients with epilepsy remain refractory or suffer significant side effects from their medications. In view of this, there is a need to not only develop more effective AED drugs but to develop safer, better tolerated drugs. To achieve this aim compounds have to be rationally designed and their effects on the target protein validated. In this study we have used CoMFA, a ligand based computer modeling program, to predict the structure of novel compounds with increased affinity for the Na channel. The resulting compound, HA, was synthesized and shown to demonstrate important state dependent blocking characteristics shared by other clinically used AED’s (Kuo, 1998). In addition, we show that these characteristics translate into anticonvulsant activity in an acute mouse seizure model with a desired side effect profile when compared with the clinically used compound phenytoin (DPH).
Comparative Molecular Field Analysis (CoMFA)
A CoMFA steric map previously designed by ourselves, was used to compare the affinities of HA and DPH (Brown et al., 1999). Biological data used in constructing the CoMFA model consisted of [3H]batrachotoxinin A 20-α -benzoate (BTX) displacement by a series of hydantoins using rat brain cerebral cortex synaptasomes. To satisfy the constraints of the model we hypothesized that substitutions at the 5 position with a large alkyl group and a substituted phenyl group would result in a ligand with increased binding to the Na channel. Furthermore, an addition of a meta chloro group to the phenyl ring was predicted to further enhance affinity. In contrast to other models used to derive novel Na channel blockers, we demonstrate that the presence of two phenyl rings, as is a common structural motif for DPH and carbamazepine, is not required for activity at Na channels or for anticonvulsant activity (Snell et al., 2000).
Comparative effects on channel gating
In general agreement with our modeling studies, HA demonstrated much greater tonic block of sodium currents as compared to DPH. However, antiepileptic drugs such as DPH are thought to be effective because of a higher affinity for the inactivated state of the channel over the resting state of the channel (Kuo and Bean, 1994). This mechanism is considered important for selective suppression of action potential bursts occurring during an epileptic seizure (Rogawski and Loscher, 2004). Thus we determined the effects of DPH and HA on steady state inactivation using a short 10 msec prepulse and longer 1 sec and 10 sec prepulses. The shorter prepulse duration allows for fast inactivation, whereas slow inactivation develops during longer depolarizing prepulses. Mechanistically, fast inactivation has been shown to involve the short intracellular loop between domains III and IV (West et al., 1992) although additionally sites within DIV S6 (McPhee et al., 1994; McPhee et al., 1995) and within the S4–S5 loop of both domains III and IV have also been implicated (McPhee et al., 1998). Fast inactivation of Na channels is thought to contribute to action potential termination and regulation of the refractory period (McCollum et al., 2003). In contrast, slow inactivation occurs on a much slower time scale of seconds and is a more complex process involving amino acids lining the S6 segments (Chen et al., 2006;O'Reilly et al., 2001). Slow inactivation is thought to contribute to overall membrane excitability by increasing action potential thresholds, thereby limiting action potential burst durations and importantly, limiting the propagation of action potentials within dendrites. Therefore, modulation of Na channel availability through enhancement of slow inactivation by either endogenous (Chen et al., 2006) or pharmacological methods could have significant effects on membrane excitability, particularly during epileptic seizures. (Nau and Wang, 2004;Rogawski and Loscher, 2004).
Consistent with previously published results, DPH reduced channel availability only at long prepulses (Kuo and Bean, 1994;Xie et al., 2001). In contrast, HA reduced channel availability at both the short and long prepulses. It has been previously suggested that DPH binds slowly to the inactivated state requiring >10 sec for binding (Kuo and Bean, 1994). This could account for the lack of DPH on fast inactivated channels since a short prepulse duration of 10 msec would not be sufficiently long enough to allow DPH to interact with its binding site. However, if this was the case then the effects of DPH on development of inactivation would not be detected until prepulse durations of >10 sec were applied. In our studies, development of inactivation for DPH was indistinguishable from HA suggesting that access to the binding site was unlikely a limiting factor, but rather the binding site for DPH is not available upon fast inactivation. Since HA clearly had an affinity for channels inactivated by the shorter prepulse these findings could suggest additional interaction sites for HA in comparison to DPH. In support of this hypothesis is the fact that the Hill slope calculated for HA was greater than 1 (1.8) suggesting more than one interaction site.
