

Abstract
KCNQ channels carry the slowly activating, voltage-dependent M-current in excitable cells such as neurons. Although the KCNQ2 homomultimer can form a functional voltage-gated K+ channel, heteromultimerization with KCNQ3 produces a > 10-fold increase in current amplitude. All KCNQ channels contain double coiled-coil domains (TCC1 and TCC2, or A-domain Head and Tail), of which TCC2 (A-domain Tail) is thought to be important for subunit recognition, channel assembly and surface expression. The mechanism by which TCC2 recognizes and associates with its partner is not fully understood, however. Our aim in the present study was to elucidate the recognition mechanism by examining the phenotypes of TCC2-deletion mutants, TCC2-swapped chimeras and point mutants. Electrophysiological analysis using Xenopus oocytes under two-electrode voltage clamp revealed that homotetrameric KCNQ3 TCC2 is a negative regulator of current expression in the absence of KCNQ2 TCC2. Recent structural analysis of KCNQ4 TCC2 revealed the presence of intercoil salt bridge networks. We therefore swapped the sign of the charged residues reportedly involved in the salt bridge formation and functionally confirmed that the intercoil salt bridge network is responsible for the subunit recognition between KCNQ2 and KCNQ3. Finally, we constructed TCC2-swapped KCNQ2/KCNQ3 mutants with KCNQ1 TCC2 or GCN4-pLI, a coiled-coil domain from an unrelated protein, and found that TCC2 is substitutable and even GCN4-pLI can work as a substitute for TCC2. Our present data provide some new insights into the role played by TCC2 during current expression, and also provide functional evidence of the importance of the intercoil salt bridge network for subunit recognition and coiled-coil formation, as is suggested by recent crystallographic data.
The KCNQ (Kv7) family of channels carries slowly activating voltage-gated K+ currents such as IKs in cardiac cells and the M-current in neurons. The genes encoding five KCNQ family members (KCNQ1–5 or Kv7.1–5) have been identified in the human genome, four of which are related to inherited diseases (Jentsch, 2000; Gutman et al. 2005). KCNQ1 is a causal gene for long-QT syndrome and deafness (Wang et al. 1996); neuronal KCNQ2 and KCNQ3 are related to inherited benign familial neonatal convulsions (BFNCs) (Biervert et al. 1998; Charlier et al. 1998; Singh et al. 1998); and KCNQ4 is related to hearing loss (Kubisch et al. 1999).
All five KCNQ subtypes are able to form homomultimeric voltage-gated K+ channels, and some will also form heteromeric channels with other KCNQ subtypes. For example, KCNQ3 barely produces a K+ current by itself; however, it associates with KCNQ2, 4 and 5, contributing to the molecular diversity of KCNQ currents (Schroeder et al. 1998; Wang et al. 1998; Kubisch et al. 1999; Lerche et al. 2000; Schroeder et al. 2000). The most extensively studied combination is the heteromultimer comprising KCNQ2 and KCNQ3. The average maximal amplitude of the current carried by KCNQ2/3 channels is 10-fold larger than that carried by homomeric KCNQ2 channels, so that this heteromultimer is thought to carry the neuronal M-current in vivo (Schroeder et al. 1998; Wang et al. 1998).
It has been proposed that current augmentation induced by heteromerization of KCNQ2 and KCNQ3 reflects three mechanisms: (1) interaction of the C-terminal regions leads to increased plasma membrane expression of the channels; (2) the negative effect on current flow of the N-terminal domain of KCNQ2 seen in homomeric channels is absent in KCNQ2/3 heteromeric channels; and (3) the critical block of current flow mediated by Ala315 in the inner vestibule of KCNQ3 in homomeric channels is absent in heteromeric channels (Etxeberria et al. 2004). Several studies related to the first mechanism have been reported so far. All KCNQ channels have a highly conserved KCNQ-specific domain, the so-called ‘A-domain’ or ‘subunit interaction (si) domain’ in the distal cytoplasmic C-terminal region (Schmitt et al. 2000; Jenke et al. 2003; Maljevic et al. 2003; Schwake et al. 2003, 2006; Kanki et al. 2004; Howard et al. 2007; Wiener et al. 2008). This domain contains two double coiled-coil domains (‘A-domain Head and Tail’ or ‘TCC1 (tetramerizing coiled-coil 1) and TCC2’) that are thought to play important roles in subunit recognition, assembly and trafficking. For instance, Schwake et al. (2006) recently showed that TCC2 is required for efficient transport of KCNQ2/3 channels to the plasma membrane. It may be therefore that an understanding of how one TCC2 recognizes the corresponding domain in another subunit is a key to understanding the mechanism by which current is augmented in heteromeric KCNQ2/3 channels.
Recent data on the crystal structure of the KCNQ4 and KCNQ1 TCC2 (A-domain Tail) homotetramer have shed light on this important topic (Howard et al. 2007; Wiener et al. 2008). One of the most important findings of those studies is that there are intercoiled-coil electrostatic contact networks within TCC2. Moreover, the patterns of these contacts in KCNQ1 differ from those in KCNQ4, which implies that this pattern may be utilized for subunit recognition and for exclusion of KCNQ1 from other KCNQ channels. Our aim in the present study was to elucidate the mechanism of current augmentation induced by heteromeric assembly of TCC2 domains. In addition, by introducing point mutations that disrupt salt bridges, we tested the hypothesis that intercoil salt bridges are important for subunit recognition. Our electrophysiological data strongly support the functional significance of TCC2's electrostatic contact network in heteromerization and imply that specific patterns within intercoil salt bridge networks are utilized to identify proper partners.
