

Abstract
Metastastic diseases cause the majority of morbidity and mortality of cancer patients. Established tumors form both physical and immunological barriers to limit immune detection and destruction. Current immunotherapy of vaccination and adoptive transfer shows limited effect at least in part due to the existing barriers in the tumors and depending on the knowledge of tumor antigens. Tumor necrosis factor superfamily member 14 (TNFSF14) LIGHT interacts with stromal cells, dendritic cells, NK cells, naïve and activated T cells and tumor cells inside the tumor tissues via its two functional receptors, HVEM and lymphotoxin β receptor (LTβR). Targeting tumor tissues with LIGHT leads to augmentation of priming, recruitment, and retention of effector cells at tumor sites, directly or indirectly, to induce strong anti-tumor immunity to inhibit the growth of primary tumors as well as eradicate metastases. Intratumor treatment would break tumor barriers and allow strong immunity against various tumors without defining tumor antigens. This review summarizes recent findings to support that LIGHT is a promising candidate for an effective cancer immunotherapy.
Keywords: tumor, metastasis, immunotherapy, TNF Superfamily, gene therapy, T cells
1. INTRODUCTION
Many cancers are antigenic and can be recognized by T cells[1]. However, mere recognition by adaptive immunity is insufficient to cause the regression of cancers. An effective anti-tumor immune response depends not only on the presence of tumor-reactive lymphocytes in the tumor-bearing host, but also requires at least three additional conditions. First, naïve tumor-specific T cells must be primed in a proper environment for their expansion and maturation to effector cells. Second, the effector T cells must be able to reach the tumor site. Finally, lymphocytes in the tumor must be able to appropriately execute effector functions to destroy cancer cells. The goal of cancer immunotherapy is to overcome tumor-associated immune-suppressive mechanisms at each of these steps, in order to induce potent anti-tumor immunity.
LIGHT [an acronym for homologous to lymphotoxins, shows inducible expression, and competes with herpes simplex virus glycoprotein D for herpesvirus entry mediator (HVEM), a receptor expressed by T lymphocytes] is a tumor necrosis factor (TNF) superfamily (TNFSF) member that is naturally expressed on immature dendritic cells (DCs) and activated T cells[2, 3]. LIGHT uniquely binds to two functional receptors, HVEM and lymphotoxin β receptor (LTβR) [2, 4], which are TNF receptor superfamily members (TNFRSF) that have distinct expression patterns. HVEM is broadly expressed on hematopoietic cells including T cells, natural killer (NK) cells, and monocytes [5, 6]. Conversely, LTβR is mainly expressed on non-hematopoietic cells such as stromal cells [7] and some monocyte cell lines [8]. Stimulation via LIGHT leads to augmentation of priming, recruitment, and retention of effector cells at tumor sites, directly or indirectly, to induce strong anti-tumor immunity. This review will discuss recent findings regarding the possible mechanisms of how LIGHT provokes anti-tumor immunity at multiple stages of T cell responses, which puts LIGHT at a unique position to be a promising candidate for cancer immunotherapy.
2. PRIMING OF TUMOR-SPECIFIC T CELLS
2.1. Hindrance of priming by tumor stroma
All solid tumors are composed of malignant cells that are embedded in a stroma containing a mixed population of nonmalignant cells: bone marrow-derived myeloid cells, non-bone marrow-derived endothelium, fibroblasts, and cells of the vasculature. Experimental evidence has demonstrated that the non-antigenic stroma may present an important immunological barrier that prevents immune recognition and destruction of tumors [9–11]. More recently, a more physiological model was developed to analyze spontaneously arising primary tumors, which originated from single transformed precursor cells embedded in nonmalignant stroma. Nascent primary solid tumors, even when highly antigenic in vitro, failed to prime antigen-specific T cells during the initial stages in vivo [12]. This observation is in concordance with other experiments that also suggest that tumor stroma may act as a barrier to antigen presentation and immune recognition [13, 14].
Anti-tumor immune response is initiated when tumor antigens are transported out of the tumor to the draining lymph nodes (DLN) [15], either perched atop an antigen-presenting cell (APC) for indirect presentation [16] or by tumor cells themselves for direct presentation [11]. However, tumor stroma may hinder T cell priming by sequestering antigenic tumor cells, thus preventing them from reaching the DLN to directly prime a T cell response [17]. In such an event, cross-presentation by professional APCs, often DCs, from the tumor tissues becomes a major pathway for naïve T cells to be primed tumor antigens. In addition, the stroma matrix may directly interfere with T cell priming by reducing the degree of available antigens. The extracellular matrix proteins can directly bind to tumor antigens [18], while the stromal endothelium and fibroblasts may compete with DCs for antigen uptake [19]. Thus, cancer cells embedded in non-malignant stroma may be ignored [11] when tumor antigens are not expressed at a high level, like in most human cancers. In order to allow priming of tumor-specific T cells and generate strong anti-tumor immunity, it is critical to: 1) penetrate and bypass the non-antigenic stroma, 2) promote cross-presentation by DCs, and 3) release more tumor antigen.
2.2. LIGHT creates lymphoid-like tissues inside the tumor to improve priming
Although cancer cells express mutant or unique proteins that the immune system can recognize as foreign [20], malignant cells are surrounded by non-malignant stroma which forms a complex multicellular “organ” resembling self. The induction of immunity against normal, non-mutated differentiation antigens expressed by tumors resembles autoimmunity. Comparable to anti-tumor immunity, it is often observed in murine autoimmune models that the presence of autoreactive cells alone is not sufficient to cause tissue destruction [21]. It has been reported recently that the organized tertiary lymphoid structure (TLS) is necessary and sufficient to induce autoimmune destruction of pancreatic islets [22]. Indeed, de novo organization of TLS is known to precede the development of a number of human autoimmune diseases [23, 24]. These observations suggest that lymphoid neogenesis within the target tissue may have a critical role in initiating and maintaining immune responses against persistent antigens. TLS are not supplied by afferent lymph vessels and are not encapsulated, which implies that they are directly exposed to signals such as stimulating antigens and cytokines from the environment. This incomplete development of TLS could potentially result in unrestricted access of DCs and lymphocytes to the TLS, favoring immune activation. Although disruption of established TLS or prevention of TLS formation may be advantageous in treating autoimmune diseases, initiation of intra-tumor lymphoid structure formation may facilitate the eradication of tumors. Considering the T cell repertoire may be less responsive against the self-antigens involved in autoimmunity than against the unique antigens involved in tumor immunity, the destruction of cancers would be further facilitated by the availability of high-affinity T lymphocytes in the presence of TLS inside the tumor.
