

Abstract
The mammalian placenta is the site of exchange of nutrients and waste between mother and embryo. In humans, placental insufficiency can result in intrauterine growth retardation, perinatal death and spontaneous abortion. We show that in C57BL/6J mice a null mutation in the gene encoding the transcriptional corepressor, Tgif, causes placental defects. The major defects are decreased vascularization of the placenta, due to a decrease in the fetal blood vessels, and decreased expression of the gap junction protein Gjb2 (Cx26). These defects result in severe growth retardation in a proportion of Tgif null embryos in Tgif heterozygous mothers, and an overall growth delay in Tgif null animals. Placental defects are much more severe if the mother also completely lacks Tgif function, and placentas from heterozygous Tgif embryos are defective in a Tgif null mother. Embryo transfer experiments show that even the placenta from a wild type embryo is compromised in the absence of maternal Tgif. These results demonstrate that Tgif functions in the normal development of the placenta, and suggest a role for maternal factors in regulating the morphogenesis of embryonically derived placental tissues.
Keywords: Tgif, placenta, morphogenesis, transcription
Introduction
The mammalian placenta provides a large surface area over which exchange of nutrients and gases between the mother and embryo occurs. In humans, placental insufficiency can result in intrauterine growth retardation (IUGR), which is the second leading cause of perinatal death, affecting up to 6% of human pregnancies. Abnormal development of the villi or extravillous trophoplasts in humans, which results in decreased feto-maternal exchange, is associated with complications including spontaneous abortion, preeclampsia and IUGR (Kingdom et al., 2000). Although there are some differences between mouse and human placentas, they are functionally equivalent and many of the important gene products, which regulate placental development, are conserved (Cross et al., 2003).
In mice, placentation initiates with the development of the trophectoderm, which differentiates to form all trophoblast lineages of the placenta (Cross et al., 2003). Following implantation, the mural trophectoderm forms primary trophoblast giant cells and penetrates the uterine stroma. The polar trophectoderm, which forms adjacent to the inner cell mass, includes progenitor cells that differentiate into secondary trophoblast giant cells, ectoplacental cone (EPC) and extraembryonic ectoderm. Both primary and secondary giant cells undergo multiple rounds of endoreduplication, resulting in cells with as high as 1024N DNA content (Varmuza et al., 1988; Zybina and Zybina, 1996). The EPC and extraembryonic ectoderm cells remain diploid, and give rise to the spongiotrophoblast and labyrinth layers of the chorio-allantoic placenta (Cross et al., 2003). In culture, the progenitor cells of both the extraembryonic ectoderm and the EPC will differentiate to form trophoblast giant cells (Tanaka et al., 1998), although their ultimate fate in vivo is to form either chorionic trophoblasts or spongiotrophoblasts. This observation has led to the concept that differentiation to the giant cell lineage may be a default pathway, and that differentiation to other lineages requires other, distinct signals (Cross et al., 1994). In support of this, several genetic mutations that result in perturbations of trophoblast differentiation, lead to over-production of trophoblast giant cells and deficiencies in spongiotrophoblast and labyrinthine trophoblasts (reviewed in (Cross et al., 1994)). For example, mutations in genes such as Mash2, Arnt and Socs3 (Guillemot et al., 1994; Kozak et al., 1997; Takahashi et al., 2003; Tanaka et al., 1997), result in an absence of spongiotrophoblast cells and loss of the labyrinth, with compensatory increases in giant cell numbers.