Methods
Molecular biology
Rat KCNQ2 (NM_133322) and KCNQ3 (AF091247) cDNAs were subcloned into the pGEMHE expression vector (Nakajo & Kubo, 2005). Point mutants were created using PCR with KOD Plus v. 2 (Toyobo, Osaka, Japan) and primers carrying a desired mutation. Deletion mutants and chimeras were made as follows. Putative TCC2 domains of KCNQ2 and KCNQ3 were initially predicted using the COILS program (http://www.ch.embnet.org/software/COILS_form.html). Based on the results, we determined regions to be deleted: amino acid residues 601–633 for KCNQ2 and 567–621 for KCNQ3. We then introduced a Not I site at the beginning of TCC2 using reverse primers (5′-GCGGCCGCCGGGGTCTTCGGGCAGT-3′ for KCNQ2; 5′-GCGGCCGCCTGGTTCATTCCTTGGA-3′ for KCNQ3) and a Spe I site at the end of TCC2 using forward primers (5′-GGCACTAGTCCACCAGCAGAGACAG-3′ for KCNQ2; 5′-GGCACTAGTCCAACAAAGGGGGCCT-3′ for KCNQ3). Ligation of the blunt ends of the PCR products yielded TCC2 deletion mutants having Not I–Spe I sites (KCNQ2ΔTCC2/KCNQ3ΔTCC2). The TCC2 domain from each KCNQ channel with Not I and Spe I sites at each end was also amplified by PCR. The entire sequence of GCN4-pLI was obtained using synthetic oligoDNA primers having Not I and Spe I sites at each end. Each coiled-coil domain was subcloned into the Not I–Spe I sites of the TCC2 deletion mutants. Using these procedures, two amino acid linker sequences, Gly–Gly–Arg and Thr–Ser, were, respectively, inserted into the chimeras at the beginning and end of the TCC2. Sequences of deletion mutants, point mutants and chimeras were subsequently confirmed by sequencing.
cRNA was prepared from the linearized plasmid cDNA using a T7 RNA transcription kit (Stratagene, La Jolla, CA, USA). The concentration and integrity of the mRNA were verified by gel electrophoresis. For the Western blotting, concentrations of the mRNA were checked again just before the injection by gel electrophoresis to confirm that the same amount of RNA was injected for each sample.
Preparation of Xenopus oocytes
Xenopus oocytes were collected from frogs anaesthetized in water containing 0.15% tricaine, after which the frogs were killed by decapitation. The isolated oocytes were treated with collagenase (2 mg ml−1, type 1, Sigma-Aldrich, St Louis, MO, USA) for 6 h to completely remove the follicular cell layer. Oocytes of similar size at stage V or VI were injected with 55 nl of cRNA solution and then incubated at 17°C in frog Ringer solution containing (mm): 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.3 Ca(NO3)2, 0.41 CaCl2 and 0.82 MgSO4 (pH 7.6) with 0.1% penicillin–streptomycin solution (Sigma-Aldrich). For coexpression of KCNQ2 and KCNQ3, the molar ratio of the mixed RNA was set at approximately 1: 1. All experiments conformed to the guidelines of the Animal Care Committee of the National Institute for Physiological Sciences.
Two electrode voltage clamp
Two days after cRNA injection, K+ currents were recorded under two-electrode voltage clamp using an OC725C amplifier (Warner Instruments, Hamden, CT, USA) and pCLAMP8 or 10 software (Axon Instruments, Union City, CA, USA). For comparison of current amplitudes, data were collected from the same batch of oocytes and recordings were made within 10 h. Data from the amplifier were digitized at 2 kHz or 10 kHz with 1 kHz filtering. The microelectrodes were drawn from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL, USA) to a resistance of 0.2–0.5 MΩ when filled with 3 m potassium acetate and 10 mm KCl (pH 7.2). The bath solution (ND96) contained (mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2 and 5 Hepes (pH 7.4). Oocytes held at −90 mV were stepped between −100 and +40 mV in 10 mV steps for 2 s each and then to −30 mV for 250 ms to obtain tail currents.
Tail current amplitudes were measured as the average value of 10–20 ms after the end of the test pulse. Using pCLAMP8 or 10 software, tail currents were fitted to a two-state Boltzmann equation: 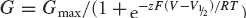 , where G is the tail current amplitude, Gmax is the maximum tail current amplitude, z is the effective charge, V1/2 is the half-activation voltage and T, F and R have their usual meanings. Gmax was always used for comparison of current amplitudes.
, where G is the tail current amplitude, Gmax is the maximum tail current amplitude, z is the effective charge, V1/2 is the half-activation voltage and T, F and R have their usual meanings. Gmax was always used for comparison of current amplitudes.
Sensitivity to tetraethylammonium (TEA; Fig. 1B) was probed using maximum tail current amplitudes by applying the step pulse protocol described above. Amplitudes were normalized for each oocyte to the value measured in the absence of TEA. The resulting dose–response profiles were fitted with the Hill equation.
Figure 1. KCNQ2ΔTCC2 cannot induce heteromerization-induced current augmentation with KCNQ3 but can form a heteromultimer with KCNQ3.

A, representative current traces for homomeric KCNQ2, heteromeric KCNQ2/KCNQ3, homomeric KCNQ2ΔTCC2 and heteromeric KCNQ2ΔTCC2/KCNQ3 channels. B, TEA sensitivity of wild-type KCNQ2 and KCNQ2ΔTCC2, with or without KCNQ3 coexpression. C, effect of a KCNQ3 dominant negative mutant (G281S) coexpressed with wild-type KCNQ2. Maximum tail current amplitudes at −30 mV are compared. ***P < 0.001 (Student's t test). D, effect of a KCNQ3 dominant negative mutant (G281S) coexpressed with KCNQ2ΔTCC2. Maximum tail current amplitudes at −30 mV are compared. **P < 0.01 (Student's t test).
All experiments were carried out at room temperature (25 ± 2°C).
Oocyte cell surface biotinylation
Cell surface proteins were biotinylated (18 oocytes for each sample) with 1.5 mg ml−1 of the membrane-impermeant N-hydroxysuccinimide-SS-biotin (NHS-SS-biotin; Pierce, Rockford, IL, USA) in ND96 for 30 min at 4°C. After washing five times in ND96 with 100 mm glycine, oocytes were homogenized by sonication in 360 μl of phosphate-buffered saline (PBS) containing 1% Triton X-100 and protease inhibitor cocktail (complete Mini EDTA-free; Roche Diagnostics, Mannheim, Germany) and maintained on ice for 30 min. The oocyte lysates were centrifuged for 5 min at 500 g to remove debris and then for 15 min at 16 000 g. After taking 10 μl of supernatant separately as total KCNQ2 proteins, 50 μl of streptavidin-coated agarose beads (Pierce) were added to the remaining supernatant, and samples were gently rocked at 4°C for 3 h to allow the streptavidin to bind biotinylated proteins. The biotin–streptavidin bead complexes were washed five times with PBS with a 10 s centrifugation (17 400 g) between washes. The final pellets were then dissolved in 30 μl of 1 × SDS sample buffer with DTT and incubated for 2 h at room temperature. The biotinylated proteins and the total proteins were separated in 7.5% SDS-PAGE gel to assess surface and total KCNQ2 proteins, respectively.