Clues to understanding the signals that lead to TLS formation come from the study of the signaling pathways involved in secondary lymphoid tissue organogenesis. Studies in mutant mice and blocking experiments have identified a key requirement for TNF-family members, mainly lymphotoxin α1β2 (LTα1β2), and to some extent TNF, in the development and organization of lymph nodes and spleen microarchitecture[25–28]. Binding of LTα1β2 and TNF to their respective receptors, LTβR and TNFR1, induces the expression of chemokines and adhesion molecules, which directly mediate lymphocyte migration and homing[29]. The first evidence that the generation of TLS could involve the same signaling pathways that regulate lymphoid organogenesis came from studies of transgenic mice [24], in which the ectopic expression of LTα and LTβ in pancreatic islets induced the formation of in-situ TLS[30–33]. Extrapolating from this earlier experiment, stimulation of LTβR or TNFR expressed by tumor stromal cells may promote the formation of lymphoid-like structures inside the tumor tissues, which in turn may facilitate tumor destruction. Systemic TNFR signaling is of course too toxic, as evidenced by other systemic TNF treatments [34]. Soluble LTα can signal through the TNFR resulting in the upregulation of chemokines. To avoid systemic toxic effects, recombinant LTα has been conjugated with tumor-specific antibody to be delivered specifically to the tumor tissue [35]. Targeting of recombinant LTα to the tumor elicits an efficient immune response associated with the induction of peripheral lymphoid-like tissue containing L-selectin+ naïve T cells and MHC class II+ antigen-presenting cells[35]. Secondary lymphoid tissue chemokine (SLC or CCL21) is among the chemokines controlled by LTβR and TNFR signaling [29]. It is normally expressed in high endothelial venules and in T-cell zones of the spleen and lymph nodes and strongly attracts naive T cells and DCs [36]. The expression of CCL21 inside the tumor resulted in a substantial, sustained influx of T cells within the mass as well as retention of DCs at the tumor site. By recruiting T cells and DCs, CCL21 in the tumor may lead to extranodal priming and inhibition of tumor growth [37].
Similar to LTα1β2, LIGHT also activates LTβR. Our data indicate that the interactions between LIGHT and LTβR restore lymphoid structures in the spleen of LTα−/ − mice [38]. In vivo data demonstrate that LIGHT mediates development of a microenvironment sufficient to break immunological tolerance to self antigens. First, ectopic expression of LIGHT in the pancreatic islets resulted in the formation of lymphoid-like structures and was necessary and sufficient for pancreatic islet destruction [22]. In addition, sustained expression of LIGHT on T cells leads to their activation, migration into peripheral tissues, and the establishment of lymphoid-like structures in situ [38, 39]. LIGHT also acts as a strong costimulatory molecule for T cell activation, possibly by binding to HVEM on T cells [3]. Therefore, LIGHT, delivered inside the tumor, is an ideal candidate for creating TLS to recruit more T cells and subsequently expand antigen-specific ones inside the tumor. In addition to the cross-presentation pathway, tumor-reactive T cells can be activated via direct presentation in the presence of antigens and costimulation. Indeed, LIGHT ectopically expressed in the tumor can effectively recruit and activate naïve T cells. The expression of LIGHT in the tumor environment induces an infiltration of naive T lymphocytes that correlates with an upregulation of both chemokine production and expression of adhesion molecules inside tumors [40]. Activation and expansion of these infiltrating T cells, likely via both cross- and direct-presentation pathways, leads to the rejection of established, highly progressive tumors at local and distal sites[40]. These experiments demonstrate that introduction of the lymphoid-like structure into the tumor stroma can be highly effective in enhancing antigen recognition and may be a potent strategy for cancer immunotherapy.
The ability to directly prime naïve T cells within the tumor itself permits several advantages. First, the efficiency and specificity of priming will be increased due to the higher tumor antigen load in situ relative to that collected in the draining lymphoid tissues [16] and second, a broader repertoire of tumor-specific naïve T cells is recruited to the site of tumor antigens, leading to a more comprehensive response [35, 41]. It has been shown that some tumor antigens may not be efficiently cross-presented due to the antigen bias in T cell cross-priming [42]. In these instances, we have demonstrated in our experimental system using the tumor cell line Ag104Ld-LIGHT that certain antigens are presented to and activate naïve T cells within the tumor via a direct presentation mechanism[40, 43]. The same tumor would have been otherwise overlooked by the host immune response had it relied solely on draining lymphoid tissues and cross-presentation. Furthermore, no additional migration steps are required for CTL to reach the site of effector function, which leads to the appearance of activated tumor-reactive T cells in a short period of time. Finally, T cell responses may react more readily to the shifting tumor antigen expression profile in situ. T cell stimulation in the absence of costimulation can induce anergy and apoptosis of antigen-specific T cells [44–46]. Costimulation has also been shown to greatly enhance tumor-specific T cell function during the effector phase [15]. Earlier studies show that LIGHT provides CD28-independent costimulation [3], which may be essential for the selective and effective activation, expansion and maintenance of tumor-specific T cells among infiltrating naïve T cells in Ag104Ld-LIGHT tumors. By working as both a costimulatory molecule and generating lymphoid-like milieus through the induction of proper chemokines and adhesion molecules, LIGHT is a potent molecule capable of generating better immune responses against established tumor. Interestingly, a recent study showed that LIGHT is constitutively expressed in some human melanoma cells and tumor-derived microvesicles, and tumors that expressed LIGHT were associated with significantly increased lymphocytic infiltration [47].
