

Summary
In this chapter, we present an approach using genomic and ribonomic profiling to investigate functional gene programs in a tumor growth model. To reach this goal, ribonomic profiling was combined with RNA interference in a tumor dormancy model. Strategies merging functional genomic technologies are outlined for the identification of novel posttranscriptionally regulated targets of p38 to show that they are functionally linked to the induction or interruption of cellular growth in cancer. In the first section of this chapter, we describe a method for the detection of mRNA subsets associated with RNA-binding proteins such as hnRNP A1 using (1) immunopurification of mRNA–protein complexes, from either whole cell lysates or subcellular fractions and (2) gene expression arrays to find those mRNAs bound to hnRNP A1. In the second section, short hairpin RNA technology was used to create a library of shRNAs that target p38 induced mRNAs expression libraries are utilized to “knockdown” the genes identified in the first section. Finally, this library of gene candidates is evaluated in vivo to address their functional role in the induction or maintenance of dormancy.
Keywords: Expression profiling, gene silencing, immunoprecipitation (IP), RNA-binding Protein (RBP), short hairpin RNA (shRNA), tumor dormancy
1. Introduction
Cancer dormancy is a stage wherein the cancer cells enter a protracted slow or nondividing state, which can persist for years until the appearance of clinically detectable metastases (1). Cellular dormancy in human cancers has been frequently suggested as a major mechanism by which cancer cells evade chemotherapy (1,2). However, lacking well-defined model systems, the molecular mechanisms underlying metastatic dormancy and their chemoresistance remain poorly understood. Here, we investigate the mechansims of tumor dormancy model using ribonomic profiling and short interfering RNA technologies for target identification and validation, respectively (Fig. 1).
Fig. 1.
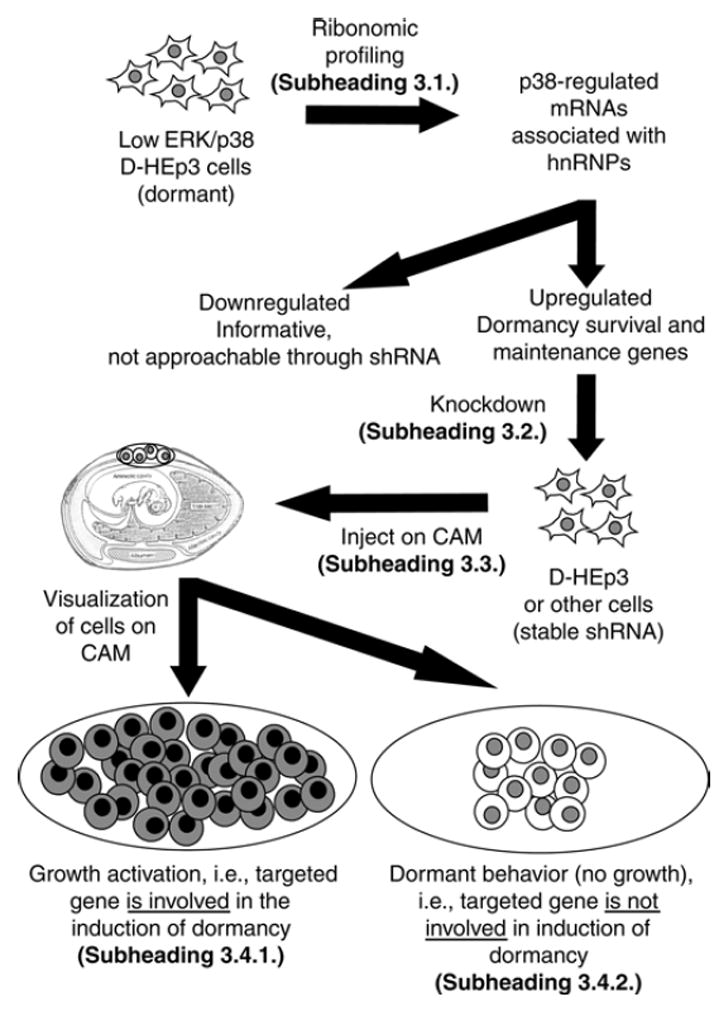
Overview of the strategy. Immunoprecipitation of hnRNPs from dormant/ non-tumorigenic cells allows for the identification of p38-target mRNAs (Subheading 3.1.). The upregulated messages may be knocked down via shRNA expressing vectors (Subheading 3.2.). The cells expressing shRNAs are introduced onto the chorioallantoinc membrame of a chick embryo (Subheading 3.3.). Growth of the shRNA cells above that of the parental cell line indicates that the knocked down gene is involved in tumor dormancy or survival (Subheading 3.4.). The other possible outcome (not shown, see Subheading 3.4.) is that the gene is required for survival and/or growth in vivo, so no tumor nodule will be detected.
A model of cancer dormancy was established in human squamous carcinoma cells (HEp3) where the ERK/p38 ratio determines their proliferative or dormant phenotype. This ratio is predictive of the proliferative behavior of various cancer cell lines (3,4). A high ERK/p38 ratio favors growth, while a low ERK/p38 ratio leads to dormancy in different cell types (3) and a strong activation of p38 dictates the induction and maintenance of tumor dormancy (3). Furthermore, we discovered that p38α regulates approx 300 genes that have been linked to the induction or interruption of dormancy. Recent evidence demonstrates that p38 regulates gene expression posttranscriptionally through p38-responsive regulatory elements in the 3′-UTRs of specific mRNAs (5). Notably, it was found that p38 activation can regulate the nuclear to cytoplasmic shuttling of mRNA binding proteins such as hnRNP A1 (i.e., heterogeneous nuclear ribonucleoproteins A1), which is implicated in the posttranscriptional regulation of mRNAs (6, Rangnathan et al., unpublished results).