Within the labyrinth, the closely interdigitated maternal and fetal blood spaces allow for the normal exchange of nutrients, oxygen and waste products between mother and embryo (Watson and Cross, 2005). The initial formation of blood spaces begins at embryonic day 9 (E9), as the primary villi develop over the chorionic surface. As these vessels develop, they undergo repeated branching, and form an intricate pattern of smaller capillaries (Cross et al., 2006). The initiation of this branching requires the Gcm1 transcription factor, which is expressed at the tips of the villi as long as they are undergoing branching (Anson-Cartwright et al., 2000; Basyuk et al., 1999). Targeted mutations of other genes have been shown to affect the formation of the fetal vasculature within the labyrinth, but it is not always clear whether this is a primary effect on vascularization or vessel branching, or secondary to other defects in placental development (Watson and Cross, 2005). The close apposition of fetal vessels and maternal blood spaces within the labyrinth allows for the transfer of nutrients between mother and embryo. This transfer requires nutrients to pass through the cells which line and separate the maternal and fetal circulation (Malassine and Cronier, 2005; Watson and Cross, 2005). Connections between these cells are formed by gap junctions, which are made up of clustered transmembrane channels, consisting of 12 subunit complexes of connexins linking one cell to the next. These intercellular connections allow the passage of small molecules and metabolites. Gap junctions formed by the connexin, Cx26, are required for transplacental glucose uptake in the mouse placenta (Gabriel et al., 1998). From E10, Cx26 is expressed in labyrinth of the placenta, in regions where maternal-fetal exchange is occurring. Deletion of the mouse Cx26 gene results in reduced glucose uptake by the embryo, embryonic growth retardation and death by E11, suggesting an essential role for Cx26 gap junctions in placental function (Gabriel et al., 1998).
TGIF (TG-interacting factor) is a transcriptional repressor, which recruits general corepressor proteins, including CtBP, mSin3 and histone deacetylases (Melhuish et al., 2001; Melhuish and Wotton, 2000; Sharma and Sun, 2001; Wotton et al., 2001; Wotton et al., 1999b). A second TGIF-like protein (TGIF2) has been identified and shown to perform many of the same functions as TGIF (Imoto et al., 2000; Melhuish et al., 2001; Melhuish and Wotton, 2006). In mice and humans, both TGIF and TGIF2 are widely expressed, suggesting possible functional redundancy (Imoto et al., 2000; Jin et al., 2005). TGIF was originally identified by its ability to bind a specific retinoid response element (Bertolino et al., 1995). More recently, TGIF has been shown to regulate transcription by binding to the retinoid receptor, RXR, and recruiting corepressors, such as CtBP (Bartholin et al., 2006). TGIF also regulates activation of gene expression by the Smad proteins in response to transforming growth factor β (TGFβ) signaling (Wotton et al., 1999a). In response to TGFβ, Smad2 and Smad3 are phosphorylated by TGFβ receptors, form a complex with the co-Smad, Smad4, and activate target gene expression (Massague et al., 2005). TGIF competes with coactivators for Smad interaction, and recruits corepressors to limit the transcriptional activation of TGFβ/Smad target genes (Wotton et al., 1999a).
In humans, mutations in TGIF are associated with holoprosencephaly (HPE), a prevalent human genetic disease affecting craniofacial development (Hayhurst and McConnell, 2003; Muenke and Beachy, 2000). Targeted deletion of Tgif in mice suggests that even in the homozygous null state, HPE-like phenotypes are not found at any significant frequency (Bartholin et al., 2006; Jin et al., 2006; Mar and Hoodless, 2006; Shen and Walsh, 2005). Although some laterality defects and altered cell cycle progression in cultured cells isolated in from Tgif null mice were observed (Mar and Hoodless, 2006), a clear role for Tgif in mouse development has not been identified. Here, we show that Tgif loss of function in a relatively pure C57BL/6J strain causes severe placental defects, primarily due to loss of Tgif function from the mother. After E9.5, a proportion of Tgif null embryos were severely growth retarded, and by E18.5 the average weights of wild type and Tgif null embryos were significantly different. Severely growth retarded embryos had placental defects, affecting both the size and vascularity of the labyrinth. Surprisingly, when the mother lacked Tgif altogether, defects in the embryo derived layers of the placenta were more frequent and more severe. The placentas of embryos from Tgif null mothers exhibited defective development of fetal blood vessels within the labyrinth, and reduced expression of Cx26. These results suggest that Tgif regulates placental development, and provide evidence for maternal regulation of the morphogenesis of embryonically derived placental tissues.