2.3. LIGHT regulates DCs at the tumor site to promote priming
Since DCs are the major APCs for antigen presentation, increased numbers of DCs may contribute to the enhanced priming of lymphocytes leading to improved anti-tumor immunity. LTβR signaling is critical for maintenance of a normal number and position of DCs in the spleen[48], which can be attributed to two major roles LTβR signaling plays in regulating of DCs.
Upon activation and maturation, DCs migrate to the T cell zones of lymphoid tissues responding to a chemokine gradient, where they initiate immune responses or induce tolerance [49–51]. This migratory process of DCs is dependent upon their ability to express chemokine receptor CCR7 [52, 53]. The ligands of CCR7 include CCL21 and CCL19, which are expressed by lymphatic endothelium and stromal cells [36]. LTβR signaling controls CCL21 and CCL19 expression in the spleen, which may be critical for the migration and/or positioning of DCs in the spleen [29]. Thus, the LTβR signaling-induced chemokine microenvironment on the stroma is critical for the recruitment of DCs.
In addition, more recent studies show that the lack of direct LTβR signaling on DCs is associated with the reduction of the number of DCs in the spleen, independent of the chemokine gradients. These studies suggest that DC proliferation is an important pathway for locally maintaining these cells in the steady state, and LTβR signaling on the DCs is critical for their proliferation [54, 55]. More importantly, LIGHT stimulation dramatically increases the number of DCs in a LTβR-dependent fashion, and intratumor expression of LIGHT can dramatically expand DCs in situ. This increase of DCs possibly contributes to the enhancement of tumor immunity in the LIGHT-mediated tumor environment, which is consistent with the one observed with inoculation of DCs into tumor tissues [55]. Therefore, LTβR signaling regulates DCs for their homeostasis during physiological and pathological conditions, and increased LIGHT- LTβR interaction could stimulate DC expansion for T cell-mediated anti-tumor immunity.
Besides quantity and localization, when antigen is cross-presented to T cells in the DLN by DCs, whether tolerance or immunity is induced depends heavily on the maturation state of the DCs presenting the antigen [56, 57]. Under normal, steady-state conditions, immature DCs cross-present antigens to induce peripheral tolerance [58]. Tolerance may be maintained at the level of T cell expansion through defective signaling pathways [59]. While steady-state conditions lead to tolerance, Signals that promote DC maturation lead to immunity [60–62]. Several characteristics that mature DCs possess including increased presentation of peptide-MHC complexes and induced expression of co-stimulatory molecules like CD80 and CD86, cytokines and chemokines are critical for the initiation and enhancement of immune responses. Thus, tumor antigens that are cross-presented by mature DCs are more likely to induce tumor immunity. A recent study has shown that global LTβR signaling is required for maximal expression of CD86 on Ag-bearing DC and for efficient priming of T cells, and that the LTβR signaling on the DCs is essential to condition them for T cell priming [63]. Moreover, the DCs that become non-functional in the absence of CD40L, an important signal for DC maturation, on Ag-specific T cells can be overcome by stimulating LTβR [63]. This observation is consistent with previous findings that LIGHT can cooperate with CD40L to induce the maturation of monocyte-derived DCs [64]. Thus, we propose that LIGHT delivered into the tumor environment can stimulate LTβR signaling to promote full conditioning of DCs during the priming of anti-tumor immune response.
2.4. Tumor cell apoptosis induced by LIGHT promotes immune recognition
Cancers often express low levels of antigens. Increasing the release of antigen from the tumor cells may break immunological ignorance. For example, apoptosis of cancer cells induced by chemotherapeutic agents or radiation releases antigen, enhancing the cross-priming of T cells [65, 66]. LIGHT has been shown to signal via LTβR and/or HVEM expressed on the tumor cells to induce apoptosis of the tumor cells, especially in the presence of interferon-γ [4, 5, 67]. Using a xenograft tumor model in which human breast cancer cells were inoculated to athymic nude mice, LIGHT directly inhibited tumor growth by causing apoptosis in the absence of T cells [68]. We have shown in multiple tumor models that LIGHT mediated tumor regression is dependent on T cells, especially CD8+ T cells [40, 69]. However, our results do not exclude the possibility that the induction of apoptosis of tumor cells by LIGHT also causes increased release of tumor antigen, leading to enhanced priming and T cell-dependent anti-tumor immunity. In addition, it would be interesting to investigate if high levels of IFN-γ in the LIGHT-mediated microenvironment [40], due to an enhanced T cell-mediated immune response, contributes to direct inhibition of tumor growth by way of LTβR signaling-induced apoptosis. In turn, the release of tumor antigen could result in a better T cell response and more IFN-γ production. It is possible that this positive loop is one of the important mechanisms of how LIGHT induces powerful anti-tumor immunity against tumors that express LTβR and/or HVEM.
3. EFFECTOR T CELLS AT THE TUMOR SITE
3.1. The immunosuppressive environment inside tumors
Even with immune recognition of cancer, which can occur in cancer patients and is evidenced by the frequent observation of T cell infiltration into cancerous tissues [70–73], it is rare for such tumor infiltrating T cells to induce the spontaneous rejection of established tumors. Accumulating evidence indicates that the tumor environment contains cells and cytokines that actively suppress primed effector T cells [74, 75]. High concentrations of transforming growth factor-β (TGF-β), produced by cancer cells or stromal cells, are frequently found in solid tumors and interfere with effective immune rejection of tumor [76]. TGF-β is a cytokine essential for the generation and survival of CD4+CD25+ regulatory T cells (Treg) [77, 78], which may themselves produce TGF-β and IL-10 to reinforce the immune suppressive loop that exists in the tumor environment. While active TGF-β does not inhibit lysis by CTLs, it can inhibit maturation of T cells to that effector state [79]. This explains, at least in part, how CD8+ T cells resistant to TGF-β1 can mediate tumor rejection [80].