Messenger RNA-binding proteins (mRBPs) play an essential role in post-transcriptional gene regulation, potentially by influencing the integrity and/or translation of cognate messages. Such posttranscriptional gene regulation has been shown to be important for partitioning messages spacially and temporally and is the basis for the functional genomic study of mRNAs, called ribonomics (7-9). For example, studies in yeast have shown that the Puf family of mRBPs differentially regulates expression of subsets of mRNAs to direct the function of their encoded proteins (10). Although 87% of the transcripts associated with Puf3 are annotated to have mitochondrial function, the mRNA population associated with Puf4 shows less than 5% annotated as mitochondrial and 27% as nucleolar in nature. The concept that mRBPs can manage functional networks has been reviewed extensively (9,11). Therefore, the immunoprecipitation of mRNA–protein complexes (mRNPs) and subsequent analysis of the associated mRNAs can provide integral information about the function of a given transcript in a particular network. Since p38 appears to regulate specific signals through hnRNP A1 posttranscriptionally, we will identify target mRNA and link their function to the induction or maintenance of dormancy.
To aid in the functional analysis of mRNAs regulated through the p38-hnRNP A1 pathway, we describe a method of using RNA interference (RNAi) to effectively “knockdown” gene expression (12,13) and thereby verify its relationship to the dormant phenotype. DNA-encoded short hairpin RNAs (shRNAs) take advantage of the cell’s endogenous RNAi machinery and offer a convenient, effective, and systematic method to knockdown differentially regulated genes identified by ribonomic approaches and thereby test their functional contribution to a dormant phenotype. Short hairpin RNAs designed against p38-upregulated targets may be used individually or they can be pooled to form a p38-target library to test the dormant phenotype.
Here, we outline a systematic approach to address the role of p38-regulated hnRNP A1 complexes and their associated transcripts in the induction/maintenance of tumor cell dormancy. First, we use ribonomic profiling to identify the hnRNP A1-associated messages that are regulated by p38 signaling in dormant cells. Second, stable cell lines were generated where genes found to be upregulated by p38 are “knocked down” by shRNA expression. By testing the growth capacity of shRNA expressing cells in the chick embryo chorioallantoic membrane (CAM) system, we access the functional contribution of knocked-downgenes to tumor dormancy in vivo to assess the functional contribution of knocked down genes to tumor dormancy in vivo (14).
2. Materials
2.1. Ribonomic Profiling
We recommend the use of distilled, DNase-RNase-free water (Invitrogen, Carlsbad, CA; cat. no. 10977-015) to prepare buffers and solutions described next (see Note 1).
All tips and tubes must be RNase free (see Note 2).
Antibody to RNA binding protein of interest.
Tissue and/or cells of interest from which RNA will be harvested.
Proteinase K. Prepare a solution at a concentration of 20 mg/mL. Store aliquots of 50 μL at −20°C and avoid freeze–thaw.
1 M Dithiothreitol. Store aliquots of 20 μL at −20°C and avoid freeze–thaw.
1X Phosphate-buffered saline (PBS).
Glycogen, molecular biology grade (Roche, cat. no. 0901393). Store at −20°C.
Complete tablets, proteinase inhibitor (Roche, cat. no. 1697498). Store at −20°C.
Bovine serum albumin (BSA)-fraction V proteinase free (Roche, cat. no. 8100350). Store at 4°C.
RNase OUT™ (Invitrogen, cat. no. 10777-019, 40 U/μL). Store at −20°C.
Superase IN™ (Ambion cat. no. 2696, 20U/μL). Store at −20°C.
Protein A Sepharose beads: either from Sigma (P3391) or Amersham Biosciences (Fast Flow, cat. no. 17-0974-01). Store at 4°C.
Protein G Agarose beads (Sigma, cat. no. P4691). Store at 4°C.
Polysome lysis buffer (PLB): 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.0, 100 mM potassium chloride, 5 mM magnesium chloride, 25 mM ethylenediaminetetraacetic acid (EDTA), 0.5%. Additional components are for lysis buffers I and II to be added to PLB immediately before use to give these concentrations: 2 mM dithiothreitol, one tablet of complete proteinase inhibitor per 50 mL of buffer, 50 U/mL RNase OUT, 50 U/mL Superase IN. Note that some of these components are added to the NT2 buffer before immunoprecipitation as noted in step 3 of Subheading 3.1.4.1.
NT2 buffer: 50 mM Tris-HCl, pH 7.4, 150 mM sodium chloride, 1 mM magnesium chloride, 0.05% Nonidet P-40.
Proteinase K digestion buffer: 100 mM Tris-HCl, pH 7.5, 10 mM EDTA, 50 mM sodium chloride, 1% sodium dodecyl sulfate.
Lysis buffer I: 40 mM Tris, pH 7.4, 100 mM sodium chloride, and 2.5 mM magnesium chloride.
Lysis buffer II: 10 mM Tris, pH 7.4, 100 mM sodium chloride, 2.5 mM magnesium chloride, and 0.5% Triton X-100.
A microtip sonicator. We use a Fisher Scientific Model F60 sonicator in these experiments.
2.2. shRNA Gene Targeting
Retroviral vector containing shRNA sequence against target mRNA (see Note 22).
Competent Escherichia coli (see Note 23).
Bacterial culture media.
Plasmid purification reagents/kits such as Qiagen midi columns (see Note 24).
Packaging cell lines capable of producing retroviral particles (several available from American-Type Culture Collection).
Media for mammalian cell lines (packaging cells and experimental cell line).
Transfection reagents (see Note 25).
Experimental cell line.