Materials and methods
Tgif gene disruption and mice
The Tgif null mutation has been described previously (Bartholin et al., 2006). Mice from the F1 generation were back-crossed 5 times to C57BL/6J to generate the N6 generation. All procedures were approved by the Animal Care and Use Committee of the University of Virginia.
DNA and RNA analyses
DNA was purified from tail snip (at P21) or yolk sac using Promega Wizard kit, or HotShot (Truett et al., 2000), and genotyped as described (Bartholin et al., 2006). RNA was isolated in Trizol, purified using an Rneasy Mini Kit (Qiagen), DNAse1 treated and repurified using Absolutely RNA kit (Stratgene). RNA was isolated from whole placenta (maternal and embryonic) from pooled litters of either wild type or Tgif-/- in-crosses. For qRT-PCR, cDNA was generated using Superscript III (Invitrogen), and analyzed in triplicate by real time PCR using a BioRad miniOpticon and Sensimix Plus SYBRgreen plus FITC mix (Quantace). Primer pairs were selected using Primer3 (http://frodo.wi.mit.edu/, see Table S1 for sequences). Expression was normalized to cyclophilin using the delta Ct (Livak) method, and is shown as mean plus standard deviation of triplicates.
Histology and embryo analyses
Placentas were fixed in 4% paraformaldehyde or Carnoy's fixative, embedded in paraffin. 5μm sections were de-paraffinized with Safeclear (Fisher) and stained with hematoxylin and eosin (H&E), or for immunofluorescence, sections were incubated with a rabbit polyclonal antibody to laminin (EY labs), Alexafluor 546-labeled goat anti-rabbit (Invitrogen) and Hoechst 33342 (Sigma). Rabbit antibodies against Cx26, Cx31 and Cx43 were from Invitrogen. For immunochemistry, antibody staining was detected using a Vectastain ABC kit (Vector Labs), developed with Impact DAB (Vector Labs), and counterstained with hematoxylin (Sigma). Images were captured using an Olympus BX51 microscope and DP70 digital camera, and manipulated in Adobe Photoshop.
Placental area and embryo and mouse weight
The relative cross-sectional areas of E10.5 placentas were determined from H&E stained Carnoy's fixed sections. Pixel areas were measured in Adobe Photoshop. A single section from the center of each placenta was used, based on the site of umbilical attachment. For analysis of maternal blood spaces and fetal blood vessels, 20× images of a central region of the labyrinth, from central placental sections were analyzed similarly. E18.5 embryos, or mice at P21, were weighed, and average weights analyzed as raw weight, or relative to the litter average. For detailed quantitative analysis of placental areas, images were captured at 20× magnification using an Aperio Scanscope.
Embryo transfer
Estrus was induced in C57BL/6J females with 2.5u of Pregnant Mare's Serum Gonadotropin followed 46h later with 2.5u of human Chorionic Gonadotropin. Females were mated with vasectomized males. 8-12 wild type ICR embryos at E0.5 were transferred into pseudopregnant females, and embryos were isolated at E11. Recipient females were between 53 and 96 days old and weighed between 10.5g and 18.5g at the time of transfer.
Results
A strain specific decrease in embryo viability in Tgif null mothers
Disruption of the mouse Tgif gene does not cause significant defects in a mixed strain background (Bartholin et al., 2006; Jin et al., 2006; Mar and Hoodless, 2006; Shen and Walsh, 2005). To test whether loss of Tgif caused defects in other genetic backgrounds, we analyzed our Tgif mutants after six sequential backcrosses to C57BL/6J. In the mixed strain background we obtained viable, normal mutants, and no significant deviations from the expected genotype frequencies were observed in the offspring from Tgif heterozygous intercrosses (Table 1, (Bartholin et al., 2006)). In contrast, when we intercrossed Tgif heterozygous mutants in the relatively pure genetic background (>98.4% C57BL/6J), 50% of the homozygous mutant mice were missing by weaning at postnatal day 21 (P21), suggesting embryonic or neonatal lethality (Table 1).