Highly antigenic tumors may fail to regress in the host due to an accumulation of regulatory T cells within the tumor microenvironment [81]. These CD4+CD25+ regulatory T cells seem not only to inhibit developing tumor-specific CD8+ T cells from gaining full effector function, but can even block transferred, fully activated tumor-specific T cells from mediating tumor rejection in vivo [82]. We have demonstrated that the local intratumoral depletion of these regulatory T cells changes the cytokine milieu of the tumor, unmasks the immunogenicity of tumor, and reverses CTL tolerization, leading to the rapid rejection of well-established tumors[81]. Our data support the idea that regulatory T cells inhibit not only the early priming events, but also the effector function of T cells inside tumors, which is supported by a recent study [82].
3.2. LIGHT sustains effector T cell functions at the tumor site
It is still not well understood if and how a CD8+ T cell that has been exposed to the immune suppressive environment inside the tumor converts back to a fully functional effector cell. The LIGHT-mediated tumor environment contains fully functional CD8+ effector T cells [40]. This might be in part contributed to the ability of LIGHT to act as a strong costimulatory molecule for T cell activation, expansion and cytokine production, possibly by binding to HVEM on T cells [2, 3, 68]. This was initially demonstrated by experiments in which immobilized recombinant LIGHT promoted T cell proliferation in the presence of anti-CD3 [5]. Reagents that blocked HVEM, like anti-HVEM antibody [6] or HVEM-Ig [83], reduced T cell expansion and their cytokine production in the same setting. This is consistent with the later findings that CD8+ T cell activation, expansion and CTL activity are defective in LIGHT-deficient mice[84–86]. It appears that endogenous LIGHT is critical for CD8+ T cell activation in vivo. Beside its critical role in initiation of T cell activation, costimulation has also been shown to greatly enhance tumor-specific T cell function during the effector phase. It would be interesting to see if LIGHT stimulation is sufficient to convert tolerized CD8+ T cell to functional effectors [15].
Besides its direct role on T cells, the additional role of LIGHT on natural killer (NK) cells may contribute to the promotion of effector T cell function for anti-tumor immunity. LIGHT is a critical ligand for activating NK cells to produce large amount of IFN-γ, possibly via HVEM expressed on the NK cells[87]. Activated NK cells facilitate the further activation of tumor-specific CD8+ T cells in an IFN-γ–dependent manner at the tumor site. The expression of LIGHT inside tumors leads to an increase in number of NK cells in the tumor, possibly through either expansion or recruitment of NK cells[87]. It would be interesting to see if LIGHT-activated NK cells also mediate direct tumor suppression in other tumor models.
T cells activated by LIGHT–HVEM produce typical T-helper cell type 1 (Th1) cytokines including IFN-γ and GM-CSF [3, 39]. Additional LIGHT stimulation can cause severe inflammation in non-lymphoid tissues, as demonstrated by murine models of constitutive expression of mouse [39] or human LIGHT [88] under a T cell promoter. We found that forced expression of LIGHT inside tumor tissues promoted a change of cytokine environment inside the tumor, possibly contributing to the generation of fully functional CD8+ T cells and leading to the eradication of highly established tumors in mice [40]. The question remains as to whether LIGHT also modulates Treg, as well as Gr-1+ CD11b+ immature myeloid cells, which also have powerful immunosuppressive effects inside a tumor environment.
4. LIGHT RECRUITS AND SUSTAINS EFFECTOR T CELLS - Combination with current strategies for cancer immunotherapy
Active immunization and adoptive T cell transfer therapy are the main strategies used thus far for cancer immunotherapy. Both of these strategies are designed to overcome the deficiency in the priming of tumor antigen-specific T cells in the cancer-bearing hosts. Cancer vaccines rely on immunization of patients with antigenic peptides, proteins or DNA expressed by the tumor directly or delivered by DC or virus, etc. Despite relative simplicity and safety, vaccine treatments have shown very limited success [89]. Although the generation in vivo of antitumor T cells in vaccinated patients could be demonstrated by techniques such as tetramer or ELISpot assays [90–92], clinical responses observed from these trials were few [89]. This was consistent with the finding in murine models that the presence of even large numbers of antigen-specific T cells is insufficient to mediate tumor regression [14, 93]. There are a multitude of explanations for this; for example, the relatively inadequate numbers or avidity of the immune cells, the inability of the tumor to recruit or activate quiescent or precursor lymphocytes, short-lived effector cells, tolerance mechanisms including the lack of costimulation, anergy, and active suppressive influences produced by the tumor environment. These obstacles must be overcome if cancer vaccines are to be effective in mediating cancer regression.
Adoptive transfer immune therapies, in which T cells are isolated from tumor or patients’ peripheral blood, are expanded in vitro in a relatively tumor antigen-specific way for adoptive transfer[94], have been used in a small number of highly selected melanoma patients[95]. Although the prerequisite knowledge of the antigens and the potential inability to isolate or expand T cells against the tumor likely limit their application to only a minority of cancer patients, it has shown promise in some of the treated patients [95].
The LIGHT-mediated tumor environment can potentially work synergistically with active immunization and adoptive transfer therapy. Signaling by LTβR expressed by tumor stromal cells, not only promotes naïve T cell recruitment by CCL21, but also upregulates chemokines and adhesion molecules, such as IP-10 and Mig [40] to potentially attract activated T cells to tumor site. Adhesion molecules such as MAdCAM-1, which are essential for lymphocyte migration into peripheral tissues, are also upregulated in the LIGHT-mediated environment [40]. The LIGHT-mediated environment promotes the recruitment of activated T cells, augmented by immunization or adoptive transfer, and improves effector cell function to potentially enhance current strategies for cancer immunotherapy.
5. GENERATION OF CTL IN THE LIGHT-MEDIATED TUMOR ENVIRONMENT -------Treatment of metastases
Micrometastases can establish early in heterogeneous primary tumor development and seed distal sites prior to clinical detection [96]. Therefore, at the time of diagnosis many cancer patients already have microscopic metastases, an observation that has led to the development of post-surgical adjuvant therapy for patients with solid tumors. Despite advances in early detection and modifications to treatment regimens, success has been limited and optimal treatment of metastatic disease continues to pose a major challenge in cancer therapy.