Hexadimethrine bromide (Polybrene, 8 mg/mL) (Sigma, cat. no. H9268).
Antibiotic for selection of retrovirus infected experimental cells.
Primers for RT-PCR analysis of mRNA of interest or antibody against corresponding protein.
2.3. Tumor Growth Assay (CAM System)
1X PBS containing 2 mM EDTA.
PBS-plus: 1X PBS supplemented to contain 1.0 mM magnesium chloride, 0.5 mM calcium chloride, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Nine- to ten-day-old embryonated eggs (Charles River Laboratories).
Collagenase solution containing BSA: 0.15 g collagenase (Sigma, cat. no. C-9891), 2.5 g BSA (Sigma, cat. no. A-2153) in 100 mL PBS-plus (see reagent 2 above).
3. Methods
3.1. Ribonomic Profiling
In this chapter we present two different protocols for the immunopurification of RNA-binding proteins. The procedure described in Subheading 3.1.4.1. is for whole cell lysate immunopurification of RNA-binding proteins, similar to that presented previously (15). Another variation of the whole cell lysate immunoprecipitation of RNA-binding proteins is presented elsewhere (16). The second method in Subheading 3.1.4.2. is tailored to specifically immunopurify hnRNP bound mRNP complexes from cellular fractions.
3.1.1. Preparation of Whole Cell RNP Lysates From Cultured Cells
Use 150 or 100 mM dishes to grow desired cell line.
Wash twice with ice-cold PBS, harvest using a scraper, and pellet the cells by centrifugation at 3000g for 5 min at 4°C.
Generate the RNP cell extract by estimating the pellet volume and adding approx 1.5 vol PLB for isolating cytoplasmic RNPs or whole cell lysis buffer II for isolating both nuclear and cytoplasmic RNPs.
Pipet several times until the extract looks uniform, and spin in a microcentrifuge at 14,000g for 10 min at 4°C.
Remove the supernatant and save.
Freeze the cell extracts in aliquots of 200–400 μL and store at −80°C (see Note 3). The extracts are ready to proceed to mRNP isolation (see Subheading 3.1.4.1.).
3.1.2. Preparation of Cellularly Fractionationed mRNP Lysates for hnRNP Immunoprecipitation
This protocol is a modified version of a previously published protocol for immunopurification of mRNP complexes from cytoplasmic and nuclear fractions (17).
Perform all fractionation steps on ice.
Grow the cells to confluence in 10-cm dish.
Wash the cells three times in ice-cold PBS and collect with a cell scraper in 0.75–1 mL of lysis buffer I (see Notes 4 and 5).
Transfer to a clean microcentrifuge tube and vortex vigorously for 5–10 s.
Incubate on ice for 10 min.
Centrifuge the sample at 2000g for 8 min at 4°C. Remove the soluble cytosolic fraction (i.e., the supernatant) from the nuclear fraction (i.e., the pellet). This cytosolic fraction can then be carried onto the mRNP immunoprecipitation (Subheading 3.1.4.2.). Proceed to step 7 with the pellet.
Resuspend the pellet from step 6 in lysis buffer II.
Incubate on ice for 5 min.
Centrifuge the sample at 2000g for 8 min at 4°C (see Note 5). Remove the Triton-extracted material by pipet. Proceed to step 10 with the pellet.
Resuspend the pellet from step 9 in lysis buffer II and sonicate on ice for 10 s using a microtip sonicator.
Centrifuge the sonicated material at 4000g for 15 min at 4°C.
The supernatant (representing the nuclear RNP fraction) can then be used (if desired) in the mRNP immunoprecipitation (Subheading 3.1.4.2.).
3.1.3. Antibody Coating of Bead Matrix
Swell or resuspend the desired amount of protein G agarose beads (for monoclonal antibodies) or protein A Sepharose beads (for rabbit serum or rabbit polyclonal antibodies) in 5–10 volumes of NT2 containing 5% BSA or lysis buffer I. IMPORTANT! (see Notes 5–8).
Add the immunoprecipitating antibody or serum and incubate for one hr at room temperature on a rotating device or from 1 to 12 h at 4°C (see Notes 9–11).
Beads coated with antibodies can be stored for several months at 4°C when buffers are supplemented to contain 0.02% sodium azide.
3.1.4. Immunoprecipitation of mRNPs
3.1.4.1. Whole Cell mRNP Isolation
Centrifuge the RNP lysate in a microcentrifuge at 14,000g for 10 min at 4°C, then transfer the supernatant to a new tube on ice.
Calculate the amount of antibody-coated beads necessary to perform the appropriate number of immunoprecipitations you are planning and wash the beads several times at room temperature with NT2 buffer (see Note 11).
Resuspend the antibody-coated beads in NT2 buffer supplemented with 50 U/mL RNase OUT, 50 U/mL Superase IN, 1 mM dithiothreitol, and 20–30 mM EDTA.
The volume of resuspended beads in NT2 buffer should correspond to approx 10 times the volume of the RNP lysate being used (see Note 12).
Mix the resuspended antibody-coated beads several times by inversion, add the RNP lysate and tumble the immunoprecipitation reactions end-over-end at room temperature for at least 2–4 h at 4°C (but preferably overnight). A sample of the supernatant can be collected at the beginning of the incubation to serve as a total RNA control, which can assess RNase activity (see Notes 13–15).
After the incubation, spin the beads down and wash with approx 10–20 bed volumes of ice-cold NT2 buffer, vigorously mixing between each rinse. Repeat four to six times (see Note 16).
Resuspend the washed beads in 600 μL proteinase K digestion buffer plus 25 μL proteinase K stock solution and incubate for 30 min at 50°C, mixing occasionally.