In addition to the hyperpolarizing shift in steady state inactivation, HA, but not DPH, shifted channel conductance in the depolarizing direction. This shift, coupled with a hyperpolarizing shift in steady-state inactivation, would result in a reduction of the voltage range where channels are still available for activation and have a finite probability of opening. This range of voltages is known as the window current and a reduction in this current would shift the action potential threshold to more depolarized potentials. An increase in the window current has been associated with increased persistent sodium current activity and epileptogenesis in animal models epilepsy. Several studies have demonstrated an increase in window current in dentate granule neurons and hippocampal CA1 neurons from rat kindling models (Ellerkmann et al., 2003;Ketelaars et al., 2001). A compounds ability to reduce this current could be quite important for antiepiletic activity.
The differential effects of HA compared with DPH may be due to additional interactions within the drug binding site. The binding site for DPH has not been fully elucidated; however, it is thought to involve the aromatic amino acids F1764 and Y1771 in the S6 of domain IV of the rNav1.2 sodium (Ragsdale et al., 1996). The increased block seen by our novel compound may be due to increased stabilization of a potential π–π stacking interaction between the phenyl ring of the compound and aromatic residues within the receptor site (McGaughey et al., 1998). Several studies suggest that the presence of two phenyl rings are sufficient and may be required for high affinity binding to sodium channels (Kuo et al., 2000;Wang et al., 2002;Yang and Kuo, 2002). In contrast to these reports, we have demonstrated that the presence of both phenyl groups is not necessary for sodium channel inhibition. Other potential binding sites for HA could include L465 and L468 in DIIIS6 since these amino acids are thought to form a portion of the AED receptor site (Yarov-Yarovoy et al., 2001). Replacement of one of the phenyl rings in DPH with an n-heptyl group resulted in increased block and unique effects on channel gating perhaps due to optimized binding with additional non-polar amino acid sites such as these.
The more pronounced frequency dependent block for HA was likely accounted for by the enhanced delay in channel repriming after inactivation, as neither compound exhibited significant open channel block or different rates of development of inactivation. In addition, the enhanced use dependent block of HA may in part be attributed to an increased affinity for ‘fast” inactivated channels. For example, fast inactivation is involved in the use dependent block exhibited by lidocaine as it is strongly attenuated in fast inactivation deficient sodium channels (Vedantham and Cannon, 1999).
HA has a greater therapeutic index than DPH
Protection against seizures was assessed using the 6 Hz seizure test at a current intensity of 32 mA. Initially described as a model of 'psychomotor seizures' (Brown et al., 1953), the 6 Hz corneal stimulation model was abandoned because of its lack of sensitivity to DPH. However, the protection afforded by levetiracetam and valproic acid in this model, in contrast to AEDs such as DPH and Lamotrigine, has led to suggestions that this could be a potential model of therapy-resistant epilepsy. In the present study, the calculated ED50 for DPH was somewhat lower (~27.5 mg/kg) perhaps because the toxicity observed at 50 mg/kg or higher prevented accurate evaluation of the behavioral response to the corneal stimulation. In fact, the TD50 value calculated for DPH in the present study was 35.6 mg/kg, with an overlapping 95% CI range. Previously, the reported ED50 for DPH in the 6Hz test at 32 mA was >60 mg/kg, i.p. (Barton et al., 2001). In contrast to DPH, the novel analogue HA differentiated itself from DPH by demonstrating clear protection at 32 mA with a P.I. value of 2.8.