Western blotting
Thirty oocytes for each sample were homogenized by sonication in 300 μl of PBS containing 1% Triton X-100 and protease inhibitor cocktail. The lysates were centrifuged for 5 min at 500 g to remove debris and then for 15 min at 16 000 g. The lysates were incubated at room temperature for 2 h in SDS-PAGE sample buffer with DTT. The proteins in sample buffer were then separated in 7.5% SDS-PAGE gel and transferred electrophoretically to nitrocellulose membrane. After blocking with 5% skimmed milk in TBS-T (20 mm Tris-HCl, 150 mm NaCl and 0.05% Tween 20) for 1 h at room temperature, the membrane was incubated overnight at 4°C with primary rabbit anti-KCNQ2 or anti-KCNQ3 antibody against the C-terminal domain (1: 500 dilution; Sigma-Aldrich). After washing, the membrane was incubated in horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin G (1: 1000 dilution; Amersham, Arlington Heights, IL, USA) for 1 h at 37°C. Immunoreactivity was visualized using an enhanced chemiluminescence detection kit (Amersham) and was detected using a LAS-3000 image analyser (Fujifilm, Tokyo, Japan).
Structural data and presentation
To facilitate understanding where electrostatic contacts can be formed on the intercoiled-coil interaction face, we made some graphical presentations for the structure of four-stranded coiled-coil domains (Figs 5 and 6). Structural data for KCNQ4 TCC2 (PDB code; 2OVC), KCNQ1 TCC2 (3BJ4) and GCN4-pLI (1GCL) were presented using PyMOL software (DeLano Scientific, Palo Alto, CA, USA). Because structural data for KCNQ2 and KCNQ3 TCC2 were not available, we drew them by PyMOL using KCNQ4 as a template, assuming that the basic structures of TCC2 of KCNQ2 and KCNQ3 were similar to that of KCNQ4 (Howard et al. 2007). We substituted each amino acid residue of KCNQ4 for the corresponding amino acid residue of KCNQ2 or KCNQ3 step by step on PyMOL. Because the state and direction of side chains were not calculated by PyMOL, we manually adjusted them so that electrostatic contacts were formed on the corresponding sites if charged residues were conserved.
Figure 5. Salt bridge networks in TCC2 are important for subunit recognition and efficient current expression.

A, speculated structures of TCC2 in homomeric KCNQ2 and KCNQ3 channels based on the crystallographic structure of KCNQ4 TCC2 (see Methods). Green and orange dotted lines represent deduced salt bridges and hydrogen bonds, respectively. Amino acid residues involved in the salt bridge/hydrogen bond network are labelled. B, speculated TCC2 structure in the heteromeric KCNQ2/3 channel. KCNQ2/3 can have two different sides having different sets of electrostatic contacts, and both are shown. Green and orange dotted lines represent putative salt bridges and hydrogen bonds, respectively. Networks 1 and 2 are circled in black and blue, respectively. C and D, speculated structures for Networks 1 (C) and 2 (D) within different combinations of charge-swapped mutants. Two possible sides are shown for each. Green dotted lines represent putative salt bridges. Mutated amino acid residues are coloured yellow and labelled. Endogenous D594 in KCNQ3 is also labelled because it can make a salt bridge with E617K in the Q2KDK mutant, as depicted in D. All other salt bridges are formed between mutated residues. E, multiple comparisons of maximum tail current amplitudes among charge-swapped mutants. **P < 0.01, ***P < 0.001 (Tukey's test). F, expression levels for the charge-swapped mutants of KCNQ2 analysed by immunoblotting with anti-KCNQ2 antibody. KCNQ3 was not coexpressed. G, expression levels for the charge-swapped mutants of KCNQ3 analysed by immunoblotting with anti-KCNQ3 antibody. KCNQ2 was not coexpressed.
Figure 6. TCC2 can be replaced with other coiled-coil domains.

A, crystallographic structure of KCNQ1 TCC2. Green and orange dotted lines represent salt bridges and hydrogen bonds, respectively. Amino acid residues involved in the salt bridge/hydrogen bond network are labelled. B, representative traces from mutants containing KCNQ1 TCC2. C, crystallographic structure of GCN4-pLI. Green dotted lines represent salt bridges. Amino acid residues involved in the salt bridges are labelled. D, representative traces from mutants containing GCN4-pLI. E, multiple comparisons of maximum tail current amplitudes among KCNQ2 and KCNQ3 mutants having KCNQ1 TCC2 expressed with a wild-type KCNQ channel or TCC2 deletion mutant. ***P < 0.001 (Tukey's test). F, multiple comparisons of maximum tail current amplitudes among KCNQ2 and KCNQ3 mutants having GCN4-pLI expressed with wild-type KCNQ channels. *P < 0.05, ***P < 0.001 (Tukey's test). G, expression levels for the TCC2-swapped KCNQ2 chimeras analysed by immunoblotting with anti-KCNQ2 antibody. KCNQ3 was not coexpressed. H, expression levels for the TCC2-swapped KCNQ3 chimeras analysed by immunoblotting with anti-KCNQ3 antibody. KCNQ2 was not coexpressed.
As the backbone structure was not changed from that of KCNQ4, the graphical presentations in Fig. 5 do not necessarily reflect the actual structure of TCC2 of KCNQ2 and KCNQ3.
Statistical analyses
The data are expressed as means ±s.e.m., with n indicating the number of samples. Differences between means were evaluated using Student's unpaired t test for comparison between two groups. Dunnett's test or Tukey's test was used for multiple comparisons. Values of P < 0.05 were considered significant.
Results
TCC2 is required for heteromerization-induced current augmentation but not necessary for the heteromultimerization
KCNQ2 homomers are able to carry voltage-dependent K+ currents; however, coexpression of KCNQ3 with KCNQ2 makes the currents much larger (Wang et al. 1998). However, as previously reported (Schwake et al. 2006), KCNQ2 lacking a second coiled-coil domain (Q2ΔTCC2) carried a voltage-dependent K+ current similar to the wild-type KCNQ2 current, but the Q2ΔTCC2 current was not augmented by coexpression of KCNQ3 (Fig. 1A).