Add 600 μL phenol:chloroform:isoamyl alcohol (v/v 25:24:1) to the bead suspension, vortex for 1 min, and centrifuge at 14,000g for 10 min at 4°C.
Remove the upper phase and repeat the extraction with one volume of chloroform.
Precipitate the RNA by adding one volume of isopropanol, 60 μL 4 M ammonium acetate, 3 μL 1 M magnesium chloride, and 8 μL glycogen.
Store samples at −80°C until ready for gene expression analysis.
To recover RNA, centrifuge samples at 14,000g for 30 min at 4°C and wash with 100 μL 80% ethanol (see Note 17).
3.1.4.2. Fractionated Cellular mRNP Isolation
This protocol is specifically designed to the immunopurification of RNP complexes bound to hnRNP proteins. The addition of protease and RNase inhibitors is to be avoided during the lysis step (see Note 18).
Typically, it is recommended to use 10 μg of antibody per immunopurification from 10-cm plate.
Wash the antibody-protein A complex five to six times with lysis buffer I.
Resuspend the antibody-protein A bead complex in lysis buffer I (see Note 6).
Mix the antibody-protein A complex gently by inverting the tube several times.
Add the RNP extracts from the cytoplasm or nucleus to the antibody-protein A-agarose complex. Incubate the complex on an orbital shaker at 4°C for no more than 10–20 min (see Note 19).
Following incubation, spin the RNP-antibody bound beads by centrifuging at 1000 rpm for 10–20 s.
Wash the beads five to six times with lysis buffer I. Mix the beads after each wash by inverting the tube several times.
Resuspend the immunopurified complexes in 200 μL Tris-EDTA buffer containing 1% SDS and incubate at 65°C for 5 min.
Purify RNA through phenol/chloroform extraction and precipitate with isopropanol (see Subheading 3.1.4.1., steps 7–12).
3.1.5. DNase Treatment of Isolated RNA (Optional)
Following RNA extraction, incubate the RNA with 4 U of RQ1 DNase (Promega, Madison, WI) for 30 min at 37°C and recover the RNA by phenol extraction and ethanol precipitation.
3.1.6. Expression Analysis of mRNA From mRNPs
Targets of mRNA binding proteins can be identified and quantified by several methods (Fig. 2). Techniques in which mRNAs can be identified directly, without amplification (15) are preferred.
Multiprobe-based RNase protection assays (PharMingen, San Diego, CA) are an ideal alternative for the optimization and high-throughput analysis of mRNP immunoprecipitations (15).
Many RNP-associated mRNAs were identified using cDNA/genomic arrays. It was found that Affymetrix tiling array platform and the BD Pharmingen Atlas Nylon cDNA Expression Array platforms were (15) excellent for conducting ribonomic analysis. Irrespective of the array platform used, apropriate controls and normalization of signals should be considered on a case by case basis (see Notes 20 and 21, and refs. 18-20).
If gene expression analysis is performed using glass arrays that use Cy3 and Cy5 labeling or on Affymetrix arrays, typically it is recommended to increase the amount of extract by three to five times that required for Atlas Nylon arrays.
Fig. 2.
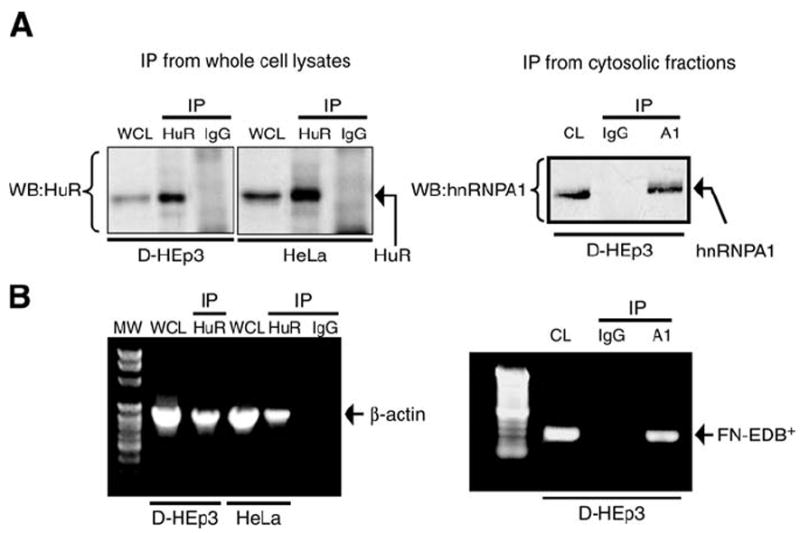
(A) Immunopurification of mRNA binding proteins HuR and hNRNP A1. Whole cell lysates (Subheading 3.1.1.) and cytoplasmic lysates (Subheading 3.1.2.) were immunopurified following the approprate procedure (Subheadings 3.1.4.1. and 3.1.4.2., respectively) using anti-HuR and anti-IgG and anti-hnRNP A1antibodies accordingly. A fraction of the immunopurified protein was analysed by Western blotting using anti-HuR antibody (left panel), or anti-hnRNP A1 antibody (right panel). Both HuR and hnRNP A1 are specifically immunopurified compared with their respective IgG controls. WCL, whole cell lysates; CL, cytoplasmic lysates; WB, Western blot. (B) RT-PCR analysis of mRNA associated with HuR and hnRNP A1 mRNP complex. RNA extracted from HuR and hnRNP A1 mRNP complexes (Subheadings 3.1.4.1. and 3.1.4.2., respectively) was subjected to RT-PCR analysis for the targets β-actin (left panel) or fibronectin-EDB (FN-EDB+, right panel). Fibronectin-EDB is the alternative spliced form of fibronectin mRNA. As hnRNP A1 is involved in alternative splicing, we tested for the association of EDB mRNA with hnRNP A1.