We have shown that expression of LIGHT in the tumor can lead to rejection not only at a local site but also at distal sites. To develop more clinically relevant approaches, adenovirus vectors that express LIGHT (Ad-LIGHT) have been constructed to deliver LIGHT into the tumor tissue. The advantages of the adenoviral delivery system are 1) high production of non-replicable virus; 2) activation of innate immunity; 3) an ability to express its carrying gene in non-dividing cells, and 4) ease of expression in most tumor cell lines. Administration of Ad-LIGHT into the tumor tissue leads to the complete rejection of an aggressive fibrosacoma Ag104Ld and retards the growth of other tumors, such as melanoma B16, colon cancer MC38, and breast cancer 4T1 in mice [69]. The poorly immunogenic 4T1 mammary carcinoma closely mimics human breast cancer in its anatomical site, immunogenicity, growth characteristics, and more importantly, metastatic properties[97–99]. We have demonstrated in this model that generating immune responses in primary tumor tissues prior to surgical resection can produce tumor-specific effector T cells sufficient to eradicate distant metastases in a CD8-dependent fashion. Local treatment with Ad-LIGHT initiated priming of tumor-specific CD8+ T cells directly in the primary tumor, with subsequent exit of CTLs which homed to distal tumors to elicit immune-mediated eradication of spontaneous metastases [69]. Recent study has also shown that attenuated Salmonella engineered to produce LIGHT inhibit primary tumor growth as well as dissemination of pulmonary metastases [100].
Conventional treatment such as surgical removal of tumor followed by radiation and chemotherapy may prevent effective immune recognition of cancers due to the loss of a major source of antigens, and damage to preexisting CTL by radiation and chemotherapy. An alternative strategy would be to utilize the primary tumor as the site of CTL priming prior to surgical resection. Delivery of LIGHT expression within the tumor environment recruits naïve T cells and generates tumor-specific CTLs that can survive and exit the microenvironment to patrol peripheral tissues and eradicate disseminated metastases.
5. CONCLUSIONS
Understanding the balance of anti-tumor effectors vs. suppressors inside the tumor may be important in determining the outcome of immune responses inside tumors. LIGHT interacts with its two receptors to stimulate different kinds of cells inside the tumor to establish a unique environment leading to augmentation of priming and recruitment of anti-tumor T cells. This may be a means of generating a dominantly pro-inflammatory environment, resulting in the rejection of both primary tumor and metastases. In addition, the LIGHT-mediated environment may possibly modulate immune suppressive mechanisms inside the tumor, which is an efficient means of converting the anti-inflammatory environment inside tumor to a pro-inflammatory one. This ideal environment would more rapidly expand the effector cells at the tumor site, while blocking local suppressive factors to form a positive loop to favor the anti-tumor immunity. Finally, it is likely that LIGHT-mediated tumor environment can work synergistically with current immunotherapeutic strategies, and other non-immune suppressive traditional therapies to achieve favorable clinical outcomes for cancer patients.
Figure 1. LIGHT enhances priming of anti-tumor T cells.
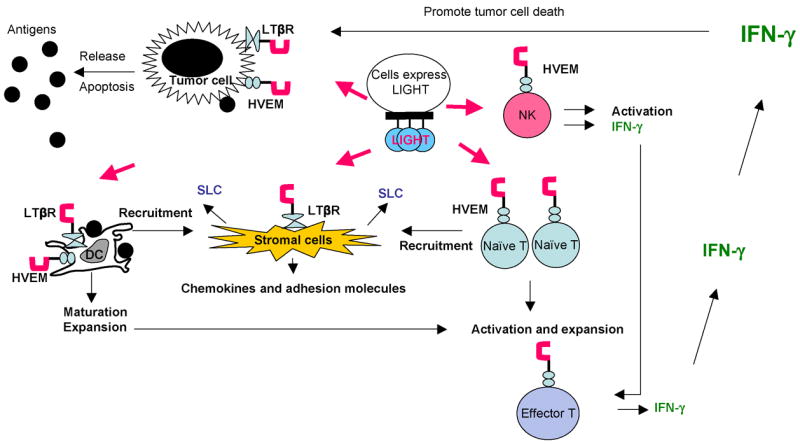
Membrane-bound LIGHT stimulates stromal cells via LTβR to upregulate chemokines, such as SLC, and adhesion molecules to recruit naïve T cells and DCs into the tumor tissue. LIGHT promotes the survival, expansion and maturation of DCs, which possibly prime the T cells in situ. LIGHT also costimulates recruited naïve T cells, possibly through HVEM, in the present of tumor antigen for their activation and expansion. NK cells, which express HVEM, can be activated by LIGHT to produce IFN-γ to facilitate the expansion and differentiation of T cells and production of abundance of IFN-γ. Tumor cells signaled by LIGHT via LTβR and/or HVEM may become apoptotic in the presence of IFN-γ leading to antigen release and better priming of anti-tumor immunity.
Figure 2. LIGHT recruits and promotes effector T cells inside the tumor tissue.
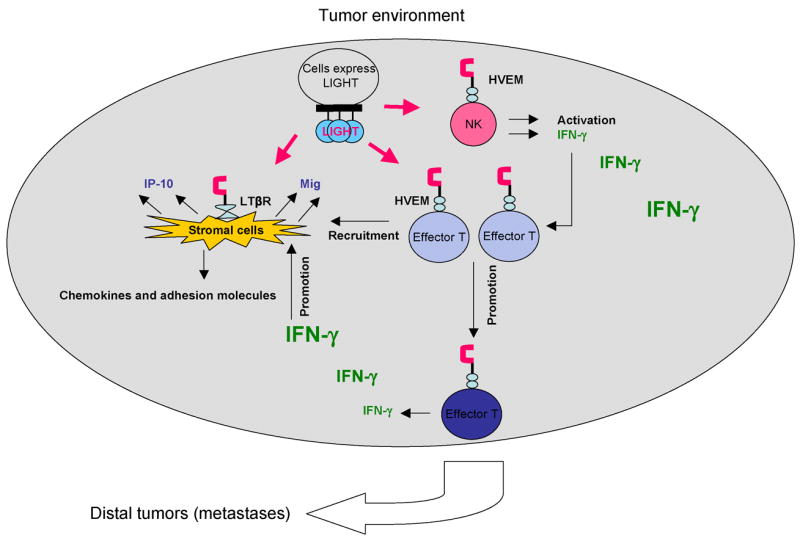
Membrane-bound LIGHT stimulates stromal cells via LTβR to upregulate chemokines, such as IP-10 and Mig to recruit activated T cells into the tumor tissue. LIGHT may also stimulate the coming T cells for their further activation and expansion. NK cells activated by LIGHT through HVEM produce IFN-γ to facilitate the expansion, differentiation and production of more abundance of IFN-γ by T cells. IFN-γ in turn promotes the production of IP-10 and Mig to form a positive loop to recruit and promote effector T cells in the tumor tissue. The functional tumor antigen-specific T cells from LIGHT-mediated environment may traffic systemically for eradication of metastases.