One possible reason for the Q2ΔTCC2 current not being augmented in the presence of KCNQ3 is that the deletion mutant was simply unable to form a heteromultimer with KCNQ3. To test that possibility, we took advantage of the fact that KCNQ2 is much more susceptible to block by TEA than KCNQ3 (Hadley et al. 2000; Schwake et al. 2006). The IC50 values for block of homomeric KCNQ2 and KCNQ3 channels by TEA are reportedly 0.3 and > 30 mm, respectively, while the IC50 value for block of heteromeric KCNQ2/3 channels is intermediate at 3.8 mm (Hadley et al. 2000). Therefore, if Q2ΔTCC2 actually forms a heteromultimer with KCNQ3, the apparent TEA sensitivity should be reduced by about one order of magnitude. When we examined the TEA sensitivities of wild-type KCNQ2 and Q2ΔTCC2, with and without KCNQ3, we found that for wild-type KCNQ2, the IC50 was increased by coexpression of KCNQ3 from 0.15 mm to 1.72 mm, indicating KCNQ2 does form a heteromultimer with KCNQ3 as previously reported (Hadley et al. 2000). For Q2ΔTCC2, the IC50 was increased by the coexpression of KCNQ3 from 0.08 mm to 0.69 mm, suggesting Q2ΔTCC2, too, forms a heteromultimer with KCNQ3 (Fig. 1B).
To further confirm the formation of heteromultimers with Q2ΔTCC2, we used a known KCNQ3 mutant (Q3(G281S)) that reportedly suppresses KCNQ2/3 currents in a dominant negative manner (Wollnik et al. 1997; Schwake et al. 2003). If KCNQ3 subunits form a heteromultimer with Q2ΔTCC2, coexpression of Q3(G281S) should suppress the Q2ΔTCC2 current. We found that the wild-type KCNQ2 current was suppressed by coexpression of Q3(G281S), as previously reported (Wollnik et al. 1997; Schwake et al. 2003); current amplitudes obtained with wild-type KCNQ2 and Q3(G281S) were significantly smaller than those obtained with the wild-type KCNQ2 current (0.94 ± 0.17 μA for KCNQ2 versus 0.08 ± 0.04 μA for KCNQ2/Q3(G281S); n = 10 for each, P < 0.001; Fig. 1C). Q2ΔTCC2 currents also were suppressed by Q3(G281S) (Fig. 1D); current amplitudes obtained with Q2ΔTCC2/Q3(G281S) were significantly smaller than those obtained with the Q2ΔTCC2 homomultimer (0.32 ± 0.06 μA for Q2ΔTCC2 versus 0.09 ± 0.01 μA for Q2ΔTCC2/Q3(G281S); n = 10 for each, P < 0.01). Thus the lack of augmentation of Q2ΔTCC2/KCNQ3 currents does not appear to reflect an inability to form a heteromultimer.
KCNQ3 TCC2 is a negative regulator of current expression in the absence of KCNQ2 TCC2
We next made a KCNQ3 TCC2 deletion mutant (Q3ΔTCC2) and examined its effect on current expression. Heteromultimers with KCNQ2/Q3ΔTCC2 carried significantly smaller current than wild-type KCNQ2/KCNQ3 did; however, KCNQ2/Q3ΔTCC2 current was significantly larger than Q2ΔTCC2/KCNQ3 current. Figure 2A and B shows that the average maximal amplitude of KCNQ2/Q3ΔTCC2 currents was 4.28 ± 0.41 μA (n = 12), which is approximately half the amplitude of wild-type KCNQ2/3 currents (8.61 ± 0.41 μA, n = 12, P < 0.001) from the same batch of oocytes but is significantly larger than the amplitude of Q2ΔTCC2/KCNQ3 (0.36 ± 0.04 μA, n = 12, P < 0.001). Interestingly, the average maximal amplitude of Q2ΔTCC2/Q3ΔTCC2 currents was 1.97 ± 0.19 μA (n = 12), which was substantially larger than the maximal Q2ΔTCC2/KCNQ3 current (P < 0.05; Fig. 2A and B). This implies that KCNQ3 TCC2 acts as a negative regulator of current expression in cases where KCNQ2 TCC2 is absent. We next quantified the surface expression levels by biotinylation using the membrane-impermeant N-hydroxysuccinimide-SS-biotin. As shown in Fig. 2C, surface expression levels were reduced when the KCNQ2/Q3 channels included either of the TCC2-deletion mutants while the whole expression levels were not different. This result indicates that the TCC2 domain is important for the efficient surface expression. Impaired surface expression could be a reason why TCC2-deletion mutants showed smaller current expression, although it does not explain why KCNQ2/Q3ΔTCC2 current was larger than Q2ΔTCC2/KCNQ3 current or why Q2ΔTCC2/Q3ΔTCC2 current was larger than Q2ΔTCC2/KCNQ3 current.
Figure 2. KCNQ3 TCC2 is a negative regulator of current expression in the absence of KCNQ2 TCC2.

A, representative current traces for KCNQ2/KCNQ3 (upper left), KCNQ2ΔTCC2/KCNQ3 (bottom left), KCNQ2/KCNQ3ΔTCC2 (upper right) and KCNQ2ΔTCC2/KCNQ3ΔTCC2 (bottom right) channels. B, multiple comparisons of maximum tail current amplitudes obtained with wild-type channels and TCC2-deletion mutants. *P < 0.05, **P < 0.01 and ***P < 0.001 (Tukey's test). C, whole and surface expression levels for TCC2-deletion mutants in oocytes. Total KCNQ2 represents KCNQ2 proteins from whole cell lysate to confirm the even expression levels for wild-type and TCC2-deletion channels. Surface KCNQ2 represents surface KCNQ2 proteins which were biotinylated before sonication (see Methods).