We have demonstrated that CoMFA can be effectively used to design a more potent analogue of DPH. Our results suggest that HA has the characteristics of an effective antiepileptic, including; use dependent block, a greater affinity for the inactivated state of the channel and an ability to reduce window currents. Taken together, these experiments demonstrate that analogues of DPH may be developed as state dependent Na channel blockers and could potentially be useful AEDs.
Acknowledgements
This work was funded by NIH RO1 CA105435–01 (MLB & MKP) and the Korean Science and Engineering Foundation (S-HK). We would like to thank the RMB Cardiovascular Research Center, University of Virginia, (PWL & PJJ) and the Dept. of Anesthesiology.
Abbreviations
- DPH
Diphenylhydantion
- HA
Hydantoin
- BTX
Batrachotoxin
- TTX
Tetrodotoxin
- CoMFA
Comparative Molecular Field Analysis
- AED
Antiepileptic drug
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 2.Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. Journal of General Physiology. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown ML, Brown GB, Brouillette WJ. Effects of log P and phenyl ring conformation on the binding of 5-phenylhydantoins to the voltage-dependent sodium channel. J. Med. Chem. 1997;40:602–607. doi: 10.1021/jm960692v. [DOI] [PubMed] [Google Scholar]
- 4.Brown ML, Zha CC, Van Dyke CC, Brown GB, Brouillette WJ. Comparative molecular field analysis of hydantoin binding to the neuronal voltage-dependent sodium channel. J. Med. Chem. 1999;42:1537–1545. doi: 10.1021/jm980556l. [DOI] [PubMed] [Google Scholar]
- 5.Brown WC, Schiffman DO, Swinyard EA, Goodman LS. Comparative assay of an antiepileptic drugs by psychomotor seizure test and minimal electroshock threshold test. J. Pharmacol. Exp. Ther. 1953;107:273–283. [PubMed] [Google Scholar]
- 6.Browne TR, Holmes GL. Epilepsy. N. Engl. J Med. 2001;344:1145–1151. doi: 10.1056/NEJM200104123441507. [DOI] [PubMed] [Google Scholar]
- 7.Camerman A, Camerman N. Diphenylhydantoin and diazepam: molecular structure similarities and steric basis of anticonvulsant activity. Science. 1970;168:1457–1458. doi: 10.1126/science.168.3938.1457. [DOI] [PubMed] [Google Scholar]
- 8.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Ellerkmann RK, Remy S, Chen J, Sochivko D, Elger CE, Urban BW, Becker A, Beck H. Molecular and functional changes in voltage-dependent Na(+) channels following pilocarpine-induced status epilepticus in rat dentate granule cells. Neuroscience. 2003;119:323–333. doi: 10.1016/s0306-4522(03)00168-4. [DOI] [PubMed] [Google Scholar]
- 11.Goldin AL. Resurgence of sodium channel research. Annu. Rev. Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 12.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. Journal of General Physiology. 1997;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hondeghem LM, Katzung BG. Time-and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochimica et Biophysica Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- 14.Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 15.Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- 16.Ketelaars SO, Gorter JA, van Vliet EA, Lopes da Silva FH, Wadman WJ. Sodium currents in isolated rat CA1 pyramidal and dentate granule neurones in the post-status epilepticus model of epilepsy. Neuroscience. 2001;105:109–120. doi: 10.1016/s0306-4522(01)00176-2. [DOI] [PubMed] [Google Scholar]
- 17.Kirsch GE, Alam M, Hartmann HA. Differential effects of sulfhydryl reagents on saxitoxin and tetrodotoxin block of voltage-dependent Na channels. Biophysical Journal. 1994;67:2305–2315. doi: 10.1016/S0006-3495(94)80716-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo CC. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Molecular Pharmacology. 1998;54:712–721. [PubMed] [Google Scholar]
- 19.Kuo CC, Bean BP. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Molecular Pharmacology. 1994;46:716–725. [PubMed] [Google Scholar]