Acknowledgments
We would like to acknowledge critical comments by Youjin Lee and Soygung Auh.
Abbreviations
- HVEM
herpesvirus entry mediator
- LIGHT
lymphotoxin-like, shows inducible expression, and competes with herpes simplex virus glycoprotein D for herpesvirus entry mediator, a receptor expressed by T lymphocytes
- LTβR
lymphotoxin-β receptor
- DC
dendritic cells
- DLN
draining lymph nodes
- TLS
tertiary lymphoid structure
- SLC
secondary lymphoid tissue chemokine
- Ad
adenovirus
Biography
 Yang-Xin Fu is a Professor in Department of Pathology and Attending Physician at the University of Chicago. Dr. Fu received his MD in Shanghai Medical University and Ph.D from University of Miami and completed his pathology residency at Washington University, St. Louis. The basic research program in his laboratory is focused on understanding the biological consequences arising from signaling by the Lymphotoxin-β receptor and related molecules in the development and function of primary, secondary, and tertiary lymphoid tissues. His recent studies have defined the roles of these molecules in autoimmunity and inflammation and developed new strategies to target tumor cells.
Yang-Xin Fu is a Professor in Department of Pathology and Attending Physician at the University of Chicago. Dr. Fu received his MD in Shanghai Medical University and Ph.D from University of Miami and completed his pathology residency at Washington University, St. Louis. The basic research program in his laboratory is focused on understanding the biological consequences arising from signaling by the Lymphotoxin-β receptor and related molecules in the development and function of primary, secondary, and tertiary lymphoid tissues. His recent studies have defined the roles of these molecules in autoimmunity and inflammation and developed new strategies to target tumor cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–9. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 3.Tamada K, Shimozaki K, Chapoval AI, Zhu G, Sica G, Flies D, et al. Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nat Med. 2000;6:283–9. doi: 10.1038/73136. [DOI] [PubMed] [Google Scholar]
- 4.Rooney IA, Butrovich KD, Glass AA, Borboroglu S, Benedict CA, Whitbeck JC, et al. The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J Biol Chem. 2000;275:14307–15. doi: 10.1074/jbc.275.19.14307. [DOI] [PubMed] [Google Scholar]
- 5.Harrop JA, McDonnell PC, Brigham-Burke M, Lyn SD, Minton J, Tan KB, et al. Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J Biol Chem. 1998;273:27548–56. doi: 10.1074/jbc.273.42.27548. [DOI] [PubMed] [Google Scholar]
- 6.Kwon BS, Tan KB, Ni J, Oh KO, Lee ZH, Kim KK, et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J Biol Chem. 1997;272:14272–6. doi: 10.1074/jbc.272.22.14272. [DOI] [PubMed] [Google Scholar]
- 7.Murphy M, Walter BN, Pike-Nobile L, Fanger NA, Guyre PM, Browning JL, et al. Expression of the lymphotoxin beta receptor on follicular stromal cells in human lymphoid tissues. Cell Death Differ. 1998;5:497–505. doi: 10.1038/sj.cdd.4400374. [DOI] [PubMed] [Google Scholar]
- 8.Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, et al. Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J Immunol. 1997;159:3288–98. [PubMed] [Google Scholar]
- 9.Woglom WH. Immunity to transplantable tumors. Cancer Rev. 1929;4:129–214. [Google Scholar]
- 10.Singh S, Ross SR, Acena M, Rowley DA, Schreiber H. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med. 1992;175:139–46. doi: 10.1084/jem.175.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci U S A. 1999;96:2233–8. doi: 10.1073/pnas.96.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–6. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 13.Ganss R, Limmer A, Sacher T, Arnold B, Hammerling GJ. Autoaggression and tumor rejection: it takes more than self-specific T-cell activation. Immunol Rev. 1999;169:263–72. doi: 10.1111/j.1600-065x.1999.tb01321.x. [DOI] [PubMed] [Google Scholar]
- 14.Wick M, Dubey P, Koeppen H, Siegel CT, Fields PE, Fitch FW, et al. Antigenic cancer cells can grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med. 1997;186:229–37. doi: 10.1084/jem.186.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai XF, Gao JX, Liu J, Wen J, Zheng P, Liu Y. On the site and mode of antigen presentation for the initiation of clonal expansion of CD8 T cells specific for a natural tumor antigen. Cancer Res. 2001;61:6860–7. [PubMed] [Google Scholar]
- 16.Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737–47. doi: 10.1016/s1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 17.Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058–64. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 18.Juprelle-Soret M, Wattiaux-De Coninck S, Wattiaux R. Subcellular localization of transglutaminase. Effect of collagen. Biochem J. 1988;250:421–7. doi: 10.1042/bj2500421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savinov AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+ T cells. J Exp Med. 2003;197:643–56. doi: 10.1084/jem.20021378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schreiber H. In: Tumor immunology. 5. WP, editor. Philadelphia: Lippincott-Raven Publishers; 2003. [Google Scholar]
- 21.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, et al. Recruitment and Activation of Naive T Cells in the Islets by Lymphotoxin beta Receptor-Dependent Tertiary Lymphoid Structure. Immunity. 2006 doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6:205–17. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 24.Ruddle NH. Lymphoid neo-organogenesis: lymphotoxin's role in inflammation and development. Immunol Res. 1999;19:119–25. doi: 10.1007/BF02786481. [DOI] [PubMed] [Google Scholar]
- 25.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 26.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–91. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 28.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 1998;9:71–9. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- 29.Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. 1999;189:403–12. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuff CA, Schwartz J, Bergman CM, Russell KS, Bender JR, Ruddle NH. Lymphotoxin alpha3 induces chemokines and adhesion molecules: insight into the role of LT alpha in inflammation and lymphoid organ development. J Immunol. 1998;161:6853–60. [PubMed] [Google Scholar]