To better understand the mechanism by which the TCC2 domain controls current expression, we swapped the TCC2 domains of KCNQ2 and KCNQ3, yielding KCNQ2(Q3) (KCNQ2 having the KCNQ3 TCC2 instead of its own) and KCNQ3(Q2). All had the same backbone (KCNQ2 and KCNQ3) but different TCC2 combinations. Figure 3 shows that the combination of KCNQ2(Q3) and wild-type KCNQ3 carried significantly less current than wild-type KCNQ2/3 (1.39 ± 0.16 μA versus 7.72 ± 0.80 μA; n = 14 for each; P < 0.001). On the other hand, there was no statistical difference between the combination of KCNQ2/KCNQ3(Q2) (5.92 ± 0.46 μA; n = 14) and wild-type KCNQ2/3. These results also support that KCNQ3 TCC2 may act as a negative regulator of current expression under circumstances where KCNQ2 TCC2 is lacking. We conclude that, in the absence of KCNQ2 TCC2, KCNQ3 TCC2 acts as a negative regulator of current expression.
Figure 3. Homomeric KCNQ3 TCC2 is a weak mediator of current expression.

A, representative traces of wild-type and TCC2-swapped mutants. KCNQ2(Q3) indicates KCNQ2 containing KCNQ3 TCC2. B, comparison of maximum tail current amplitudes obtained with wild-type and TCC2-swapped mutants. Q2(Q3) indicates KCNQ2(Q3). ***P < 0.001 (Tukey's test).
Inefficiency of current expression is determined by some amino acid residues in KCNQ3 TCC2
Comparison of KCNQ2 and KCNQ3 revealed seven amino acids to be different in the region between residues 601 and 617 in KCNQ2 TCC2 (Fig. 4A). According to the report by Howard et al. (2007), KCNQ3 amino acids F585 (L606 in KCNQ2), D594 (S615 in KCNQ2) and G596 (E617 in KCNQ2) are likely to be responsible for the instability of tetrameric coiled-coil formation. F585 is located in the ‘a’ position within the heptad repeat of the coiled-coil. The side chains in the ‘a’ and ‘d’ positions of the domain must be hydrophobic and face the centre of the coiled-coil, and the side chain of F585 could be too bulky to form a coiled-coil structure. On the other hand, D594 and G596 are located in a hydrophilic environment. Corresponding amino acids in KCNQ4 participate in salt-bridge/hydrogen-bond networks which may stabilize the tetrameric coiled-coil formation (Howard et al. 2007). To determine whether these amino acids are truly responsible for the diminished current expression in homomeric KCNQ3 TCC2, we replaced each amino acid in KCNQ2 TCC2 with the corresponding residue from KCNQ3 TCC2 (L606F, G607V, L614H, S615D and E617G substitutions). We found that all these point mutants carried a normal KCNQ2/3 current, except S615D; it was not functional at all, though introduction of a second L614H mutation fully restored the current (data not shown). From these results, we could not conclude that a particular amino acid within KCNQ3 is responsible for the current suppression.
Figure 4. Three amino acid residues of KCNQ3 TCC2 are responsible for the inefficient current expression.

A, amino acid sequences of TCC2 of rat KCNQ2 (601st–626th amino acid residues) and KCNQ3 (580–605th amino acid residues) are shown along with the sequences of human KCNQ4 and human KCNQ1. Conserved and similar amino acids in KCNQ2 and KCNQ3 are indicated by asterisks and dots, respectively. The first and fourth amino acids in the heptad repeats (a–b–c–d–e–f–g) of the coiled-coil domain are indicated by the letters ‘a’ and ‘d’. B, representative current traces for Q2(Q3) and its mutants in the presence of KCNQ3. LSE indicates the F585L/D594S/G596E triple mutant. C, comparison of maximum tail current amplitudes for point mutants. SE indicates the D594S/G596E double mutant. ***P < 0.001 (Dunnett's test). D, expression levels for the Q2(Q3) mutants analysed by immunoblotting with anti-KCNQ2 antibody. KCNQ3 was not coexpressed. E, expression levels for the Q3 mutants analysed by immunoblotting with anti-KCNQ3 antibody. Q2(Q3) was not coexpressed.
In that context, we next tried to recover the KCNQ2(Q3)/KCNQ3 current by introducing KCNQ2 amino acid substitutions. We introduced an F585L mutation, D594S/G596E double mutation or F585L/D594S/G596E triple mutation into KCNQ2(Q3) or wild-type KCNQ3. The average maximal amplitude of KCNQ2(Q3)/KCNQ3 currents was 0.54 ± 0.08 μA (n = 16; Fig. 4B and C). Introducing an F585L mutation into KCNQ2(Q3) significantly increased current amplitude (2.20 ± 0.24 μA; n = 16; P < 0.001, Fig. 4B and C), as did introducing a F585L/D594S/G596E triple mutation (2.57 ± 0.34 μA; n = 16; P < 0.001). Introducing a D594S/G596E double mutation into KCNQ2(Q3) did not significantly increase current amplitude (1.19 ± 0.15 μA; n = 16; P = 0.07, Fig. 4C). Unexpectedly, none of the KCNQ3 mutations led to current recovery (Fig. 4C, right three bars). Still, the results obtained with the KCNQ2(Q3) mutants convinced us that F585 is at least partially responsible for the diminished amplitude of currents through the KCNQ2(Q3)/KCNQ3 channel. We also checked expression levels for the KCNQ2(Q3) mutants by Western blotting. Interestingly, the protein expression levels turned out to be parallel to the current expression levels (Fig. 4D). Introducing the point mutations, which increased current amplitude, also increased protein expression level of KCNQ2(Q3). This result implies that these three amino acids might be not only responsible for the stability of coiled-coil formation as Howard et al. (2007) reported, but also important for the protein expression level or whole protein stability. On the other hand, the protein expression levels for Q3 mutants were comparable to that of wild-type KCNQ3 (Fig. 4E). This result is also parallel to the current expression levels for KCNQ3 mutants, which failed to increase the current amplitude (Fig. 4C, right three bars).