- 20.Kuo CC, Huang RC, Lou BS. Inhibition of Na+ current by diphenhydramine and other diphenyl compounds: Molecular determinants of selective binding to the inactivated channels. Molecular Pharmacology. 2000;57:135–143. [PubMed] [Google Scholar]
- 21.Macdonald RL, Kelly KM. Mechanisms of action of currently prescribed and newly developed antiepileptic drugs. Epilepsia. 1994;35 Suppl 4:S41–S50. doi: 10.1111/j.1528-1157.1994.tb05955.x. [DOI] [PubMed] [Google Scholar]
- 22.McCollum IJ, Vilin YY, Spackman E, Fujimoto E, Ruben PC. Negatively charged residues adjacent to IFM motif in the DIII-DIV linker of hNa(V)1.4 differentially affect slow inactivation. FEBS Lett. 2003;552:163–169. doi: 10.1016/s0014-5793(03)00912-8. [DOI] [PubMed] [Google Scholar]
- 23.McGaughey GB, Gagne M, Rappe AK. pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 1998;273:15458–15463. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 24.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A mutation in segment IVS6 disrupts fast inactivation of sodium channels. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12346–12350. doi: 10.1073/pnas.91.25.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. Journal of Biological Chemistry. 1995;270:12025–12034. doi: 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- 26.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel alpha-subunit in fast inactivation. J. Biol. Chem. 1998;273:1121–1129. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- 27.Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J. Membr. Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- 29.O'Reilly JP, Wang SY, Wang GK. Residue-specific effects on slow inactivation at V787 in D2-S6 of Na(v)1.4 sodium channels. Biophysical Journal. 2001;81:2100–2111. doi: 10.1016/S0006-3495(01)75858-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt D, Rogawski MA. New strategies for the identification of drugs to prevent the development or progression of epilepsy. Epilepsy Res. 2002;50:71–78. doi: 10.1016/s0920-1211(02)00070-0. [DOI] [PubMed] [Google Scholar]
- 33.Snell LD, Claffey DJ, Ruth JA, Valenzuela CF, Cardoso R, Wang Z, Levinson SR, Sather WA, Williamson AV, Ingersoll NC, Ovchinnikova L, Bhave SV, Hoffman PL, Tabakoff B. Novel structure having antagonist actions at both the glycine site of the N-methyl-D-aspartate receptor and neuronal voltage-sensitive sodium channels: biochemical, electrophysiological, and behavioral characterization. J. Pharmacol. Exp. Ther. 2000;292:215–227. [PubMed] [Google Scholar]
- 34.Vedantham V, Cannon SC. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J. Gen. Physiol. 1999;113:7–16. doi: 10.1085/jgp.113.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang ZJ, Snell LD, Tabakoff B, Levinson SR. Inhibition of neuronal Na+ channels by the novel antiepileptic compound DCUKA: Identification of the diphenylureido moiety as an inactivation modifier. Experimental Neurology. 2002;178:129–138. doi: 10.1006/exnr.2002.8029. [DOI] [PubMed] [Google Scholar]
- 36.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie X, Dale TJ, John VH, Cater HL, Peakman TC, Clare JJ. Electrophysiological and pharmacological properties of the human brain type IIA Na+ channel expressed in a stable mammalian cell line. Pflugers Arch. 2001;441:425–433. doi: 10.1007/s004240000448. [DOI] [PubMed] [Google Scholar]
- 38.Yang YC, Kuo CC. Inhibition of Na+ current by imipramine and related compounds: Different binding kinetics as an inactivation stabilizer and as an open channel blocker. Molecular Pharmacology. 2002;62:1228–1237. doi: 10.1124/mol.62.5.1228. [DOI] [PubMed] [Google Scholar]
- 39.Yarov-Yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall WA. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J. Biol. Chem. 2001;276:20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- 40.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]