3.2. shRNA Gene Silencing for Functional Analysis of Cancer Cell Growth
The results from ribonomic profiling will provide a list of putative genes involved in the induction of cancer cell dormancy or survival. Plasmid encoded shRNAs may be tested individually or, alternatively, the system can be easily expanded by pooling the plasmids to form a library of shRNAs targeting genes upregulated by p38. Modifiers of tumor dormancy are then screened in a high throughput manner and targets are identified via a unique barcode sequence associated with each shRNA (21).
Hairpin sequences for a gene are designed and inserted into a plasmid capable of retroviral packaging (Fig. 3, see alsoNote 26). For space and simplicity, the single target approach is presented here and the library approach has been described previously in ref.21.
Fig. 3.
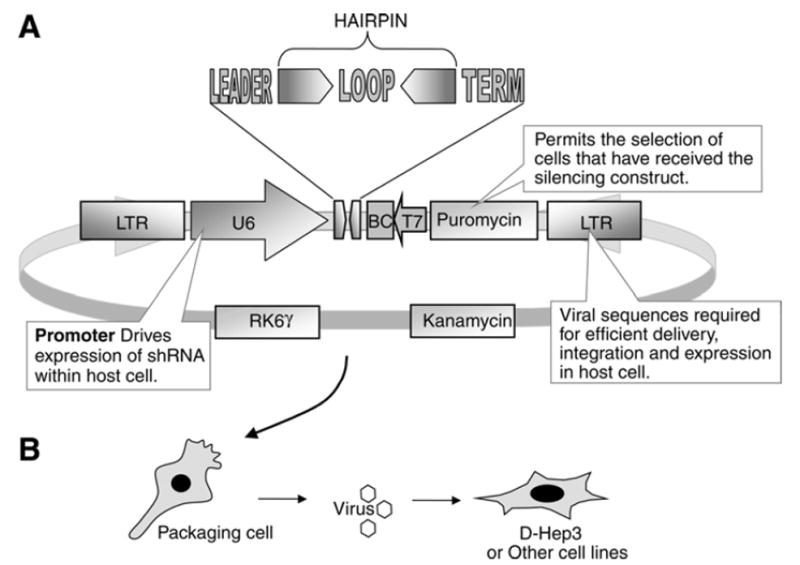
Schematic representation of the retrovirus producing pSHAG-MAGIC vector. (A) Hairpin constructs are designed and ligated into pSHAG-MAGIC. The region flanked by two long terminal repeats contains the shRNA and the puromycin selection cassette. The vector contains the RK6γ origin which requires expression of PIR1 for replication. (B) Once the hairpin has been inserted into pSHAG-MAGIC, the vector is used to transfect packaging cells capable of producing retrovirus. Viral particles are then used to infect the target cell line. The barcode sequence can be used to follow hairpin construct in complex population via microarray hybridization as described in ref.21.
3.2.1. Growth and Preparation of shRNA-Encoding Plasmid DNA
Once an oligonucleotide has been designed that will form a short hairpin structure complimentary to a target message it can be ligated into a plasmid capable of retroviral packaging (21).
Transform competent E. coli. with shRNA containing plasmid following standard protocols.
Pick a single colony and seed 250 mL liquid culture for plasmid preparation (see Note 24).
3.2.2. Production of Stable Cell Lines Expressing shRNAs
After the plasmid containing the appropriate shRNA has been purified, we may proceed to the production of stable cell lines.
Day 1. Plate exponentially growing packaging cells (several lines are available from American Type Culture Collection) so that they will be approx 30–50% confluent the following day (see Note 27).
Day 2. One to two hours before transfection replace the old media with fresh serum containing media.
For transfection use about 15 μg plasmid DNA per 10-cm plate or 1–4 μg per well in a six-well dish, see Note 28). Several methods are available for the introduction of plasmid DNA into cells (see Note 25).
Following transfection incubate the cells at 37°C for 72 h (see Note 29).
Day 4. Plate experimental cell line such that they can continue to grow for 3 d without reaching confluency.
Day 5. Collect the viral supernatant from packaging cells (see Note 30) and filter through a 0.45-μm syringe filter. Store at 4°C if supernatant will be used immediately or −80°C for later use (see Note 31).
Replace experimental cell line media with 0.5 mL fresh media.
Add supernatant/polybrene mixture (8 μg polybrene per milliliter viral supernatant) to each well (see Notes 32–34).
Change media 12–24 h after infection.
Begin antibiotic selection 3 d postinfection (see Note 35).
Perform the following steps in order to assess the degree of shRNA-mediated reduction of expression.
Prepare RNA from the stable cell line clone, and then perform RT-PCR.
Alternatively, immunoblots may be performed to check for the presence and/or level of the corresponding protein.
3.3. Tumor Growth Assay (CAM System)
In this section we present a method to successfully knockdown the expression of those messages that were found to be upregulated and associated with hnRNP A1. After having obtained dormant/non-tumorigenic (D-HEp3) cells stably expressing the shRNA (see Note 36), they will be inoculated onto the CAM of embryonated eggs (see Note 37). The functional assay to test the role of these messages in the model of cancer dormancy is tumor formation after innoculation. Although expression of some of these genes may be involved in the modulation of the dormant phenotype, others may be essential for the survival of the dormant tumor cells in vivo. There may be other genes whose increase or decrease in expression may not have any effect on the tumor cell dormancy, but they cannot be tested.
Using 9- or 10-d-old embryonated eggs are recommended.
The CAMs were prepared as previously described in ref.14. Puncture the egg shell on the long side of the egg (see Note 39).