Intercoil salt bridges are important for specific binding between different coils
Recent determination of the crystal structure of the KCNQ4 coiled-coil revealed the presence of several intra- and intercoil electrostatic interactions (Howard et al. 2007). Two adjacent KCNQ4 TCC2 domains make five intercoil electrostatic contacts, including three salt bridges (R618–E623, Q625–E630, S628–E630, K632–E630 and K632–D634). All five of those contacts are conserved in KCNQ2 (R605–E610, Q612–E617, S615–E617, K619–E617 and K619–D621; Fig. 5A left), but only two are conserved in KCNQ3 (K584–E589 and K598–D600; Fig. 5A right) (Howard et al. 2007). Within a KCNQ2/3 channel, in any stoichiometry or combination of KCNQ2 and KCNQ3, at least one KCNQ2–KCNQ3 interaction face at TCC2 would make four intercoil contacts, including three salt bridges (Fig. 5B left), and at least one other KCNQ2–KCNQ3 interaction face would contribute to two intercoil salt bridges (Fig. 5B right). Comparing the salt bridge networks of KCNQ2 and KCNQ3, E617 of KCNQ2 appears to have a huge impact on the difference between KCNQ2 and KCNQ3, as it is involved in three electrostatic contacts (Q612–E617, S615–E617 and K619–E617) and is absent from KCNQ3 (G596 instead). As we have already described, however, currents carried by the KCNQ2 E617G were unimpaired and comparable to those seen with wild-type KCNQ2.
With that in mind, we decided to disrupt all of the salt bridges in network 1 and/or network 2 between KCNQ2 and KCNQ3 (Fig. 5B) by reversing the sign of the charges of the participating amino acid residues. In that way the intercoil salt bridges between the mutant and wild-type KCNQ channels would be disrupted, while those between mutants would be retained. We introduced a double mutation into KCNQ2 (R605E and E610R; ‘Q2ER’ in Fig. 5C) and a double mutation into KCNQ3 (K584E and E589K; ‘Q3EK’ in Fig. 5C) for network 1, and a triple mutation into KCNQ2 (E617K, K619D and D621K; ‘Q2KDK’ in Fig. 5D) and a double mutation into KCNQ3 (K598D and D600K; ‘Q3DK’ in Fig. 5D) for network 2. Q2ERKDK and Q3EKDK possessed all the mutations described above in networks 1 and 2. As summarized in Fig. 5E, currents carried by any of the KCNQ3 mutants (Q3EK, Q3DK and Q3EKDK) with wild-type KCNQ2 were significantly smaller than wild-type KCNQ2/3 currents. For example, the average maximal amplitude of Q3EKDK/KCNQ2 currents was 1.59 ± 0.29 μA (n = 15), while Q3EKDK/Q2ERKDK current amplitude was 6.66 ± 0.58 μA (n = 14; P < 0.001 versus Q3EKDK/KCNQ2). Unexpectedly, all KCNQ2 mutants (Q2ER, Q2KDK and Q2ERKDK) carried substantial current with wild-type KCNQ3 (Fig. 5E). The average maximal Q2ERKDK/KCNQ3 current was 3.98 ± 0.41 μA (n = 15), which was comparable to the wild-type KCNQ2/3 current (5.52 ± 0.85 μA; n = 15, P > 0.05). This might be due to a newly formed salt bridge between a mutated amino acid and an endogenous charged amino acid residue, such as between E617K and endogenous D594 (Fig. 6D), but we still do not have a clear answer explaining why this happened. However, our finding that KCNQ2 mutants could restore currents carried by KCNQ3 mutants to wild-type levels (red lines in Fig. 5E) suggests the charged residues within TCC2 play a key role in forming the coiled-coil domain. We also checked the protein expression levels of the wild-type and the mutants of KCNQ2 and KCNQ3 (Fig. 5F and G). In this case, the expression levels for all mutants were similar to those of wild-type channels. This denies the possibility that the larger currents seen in the KCNQ mutants are merely due to the enhanced protein expression. It implies that not whole protein expression level but surface expression level is controlled by the proper combination of the coiled-coil domain. Salt bridge networks may be responsible for finding the appropriate partner.
Coiled-coil domains from different molecules can be substitutes for TCC2
We next tested whether coiled-coil domains from other molecules could serve as substitutes for KCNQ2/3 TCC2. We first replaced the TCC2 domains from KCNQ2 and KCNQ3 with that from KCNQ1 (Q2(Q1) and Q3(Q1); Fig. 6A and B). KCNQ1 also contains double coiled-coil domains at its cytoplasmic C-terminus (Jenke et al. 2003; Kanki et al. 2004; Wiener et al. 2008), but the degree of sequence similarity between KCNQ1 and other KCNQ channels, especially within TCC2, is not very high (see Fig. 4A). KCNQ1 has a different set of salt bridges (Wiener et al. 2008) and does not form heterotetramers with other KCNQ channels (Maljevic et al. 2003; Schwake et al. 2003, 2006). As we expected, neither Q2(Q1) nor Q3(Q1) carried a large current with wild-type KCNQ2 or KCNQ3 (Fig. 6E). However, the current was restored by combining Q2(Q1) with Q3(Q1) (Fig. 6B and E). The average maximal amplitude of Q2(Q1)/Q3(Q1) currents was 5.26 ± 0.22 μA (n = 12), which is significantly larger than either Q2/Q3(Q1) (1.47 ± 0.06 μA; n = 12; P < 0.001) or Q2(Q1)/Q3 (2.27 ± 0.27 μA; n = 12; P < 0.001) currents. To assess the specificity of the interaction with KCNQ1 TCC2, we also examined the effectiveness of TCC2 deletion mutants (Q2ΔTCC2 and Q3ΔTCC2). The average maximal amplitudes of Q2ΔTCC2/Q3(Q1) (0.32 ± 0.01 μA; n = 12) and Q2(Q1)/Q3ΔTCC2 (2.97 ± 0.45 μA; n = 12) currents were significantly smaller than those of Q2(Q1)/Q3(Q1) currents (P < 0.001 versus each; Fig. 6E blue lines). The relatively large amplitudes of Q2(Q1)/Q3ΔTCC2 currents reflects the same tendency seen in wild-type KCNQ2 with Q3ΔTCC2 (see Fig. 2). These results confirm that there is a specific interaction within KCNQ1 TCC2, and that the TCC2 domain of KCNQ2/3 complex is substitutable with TCC2 from other types of KCNQ channels.