Table 1.
Genotyping data from Tgif mutant intercrosses
| Strain | Mo × Faa | Stageb | +/+c | +/- | -/- | Total | Litters | Ave | Chi2 | Sigd |
|---|---|---|---|---|---|---|---|---|---|---|
| C57/129 mixe | +/- × +/- | P21 | 59 | 90 | 53 | 202 | 24 | 8.4 | 2.75 | no |
| 29% | 45% | 26% | ||||||||
| C57BL/6 (N6) | +/- × +/- | P21 | 183 | 332 | 97 | 612 | 101 | 6.1 | 28.6 | 0.001 |
| 30% | 54% | 16% | ||||||||
| C57/129 mix | +/- × -/- | P21 | 37 | 35 | 72 | 10 | 7.2 | 0.06 | no | |
| 51% | 49% | |||||||||
| C57/129 mix | -/- × +/- | P21 | 33 | 22 | 55 | 10 | 5.5 | 2.2 | no | |
| 60% | 40% | |||||||||
| C57BL/6 (N6) | +/- × -/- | P21 | 81 | 43 | 124 | 24 | 5.2 | 11.7 | 0.001 | |
| 65% | 35% | |||||||||
| C57BL/6 (N6) | -/- × +/- | P21 | 18 | 3 | 21 | 4f | 5.3 | 10.7 | 0.01 | |
| 86% | 14% | |||||||||
| C57BL/6 (N6) | +/- × -/- | E10.5 | 22 | 22 | 44 | 6 | 7.3 | 0 | no | |
| 50% | 50% | |||||||||
| C57BL/6 (N6) | -/- × +/- | E10.5 | 29 | 22 | 51 | 8 | 6.4 | 0.96 | no | |
| 59% | 41% |
the Tgif genotype of the mother and father are shown
Stage at which embryos or mice were analyzed (E; embryonic day, P21; postnatal day 21)
The number of offspring of each genotype is shown, with the percentage below.
The significance level, by Chi squared is shown
The strain backgrounds are as follows: C57/129 mix = mixed C57BL/6Jx129Sv/J, N6 = six back-crosses to C57BL/6J.
Tgif null females in the C57BL/6J background gave very few viable litters.
We next crossed Tgif null animals with heterozygotes. In the C57BL/6J background, the number of homozygous null animals born to heterozygous mothers was reduced by about half (Table 1). However, when the mother was null for Tgif and the father heterozygous we obtained only four viable litters, and only three Tgif null mice were weaned from them. In the mixed strain background, there was some decrease in the number of homozygous mutants weaned from homozygous null mothers, but with this number of animals the decrease was not significant (Table 1). We were able to isolate embryos at E10.5 from heterozygous by homozygous null crosses, even when the mother was the null. This suggests that the lack of weaned mice from C57BL/6J Tgif null mothers was due to a failure to maintain the pregnancy.
Growth defects in Tgif null mice
We noticed that a proportion of the embryos at E9.5-E18.5 from heterozygous intercrosses were significantly smaller than the average. Almost all the smaller embryos were Tgif-/-, suggesting that there may be a growth delay in a proportion of Tgif null embryos from as early as E9.5. To test whether there was a more general growth retardation in Tgif null embryos, we weighed 152 embryos at E18.5 from heterozygous intercrosses. Embryo weights were normalized by litter and expressed as a percentage of the litter average. The average weight of wild type and heterozygous embryos was almost identical, whereas the homozygous null embryos were on average lighter (Figure 1A). Analysis of 102 mice weaned at P21 from heterozygous intercrosses, (excluding severely runted animals; <50% of the average litter weight), revealed that the Tgif null mice were on average about 20% lighter than their littermates (Figure 1B). At both E18.5 and P21, the difference between wild type and homozygous null animals was highly statistically significant by Student's T test, while no significant difference between the wild type and heterozygous mutants was seen at either stage.
Fig. 1. Tgif null mice are growth retarded.