Apply suction though an opening over the eggshell of the natural air sac, so as to create an artificial air chamber. This results in the separation of the CAM from the eggshell (see Note 39).
Open a window over the displaced CAM and seal it with a piece of sterile tape.
Once the CAM is ready, detach stable shRNA expressing dormant/non-tumorigenic D-HEp3 cells from culture using PBS containing 2 mM EDTA (see Notes 36 and 40).
Pellet the cells by centrifuging at 3000g for 10 min at 4°C. Discard supernatant.
Wash the cells twice with 1X PBS.
Use 5 × 105 cells per inoculation per CAM (see Note 41).
Resuspend cells in step 6 in 50 μL of PBS-plus.
Inoculate the cells gently onto the CAM using a sterile pipet (see Note 39).
Seal the opening of the eggshell over the CAM with transparent tape.
Incubate the eggs (tape window up) in a stationery incubator at 37°C for 3–7 d.
Excise the tumor nodule and weigh it.
At the end point of the assay (see Note 42), mince the tumor into small pieces in a clean Petri dish.
Incubate in type IA collagenase for 30 min at 37°C (see Note 43).
Count the number of tumor cells with a hemocytometer (see Note 44).
Analyze cell viability using Trypan blue exclusion test.
3.4. Functional Analysis of shRNA Mediated Knockdown of Messages
shRNA mediated downregulation of genes that were found to be upregulated in the dormant cells may have three phenotypic outcomes upon inoculation on the CAM (Fig. 4).
Fig. 4.
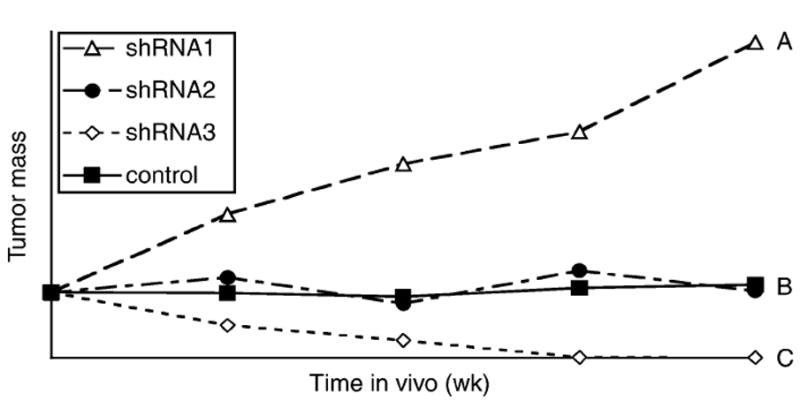
A graph portraying three possible phenotypic outcomes of shRNA mediated downregulation of gene expression. (A) Growing nodule (open triangles, shRNA1): Genes are required for induction/maintenance of dormancy. (B) Dormant nodule (closed circles, shRNA2): genes are not required for induction/maintenance of dormancy. (C) No detectable growth (open diamonds, shRNA3): genes are required for survival in vivo.
Increased tumor growth: if the expression of a gene required for the induction/ maintenance of dormancy is downregulated using shRNA, this would result in interruption of dormancy and enhanced tumor growth. Therefore, a tumor growth rate higher than the control sample suggests the involvement of that gene’s expression in the dormancy process (see Note 45).
Dormant nodule: some genes identified by our technique could be dispensable either to the dormancy program or in vivo viability. Hence downregulation of these would show no effect on tumor growth (see Note 45).
No detectable tumor: some of the genes that were found to be upregulated by ribonomic profiling could be essential for the survival of the tumor cells in vivo. Therefore, downregulation of these gene products in the dormant cells would lead to small or no tumor growth (see Note 46).
4. Notes
Generally, solutions which are certified DNase-free and RNase-free from the manufacturer will make for easier solution preparation and allow for faster troubleshooting if they are handled properly. Ambion’s buffer kit (cat. no. 9010) contains concentrated solutions of Tris (pH 7.0 and pH 8.0), EDTA, sodium chloride, magnesium chloride, potassium choride, ammonium acetate, and DEPC-treated water.
All instruments, glassware and plasticware that touch cells or cell lysates should be certified DNase-free and RNase-free or should be prewashed with RNaseZap (Ambion, cat. no. 9780; 9782) or RNaseAway (Molecular BioProducts cat. no. 7001) followed by DEPC water and allowed to air-dry.
Extracts typically range in concentration from 10 to 50 μg/mL of total protein, depending on the cytoplasmic volume of the cell type being used.
The number of cells required for each immunopurification can vary from cell type to cell type. Typically we recommend one confluent 10-cm dish per immunopurification. In the case of HEp3 cells, this corresponds to approx 8 × 106 cells.
Again, the amount of lysis buffer used typically depends on the amount of protein. Typically we recommend resuspending in a volume of lysis buffer that will have a protein concentration of 1–2 mg/mL. Lysis buffer I is used to isolate cytoplasmic fraction while lysis buffer II is used to isolate nuclear material upon sonication. However in the process we recommend an intermediate step of lysis with lysis buffer II, which partially solubilizes the nuclear membrane. This selectively releases soluble nuclear components as well as Triton X-100 soluble organellar material, leaving behind a purely nuclear pellet.
Important! The whole cell variant (Subheading 3.1.1.) uses NT2 buffer from Subheading 2.1. to resuspend and wash the antibody coated beads. The cellular fractionation variant (Subheading 3.1.2.) uses lysis buffer I from Subheading 2.1. for bead resuspension and washing.
The antibody that is used for immunopurification of hnRNP A1 is a mouse monoclonal antibody (4B10 [22]). However, it functions best with protein A and not protein G agarose.