We then replaced KCNQ2/3 TCC2 with GCN4-pLI, an artificial four stranded coiled-coil domain originally from a leucine zipper (Harbury et al. 1993; Minor et al. 2000; Zerangue et al. 2000). GCN4-pLI has two sets of salt bridges (total eight in one tetramer), Lys8-Glu10 and His18-Glu20 (Harbury et al. 1993), and the salt bridge pattern is completely different from those of KCNQ channels (Fig. 6C). Neither Q2/Q3(GCN4) nor Q2(GCN4)/Q2 carried currents as large as in the wild-type KCNQ2/3 channel, but the Q2(GCN4)/Q3(GCN4) combination restored the current to some extent (Fig. 6D and F). The average maximal amplitude of Q2(GCN4)/Q3(GCN4) currents was 2.41 ± 0.28 μA (n = 15), which is significantly larger than that of the Q2/Q3(GCN4) (1.08 ± 0.53 μA; n = 15; P < 0.05) or Q2(GCN4)/Q3 (1.11 ± 0.91 μA; n = 12; P < 0.05) current. The fact that KCNQ2/3 TCC2 can be replaced even with a different coiled-coil domain from a different protein indicates that TCC2 includes no specific trafficking signal and is not specifically indispensable to current augmentation. We confirmed that the protein expression levels of Q2(Q1) and Q2(GCN4) were as large as that of wild-type KCNQ2 (Fig. 6G). We also tried to confirm that the protein expression levels of Q3(Q1) and Q3(GCN4) were as large as that of wild-type KCNQ3 but it was difficult because Q3(Q1) and Q3(GCN4) appeared to be degraded to some extent. Still as both Q3(Q1) and Q3(GCN4) could up-regulate the current of Q2(Q1) or Q2(GCN4) better than wild-type KCNQ3, it is likely that Q3(Q1) and Q3(GCN4) were functional in Xenopus oocytes.
Discussion
It is well established that the second coiled-coil domain (TCC2 or A-domain Tail) in the cytoplasmic C-terminus of KCNQ channels plays an important role in the enhanced current expression induced by heteromultimerization of KCNQ2 and KCNQ3 (Maljevic et al. 2003; Schwake et al. 2003, 2006). In the present study, we aimed to elucidate the mechanism underlying the enhanced current expression by taking advantage of the recently revealed crystal structure of KCNQ4 TCC2 (Howard et al. 2007). Our three main findings are that (1) KCNQ3 TCC2 is a negative regulator of current expression in the absence of KCNQ2 TCC2; (2) intercoil salt bridges play an important role in heteromultimerization and subunit recognition; and (3) TCC2 is replaceable with any coiled-coil domain. Here we discuss the implications of our results, including a couple of discordant aspects, in the context of other recent reports.
The role of TCC2 for current expression
Heteromerization of KCNQ2 and KCNQ3 induces robust current expression (Wang et al. 1998). As described in the introduction, at least three mechanisms have been proposed (Etxeberria et al. 2004), and one of them is that the interaction of the C-terminal regions leads to increased plasma membrane expression of the channels. TCC2 (A-domain Tail) is now considered to be an interaction site within the C-terminal region and a molecular determinant for the enhanced current expression (Maljevic et al. 2003; Schwake et al. 2003, 2006; Kanki et al. 2004, 2006). Our present study focused solely on TCC2, so to exclude the effects of other mechanisms on current augmentation, we compared current amplitudes among TCC2 chimeras and mutants having the same backbone (heteromer of KCNQ2 and KCNQ3).
Using TCC2 chimeras and deletion mutants, Schwake et al. (2006) showed that tetramerizing TCC2 is important for efficient transport of KCNQ2/3 channels to the plasma membrane. They also showed that currents through a KCNQ2 TCC2 deletion mutant (KCNQ2ΔTCC2) were not facilitated by coexpression with KCNQ3, which we also observed in the present work (Fig. 1). They hypothesized two scenarios for the possible mechanism, that an efficient forward trafficking motif is formed or exposed by the interaction of two TCC2s, and that an endoplasmic reticulum (ER) retention motif is masked by the interaction. In the present study, we found that even an unrelated coiled-coil domain (GCN4-pLI) can function as a substitute for TCC2 (Fig. 6). This means that the formed forward trafficking motif suggested in the first scenario is not necessary as the amino acid sequence of GCN4-pLI does not resemble TCC2 of KCNQ channels at all. We think this makes the first scenario unlikely, though it is still possible that a forward trafficking motif outside TCC2 might be somehow formed or exposed when a four stranded coiled-coil is formed. If the second scenario is the case, KCNQ3 TCC2 might be a good candidate for an endoplasmic retention motif. We noticed that the KCNQ3 TCC2 deletion mutant (KCNQ3ΔTCC2) was not as an effective current suppressor as KCNQ2ΔTCC2 was (Fig. 2). We also observed that the current from coexpression of KCNQ2ΔTCC2 and KCNQ3ΔTCC2 was significantly larger than that of KCNQ2ΔTCC2 and KCNQ3 (Fig. 2). In addition, as shown in Fig. 3, current expression was significantly impaired in the absence of KCNQ2 TCC2. These results imply a possible role of KCNQ3 TCC2 for the current expression: KCNQ3 TCC2 might be a negative current regulator in the absence of KCNQ2 TCC2. If so, one possible scenario is that KCNQ3 TCC2 serves as an ER retention signal in the absence of KCNQ2 TCC2 as Schwake et al. (2006) proposed. Forming tetramer with KCNQ2 TCC2 might release KCNQ3 from ER to the plasma membrane.
Using size exclusion chromatography, Howard et al. (2007) showed that KCNQ3 TCC2 could not form homotetramer while other KCNQ TCC2 could. Their data also explain why homomeric KCNQ3 TCC2 does not efficiently carry current. They identified three amino acids (F585, D594 and G596) responsible for the inability to form homotetramer (Howard et al. 2007). Our findings that current amplitude was effectively recovered when the Q2(Q3) F585L mutant or Q2(Q3) F585L/D594S/G596E triple mutant was coexpressed with wild-type KCNQ3 (Fig. 4B and C) support their results. Interestingly, the introduction of mutations on these amino acid residues (F585L/D594S/G596E) increased the protein expression level in KCNQ2(Q3) chimera constructs (Fig. 4D) while expression levels of KCNQ2 were not affected by the deletion of TCC2 (KCNQ2ΔTCC2, Fig. 2C), by the introduction of opposite charges in TCC2 (Fig. 5F) and by the introduction of KCNQ1 TCC2 or GCN4-pLI (Fig. 6C). Having KCNQ3 TCC2 therefore might have a negative effect on the protein expression level, although the lower protein expression level might be an indirect result of ER retention. This was not the case, however, for the protein expression levels of KCNQ3 and KCNQ3 LSE (F585L/D594S/G596E) mutants (Fig. 4C and E). This is possibly due to KCNQ2(Q3) protein being more vulnerable than KCNQ3 protein so that the effect of mutation for stabilizing coiled-coil was more apparent on KCNQ2(Q3) than on KCNQ3.