- 31.Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LT alpha beta directs lymphoid organ neogenesis with concomitant expression of peripheral node addressin and a HEV-restricted sulfotransferase. J Exp Med. 2003;197:1153–63. doi: 10.1084/jem.20021761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol. 2000;156:1133–8. doi: 10.1016/S0002-9440(10)64981-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. 1996;183:1461–72. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spriggs DR, Sherman ML, Michie H, Arthur KA, Imamura K, Wilmore D, et al. Recombinant human tumor necrosis factor administered as a 24-hour intravenous infusion. A phase I and pharmacologic study. J Natl Cancer Inst. 1988;80:1039–44. doi: 10.1093/jnci/80.13.1039. [DOI] [PubMed] [Google Scholar]
- 35.Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfeld RA, et al. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity. 2001;14:111–21. doi: 10.1016/s1074-7613(01)00094-2. [DOI] [PubMed] [Google Scholar]
- 36.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- 37.Kirk CJ, Hartigan-O'Connor D, Nickoloff BJ, Chamberlain JS, Giedlin M, Aukerman L, et al. T cell-dependent antitumor immunity mediated by secondary lymphoid tissue chemokine: augmentation of dendritic cell-based immunotherapy. Cancer Res. 2001;61:2062–70. [PubMed] [Google Scholar]
- 38.Wang J, Foster A, Chin R, Yu P, Sun Y, Wang Y, et al. The complementation of lymphotoxin deficiency with LIGHT, a newly discovered TNF family member, for the restoration of secondary lymphoid structure and function. Eur J Immunol. 2002;32:1969–79. doi: 10.1002/1521-4141(200207)32:7<1969::AID-IMMU1969>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, et al. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest. 2001;108:1771–80. doi: 10.1172/JCI13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. 2004;5:141–9. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- 41.Sharma S, Stolina M, Luo J, Strieter RM, Burdick M, Zhu LX, et al. Secondary lymphoid tissue chemokine mediates T cell-dependent antitumor responses in vivo. J Immunol. 2000;164:4558–63. doi: 10.4049/jimmunol.164.9.4558. [DOI] [PubMed] [Google Scholar]
- 42.Wolkers MC, Brouwenstijn N, Bakker AH, Toebes M, Schumacher TN. Antigen bias in T cell cross-priming. Science. 2004;304:1314–7. doi: 10.1126/science.1096268. [DOI] [PubMed] [Google Scholar]
- 43.Yu P, Spiotto MT, Lee Y, Schreiber H, Fu YX. Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J Exp Med. 2003;197:985–95. doi: 10.1084/jem.20021804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen L. Overcoming T cell ignorance by providing costimulation. Implications for the immune response against cancer. Adv Exp Med Biol. 1998;451:159–65. [PubMed] [Google Scholar]
- 45.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–58. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 46.Pardoll DM. Spinning molecular immunology into successful immunotherapy. Nat Rev Immunol. 2002;2:227–38. doi: 10.1038/nri774. [DOI] [PubMed] [Google Scholar]
- 47.Mortarini R, Scarito A, Nonaka D, Zanon M, Bersani I, Montaldi E, et al. Constitutive expression and costimulatory function of LIGHT/TNFSF14 on human melanoma cells and melanoma-derived microvesicles. Cancer Res. 2005;65:3428–36. doi: 10.1158/0008-5472.CAN-04-3239. [DOI] [PubMed] [Google Scholar]
- 48.Wu Q, Wang Y, Wang J, Hedgeman EO, Browning JL, Fu YX. The requirement of membrane lymphotoxin for the presence of dendritic cells in lymphoid tissues. J Exp Med. 1999;190:629–38. doi: 10.1084/jem.190.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–6. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 50.Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106:259–62. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 51.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–8. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 52.Randolph GJ, Sanchez-Schmitz G, Angeli V. Factors and signals that govern the migration of dendritic cells via lymphatics: recent advances. Springer Semin Immunopathol. 2005;26:273–87. doi: 10.1007/s00281-004-0168-0. [DOI] [PubMed] [Google Scholar]
- 53.Sozzani S, Allavena P, Vecchi A, Mantovani A. Chemokines and dendritic cell traffic. J Clin Immunol. 2000;20:151–60. doi: 10.1023/a:1006659211340. [DOI] [PubMed] [Google Scholar]
- 54.Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005;22:439–50. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Wang YG, Kim KD, Wang J, Yu P, Fu YX. Stimulating Lymphotoxin {beta} Receptor on the Dendritic Cells Is Critical for Their Homeostasis and Expansion. J Immunol. 2005;175:6997–7002. doi: 10.4049/jimmunol.175.10.6997. [DOI] [PubMed] [Google Scholar]
- 56.Inaba K, Schuler G, Witmer MD, Valinksy J, Atassi B, Steinman RM. Immunologic properties of purified epidermal Langerhans cells. Distinct requirements for stimulation of unprimed and sensitized T lymphocytes. J Exp Med. 1986;164:605–13. doi: 10.1084/jem.164.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Labeur MS, Roters B, Pers B, Mehling A, Luger TA, Schwarz T, et al. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol. 1999;162:168–75. [PubMed] [Google Scholar]
- 58.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohlen C, Kalos M, Cheng LE, Shur AC, Hong DJ, Carson BD, et al. CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J Exp Med. 2002;195:1407–18. doi: 10.1084/jem.20011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhodapkar MV, Steinman RM, Sapp M, Desai H, Fossella C, Krasovsky J, et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J Clin Invest. 1999;104:173–80. doi: 10.1172/JCI6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Probst HC, Lagnel J, Kollias G, van den Broek M. Inducible transgenic mice reveal resting dendritic cells as potent inducers of CD8+ T cell tolerance. Immunity. 2003;18:713–20. doi: 10.1016/s1074-7613(03)00120-1. [DOI] [PubMed] [Google Scholar]