As a general rule, we recommend Protein-A Sepharose 4 Fast Flow beads (Amersham Biosciences) or Protein-A Sepharose CL-4B (Sigma) if you plan to use rabbit serum and protein-6 Agarose beads (Sigma) if you plan to use murine monoclonal antibodies.
Check the binding capacity of the beads and the antibody concentration in order to introduce the antibody in excess and therefore minimize background-binding problems. Typically 2–20 μL serum per immunoprecipitation reaction are used, depending on the concentration and specificity of the antibody.
Antibody-coated beads can be prepared in bulk and stored at 4°C with 0.02% sodium azide.
Depending on antibody titer and mRNP concentration, use 50–100 μL packed antibody-coated beads and 100–400 μL RNP lysate (~2–5 mg total protein) for each immunoprecipitation reaction.
Performing the immunoprecipitation reactions in larger volumes can decrease background problems.
We have noted that the temperature and duration of incubation can influence the efficacy and/or quality of the immunoprecipitation reaction. Longer incubation times may result in better RNP recovery. However, carrying the reaction for too long may result in RNA repartitioning and/or degradation (23).
Provided there is no RNA degradation or problems related to postlysis protein mRNA exchange (23), immunoprecipitations should be performed for a minimum of 2–3 h at room temperature or overnight at 4°C. A low background is occasionally observed, which is presumably the result of nonspecific binding to the beads.
A concern when isolating mRNP complexes is the possibility of exchange of proteins and mRNAs. In principle, crosslinking agents, such as formaldehyde, could prevent this (24). However, we have found mRNA exchange to occur at a minimal level using these methods and crosslinking therefore to be unnecessary. In some cases, formaldehyde actually can interfere with subsequent mRNA detection methods (25).
Several additional washes with NT2 buffer supplemented with 1–3 M urea can increase specificity and reduce background (8). However, it is important to first determine whether urea disrupts binding of the antibody to the target protein.
RNA pellets from isopropanol precipitations can detach from the centrifuge tube very easily. Extra care should be taken when resuspending the RNA pellet.
The addition of RNase inhibitors may interfere with the binding of RBPs to their cognate targets (communication between J.A. Aguirre-Ghiso and S. Piñol-Roma).
Immunopurification of hnRNP bound mRNP complexes are typically performed for a maximum of 30 min as longer immunopurification times can result in the disruption of the mRNP complex (23).
Depending on the quality of the antibody being used for ribonomic profiling, results and background can vary. Although nonspecific binding can occur, background also may arise from specific RNA–antibody interactions (26).
Informative comparisons between total mRNA profiles and mRNP-associated RNAs are frequently limited by the dramatic differences in signal intensity. There can be a large difference in the number of mRNA species detected in the total RNA as compared with mRNP complexes, which makes most normalization approaches misleading. For this reason, we have typically not compared mRNP profiles with total RNA and suggest that totals be compared with other totals and mRNP immunoprecipitations compared with other mRNP immunoprecipitations. We have found that poly-A binding protein is an useful control RBP.
We recommend constructing at least three hairpins per gene. Hairpins may be purchased commercially from vendors such as Open Biosystems; Ambion, Inc.; or Santa Cruz Biotechnology, Inc. Alternatively, shRNA sequences may be generated using the software on their websites.
The strain of competent E. coli and corresponding cell culture media will vary depending on which plasmid is chosen.
Purity of plasmid DNA is crucial for optimal results. We suggest preparing the DNA using Qiagen columns or cesium chloride gradients.
Lipid based transfection reagents, like FuGENE (Roche, cat. no. 1 814 443) or Lipofectamine (Invitrogen, cat. no. 18324-111) were used. Calcium chloride transfection methods also work well.
Plasmid design and construction for shRNA expression is beyond the scope of this manuscript and has been previously reviewed in ref.21.
In our expericence, transfections generally work better when packaging cells are freshly passaged. General protocols and notes for retrovirus production can be found at the following website http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html.
We also recommend performing in parallel some form of positive control transfection using an easily identifiable marker such as green fluorescent protein as a means of assessing the degree of transfection. If the plasmid is capable of being packaged as a retrovirus, it can also aid in assessing the degree of infection.
Viral production is greatest 48–72 h after addition of transfection mixture.
Retroviruses are potentially dangerous and infectious to humans. Follow proper procedures and strictly observe biosafety guidelines when using retroviruses.
Repeated freeze–thaw cycles significantly reduce viral titer. We recommend freezing in 1-mL aliquots if multiple uses are required.
Reducing the total volume of media (for the first 24 h) or centrifuging the cell culture dishes may help improve efficiency of infection.
Remember to always include an uninfected control. This is especially important when you are performing antibiotic selections.
Infection of experimental cell lines with retrovirus essentially involves mixing a cationic polymer such as polybrene with viral supernatant and adding it to cells.
Allow 24 h for infection and 48 h to allow for the expression of the shRNA before antibiotic addition. The concentration of antibiotic needed to kill cells varies widely. We recommend testing a range of antibiotic concentrations.
The central hypothesis of these experiments is that cancer dormancy program is modulated when expression of genes in the program are disrupted. Therefore, testing of the dormancy program must be performed with cell lines shown to have a dormant/non-tumorigenic or tumorigenic behavior on CAM such as D-HEp3, MCF-7, MDA-MB-231, PC3, and so on (27).
The CAM is a very efficient, timesaving and cost-effective system for the purpose of intital screening purposes. Further, owing to the absence of a mature immune system and the presence of a rich vascular bed, the CAM provides a favorable mileu for the growth of several human tumor cell lines (14).
Usually the eggshell is punctured over the area of the CAM rich in embryonic blood vessels.