We noticed that four-stranded TCC2 is not ultimately required for current expression: KCNQ2ΔTCC2/KCNQ3ΔTCC2 carried a substantial current, though the amplitude was not as large as the wild-type KCNQ2/3 current (Fig. 2). Similarly, KCNQ2/KCNQ3ΔTCC2, which should not contain a four-stranded coiled-coil domain, also carried a relatively large current (Fig. 2). Nevertheless, a necessary condition for the most efficient current expression is the formation of a stable four-stranded coiled-coil domain as shown in Fig. 2C.
The role of intercoil salt bridges for heteromultimerization
The recent report on the crystal structure of the homotetrameric KCNQ4A-domain Tail (TCC2) provides significant insight into the formation of heterotetramers (Howard et al. 2007). There are two networks of electrostatic contacts in KCNQ4A-domain Tails (TCC2). Howard et al. (2007) proposed that these electrostatic contacts may stabilize the coiled-coil structure and also underlie a specific association between different coils. To test their ideas, we swapped the sign of charges of the residues of KCNQ2 and KCNQ3 involved in the electrostatic contact networks (Fig. 5). Charge-swapped mutants for KCNQ3 (Q3EK, Q3DK and Q3EKDK) worked as we expected; they failed to produce large current with wild-type KCNQ2 while they showed large current with KCNQ2 charge-swapped mutants (Q2ER, Q2KDK and Q2ERKDK) (Fig. 5). However, KCNQ2 charge-swapped mutants similarly produced large current both with KCNQ3 and with KCNQ3 charge-swapped mutants. One possible reason is that mutated amino acid residues might form unexpected electrostatic contacts with endogenous KCNQ3 amino acid residues. For example, E617K in Q2KDK can form salt bridges with endogenous D594 in KCNQ3 as shown in Fig. 5D. R605E in Q2ER might form salt bridges with endogenous R590 in KCNQ3. These unexpected electrostatic contacts might explain the results. As a second possibility, we also have to consider the contribution from hydrophobic amino acid residues located at ‘a’ or ‘d’ positions. Salt bridges might be less important for coiled-coil formation in KCNQ2 while salt bridges are important for coiled-coil formation in KCNQ3 because it has an ‘unfavourable’ amino acid residue (F585) at the ‘a’ position which prevents TCC2 from forming four stranded coiled-coil (Howard et al. 2007). Unequal contributions from different subunits due to these possible mechanisms might explain the results of Figs 4 and 5.
The patterns of the network of electrostatic contacts differ among KCNQ channels. All electrostatic contacts seen in KCNQ4 are also conserved in KCNQ2 and 5, but KCNQ3 lacks three contacts in network 2 (Howard et al. 2007), and different types of amino acid residues are involved in the network in KCNQ1 (Wiener et al. 2008). These different patterns might contribute to the discrimination of proper partners for the formation of a tetrameric coiled-coil as implied in Fig. 6. This mechanism could be important not only for regulating the specific interactions between KCNQ channel subunits, but also for the interaction between channels and other proteins having coiled-coil domains such as yotiao (Marx et al. 2002).
Acknowledgments
We thank Dr D. McKinnon for providing us with cDNA encoding rat KCNQ2 and rat KCNQ3. We thank Dr M. Ito for help in Western blotting and Ms Y. Asai for preparation of oocytes. This work was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan to K.N. (18790164) and to Y.K., and from the Japan Society for the Promotion of Science to Y.K.
References
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Etxeberria A, Santana-Castro I, Regalado MP, Aivar P, Villarroel A. Three mechanisms underlie KCNQ2/3 heteromeric potassium M-channel potentiation. J Neurosci. 2004;24:9146–9152. doi: 10.1523/JNEUROSCI.3194-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1–4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Howard RJ, Clark KA, Holton JM, Minor DL., Jr Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron. 2007;53:663–675. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenke M, Sanchez A, Monje F, Stuhmer W, Weseloh RM, Pardo LA. C-terminal domains implicated in the functional surface expression of potassium channels. EMBO J. 2003;22:395–403. doi: 10.1093/emboj/cdg035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Kanki H, Kupershmidt S, Yang T, Wells S, Roden DM. A structural requirement for processing the cardiac K+ channel KCNQ1. J Biol Chem. 2004;279:33976–33983. doi: 10.1074/jbc.M404539200. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Lerche C, Seebohm G, Alekov AK, Busch AE, Lerche H. C-terminal interaction of KCNQ2 and KCNQ3 K+ channels. J Physiol. 2003;548:353–360. doi: 10.1113/jphysiol.2003.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- Minor DL, Lin YF, Mobley BC, Avelar A, Jan YN, Jan LY, Berger JM. The polar T1 interface is linked to conformational changes that open the voltage-gated potassium channel. Cell. 2000;102:657–670. doi: 10.1016/s0092-8674(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Nakajo K, Kubo Y. Protein kinase C shifts the voltage dependence of KCNQ/M channels expressed in Xenopus oocytes. J Physiol. 2005;569:59–74. doi: 10.1113/jphysiol.2005.094995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N, Schwarz M, Peretz A, Abitbol I, Attali B, Pongs O. A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO J. 2000;19:332–340. doi: 10.1093/emboj/19.3.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- Schwake M, Athanasiadu D, Beimgraben C, Blanz J, Beck C, Jentsch TJ, Saftig P, Friedrich T. Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J Neurosci. 2006;26:3757–3766. doi: 10.1523/JNEUROSCI.5017-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M, Jentsch TJ, Friedrich T. A carboxyterminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, De Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, Hirsch JA. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem. 2008;283:5815–5830. doi: 10.1074/jbc.M707541200. [DOI] [PubMed] [Google Scholar]
- Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathophysiological mechanisms of dominant and recessive KVLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet. 1997;6:1943–1949. doi: 10.1093/hmg/6.11.1943. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Jan YN, Jan LY. An artificial tetramerization domain restores efficient assembly of functional Shaker channels lacking T1. Proc Natl Acad Sci U S A. 2000;97:3591–3595. doi: 10.1073/pnas.060016797. [DOI] [PMC free article] [PubMed] [Google Scholar]