- 62.Schuler G, Steinman RM. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J Exp Med. 1997;186:1183–7. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Summers-DeLuca LE, McCarthy DD, Cosovic B, Ward LA, Lo CC, Scheu S, et al. Expression of lymphotoxin-alphabeta on antigen-specific T cells is required for DC function. J Exp Med. 2007;204:1071–81. doi: 10.1084/jem.20061968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morel Y, Truneh A, Sweet RW, Olive D, Costello RT. The TNF superfamily members LIGHT and CD154 (CD40 ligand) costimulate induction of dendritic cell maturation and elicit specific CTL activity. J Immunol. 2001;167:2479–86. doi: 10.4049/jimmunol.167.5.2479. [DOI] [PubMed] [Google Scholar]
- 65.Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003;170:4905–13. doi: 10.4049/jimmunol.170.10.4905. [DOI] [PubMed] [Google Scholar]
- 66.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Browning JL, Miatkowski K, Sizing I, Griffiths D, Zafari M, Benjamin CD, et al. Signaling through the lymphotoxin beta receptor induces the death of some adenocarcinoma tumor lines. J Exp Med. 1996;183:867–78. doi: 10.1084/jem.183.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, et al. LIGHT, a novel ligand for lymphotoxin beta receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest. 1998;102:1142–51. doi: 10.1172/JCI3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu P, Lee Y, Wang Y, Liu X, Auh S, Gajewski TF, et al. Targeting the primary tumor to generate CTL for the effective eradication of spontaneous metastases. J Immunol. 2007;179:1960–8. doi: 10.4049/jimmunol.179.3.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clark WH, Jr, Elder DE, Guerry Dt, Braitman LE, Trock BJ, Schultz D, et al. Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst. 1989;81:1893–904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- 71.Clemente CG, Mihm MC, Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–10. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 72.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 73.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 74.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu P, Rowley DA, Fu YX, Schreiber H. The role of stroma in immune recognition and destruction of well-established solid tumors. Curr Opin Immunol. 2006;18:226–31. doi: 10.1016/j.coi.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 76.Torre-Amione G, Beauchamp RD, Koeppen H, Park BH, Schreiber H, Moses HL, et al. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc Natl Acad Sci U S A. 1990;87:1486–90. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huber S, Schramm C, Lehr HA, Mann A, Schmitt S, Becker C, et al. Cutting edge: TGF-beta signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol. 2004;173:6526–31. doi: 10.4049/jimmunol.173.11.6526. [DOI] [PubMed] [Google Scholar]
- 79.Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174:5215–23. doi: 10.4049/jimmunol.174.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Q, Yang X, Pins M, Javonovic B, Kuzel T, Kim SJ, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res. 2005;65:1761–9. doi: 10.1158/0008-5472.CAN-04-3169. [DOI] [PubMed] [Google Scholar]
- 81.Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, et al. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779–91. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, et al. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–41. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 83.Harrop JA, Reddy M, Dede K, Brigham-Burke M, Lyn S, Tan KB, et al. Antibodies to TR2 (herpesvirus entry mediator), a new member of the TNF receptor superfamily, block T cell proliferation, expression of activation markers, and production of cytokines. J Immunol. 1998;161:1786–94. [PubMed] [Google Scholar]
- 84.Liu J, Schmidt CS, Zhao F, Okragly AJ, Glasebrook A, Fox N, et al. LIGHT-deficiency impairs CD8+ T cell expansion, but not effector function. Int Immunol. 2003;15:861–70. doi: 10.1093/intimm/dxg082. [DOI] [PubMed] [Google Scholar]
- 85.Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195:1613–24. doi: 10.1084/jem.20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tamada K, Ni J, Zhu G, Fiscella M, Teng B, van Deursen JM, et al. Cutting edge: selective impairment of CD8+ T cell function in mice lacking the TNF superfamily member LIGHT. J Immunol. 2002;168:4832–5. doi: 10.4049/jimmunol.168.10.4832. [DOI] [PubMed] [Google Scholar]
- 87.Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. 2006;107:1342–51. doi: 10.1182/blood-2005-08-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shaikh RB, Santee S, Granger SW, Butrovich K, Cheung T, Kronenberg M, et al. Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J Immunol. 2001;167:6330–7. doi: 10.4049/jimmunol.167.11.6330. [DOI] [PubMed] [Google Scholar]
- 89.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest. 2003;21:873–86. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- 91.Stift A, Friedl J, Dubsky P, Bachleitner-Hofmann T, Schueller G, Zontsich T, et al. Dendritic cell-based vaccination in solid cancer. J Clin Oncol. 2003;21:135–42. doi: 10.1200/JCO.2003.02.135. [DOI] [PubMed] [Google Scholar]
- 92.Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, Jonuleit H, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190:1669–78. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–73. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 95.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 97.Miller FR, Miller BE, Heppner GH. Characterization of metastatic heterogeneity among subpopulations of a single mouse mammary tumor: heterogeneity in phenotypic stability. Invasion Metastasis. 1983;3:22–31. [PubMed] [Google Scholar]
- 98.Parviz M, Chin CS, Graham LJ, Miller C, Lee C, George K, et al. Successful adoptive immunotherapy with vaccine-sensitized T cells, despite no effect with vaccination alone in a weakly immunogenic tumor model. Cancer Immunol Immunother. 2003;52:739–50. doi: 10.1007/s00262-003-0405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998;58:1486–93. [PubMed] [Google Scholar]
- 100.Loeffler M, Le'Negrate G, Krajewska M, Reed JC. Attenuated Salmonella engineered to produce human cytokine LIGHT inhibit tumor growth. Proc Natl Acad Sci U S A. 2007;104:12879–83. doi: 10.1073/pnas.0701959104. [DOI] [PMC free article] [PubMed] [Google Scholar]