Be extremely careful during the preparation of CAM. Use of excessive pressure during this process can result in the breakage of the eggs. In addition, care should be taken while inoculating the cells on the CAM. Piercing and damaging of the CAM with the pipet tip can be avoided by approaching the CAM at a low angle to inoculate the cells.
Detachment of cells using 2 mM EDTA in PBS instead of trypsin is important. We do not recommend the use of trypsin as it may disrupt cell surface molecules required for cell–cell interaction.
We recommend using this number of cells, but you may use anywhere from 2 × 105 to 10 × 105 cells, depending on the growth rate of the cell line used.
The tumor cells can be grown on a given CAM for a maximum period of 7 d from the time of inoculation. However, after 1 wk of incubation the tumor nodule formed at the site of inoculation can be excised, minced, inspected microscopically for the presence of tumor cells and reinoculated on a fresh CAM. Thus, the same tumor population can be serially passaged week after week from one CAM to another.
The treatment with collagenase causes the dissociation of the CAM tissue and the tumor nodule to produce a single cell suspension.
Tumor cells are recognized by their larger diameter compared to chicken cells.
Normally in our system a dormant tumor nodule weighs approx 25–50 mg. Therefore tumors whose size is >50 mg and continues to grow upon serial passaging, it is indicative of a gene required for the induction/maintenance of the dormancy process.
Although some of the genes found to be upregulated by ribonomic profiling could be essential for the survival of the tumor cells in vivo, others may be necessary for its viability in vitro. Downregulation of these genes using shRNA would result in loss of viability in culture. This can be overcome by using shRNA controlled by an inducible promoter system.
References
- 1.Pantel K, Otte M. Occult micormetastasis: enrichment, identification and characterization of single disseminated tumor cells. Semin Cancer Biol. 2001;11:327–337. doi: 10.1006/scbi.2001.0388. [DOI] [PubMed] [Google Scholar]
- 2.Naumov GN, Townson JL, MacDonald IC, et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 3.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- 5.Clark AR, Dean JL, Saklatvala J. Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 2003;546:37–44. doi: 10.1016/s0014-5793(03)00439-3. [DOI] [PubMed] [Google Scholar]
- 6.van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Caceres JF. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol. 2000;149:307–316. doi: 10.1083/jcb.149.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tenenbaum SA, Carson CC, Lager PJ, Keene JD. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci USA. 2000;97:14,085–14,090. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tenenbaum SA, Lager PJ, Carson CC, Keene JD. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002;26:191–198. doi: 10.1016/S1046-2023(02)00022-1. [DOI] [PubMed] [Google Scholar]
- 9.Hieronymus H, Silver PA. A systems view of mRNP biology. Genes Dev. 2004;18:2845–2860. doi: 10.1101/gad.1256904. [DOI] [PubMed] [Google Scholar]
- 10.Gerber AP, Herschlag D, Brown PO. Extensive association of functionally and cytotpically related mRNAs with Puf family RNA-binding proteins in yeast. PLOS Biol. 2004;2:342–354. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keene JD, Tenenbaum SA. Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell. 2002;9:1161–1167. doi: 10.1016/s1097-2765(02)00559-2. [DOI] [PubMed] [Google Scholar]
- 12.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 13.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 14.Scher C, Haudenschild C, Klagsbrun M. The chick chorioallantoic membrane as a model system for the study of tissue invasion by viral transformed cells. Cell. 1976;8:373–382. doi: 10.1016/0092-8674(76)90149-5. [DOI] [PubMed] [Google Scholar]
- 15.Penalva LO, Tenenbaum SA, Keene JD. Gene expression analysis of messenger RNP complexes. Methods Mol Biol. 2004;257:125–134. doi: 10.1385/1-59259-750-5:125. [DOI] [PubMed] [Google Scholar]
- 16.Tenenbaum SA, Lager PJ, Carson CC, Keene JD. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002;26:191–198. doi: 10.1016/S1046-2023(02)00022-1. [DOI] [PubMed] [Google Scholar]
- 17.Mili S, Shu HJ, Zhao Y, Pinol-Roma S. Distinct RNP complexes of shuttling hnRNP proteins with pre-mRNA and mRNA: candidate intermediates in formation and export of mRNA. Mol Cell Biol. 2001;21:7307–7319. doi: 10.1128/MCB.21.21.7307-7319.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 19.Hieronymus H, Silver PA. Genome-wide analysis of RNA-protein interactions illustrates specificity of the mRNA export machinery. Nat Genet. 2003;13:13. doi: 10.1038/ng1080. [DOI] [PubMed] [Google Scholar]
- 20.Roy PJ, Stuart JM, Lund J, Kim SK. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 21.Hannon GJ, Sun P, Carnero A, et al. MaRX: an approach to genetics in mammalian cells. Science. 1999;283:1129–1130. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- 22.Piñol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 1988;2:215–227. doi: 10.1101/gad.2.2.215. [DOI] [PubMed] [Google Scholar]
- 23.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. Reversible cross-linking combined with immunoprecipitation to study RNA–protein interactions in vivo. Methods. 2002;26:182–190. doi: 10.1016/S1046-2023(02)00021-X. [DOI] [PubMed] [Google Scholar]
- 25.Penalva LO, Burdick MD, Lin SM, Sutterluety H, Keene JD. RNA-binding proteins to assess gene expression states of co-cultivated cells in response to tumor cells. Mol Cancer. 2004;3:24–35. doi: 10.1186/1476-4598-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipes BD, Keene JD. Autoimmune epitopes in messenger RNA. RNA. 2002;8:762–771. doi: 10.1017/s1355838202021507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naumov GN, Bender E, Zurakowski D, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]