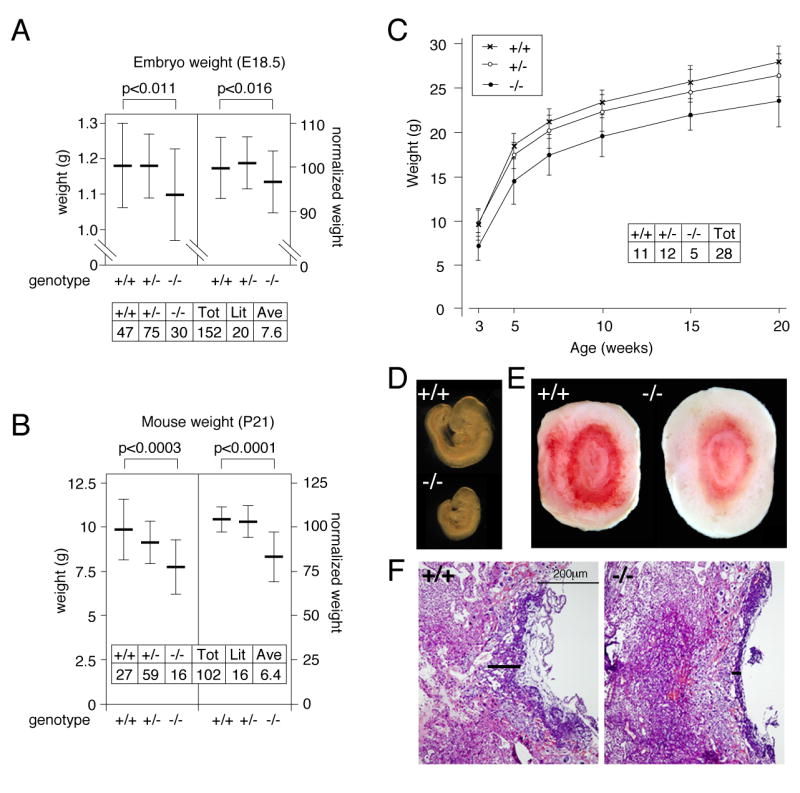
A) The weights of 152 embryos at E18.5 from 20 litters of Tgif heterozygous intercrosses were determined. The average weight (+/- s.d.) is plotted for each genotype. B) 102 mice were weighed at P21, and the average weight for each genotype is plotted. In the left hand panels for A and B, raw weights in grams are plotted and in the right hand panels, the normalized weights are shown as a percentage of the average litter weight. The significance for the difference between wild type and homozygous mutants, as determined by a Student's T test, is shown above each graph. C) 50 mice from 8 litters of heterozygous intercrosses were weighed at P21, and at intervals thereafter, up to 20 weeks of age. The average (+/- s.d.) weight of male mice for each genotype is plotted. D) A wild type and a growth retarded Tgif-/- E9.5 embryo are shown. E) Wild type and mutant placentas are shown. F) An H&E stained section through the center of each placenta is shown (10× magnification). Black bars indicate the thickness of the labyrinth.
To test whether Tgif null mice catch up with their littermates following weaning, we measured the weights of 28 male mice over a period of 20 weeks. These mice were obtained from 8 litters from heterozygous intercrosses, and were housed in equal numbers per cage, grouped by litter. The average weight of homozygous mutants remains below that of either wild types or heterozygotes, and as a group they do not catch up by 20 weeks of age (Figure 1C). Comparison of each individual mutant to the average shows that most mutants are smaller at weaning and do not fully catch up with their littermates by 20 weeks of age. Similar results were obtained with female mice (data not shown). Thus, it appears that the growth retardation suffered in utero persists as the mice mature.
Labyrinth defects in Tgif mutants
In cases where Tgif null embryos were severely reduced in size, the placenta was also incompletely formed, and often appeared to have reduced vascularization (Figure 1D, E). When these placentas were sectioned and stained with hematoxylin and eosin (H&E), it appeared that in the mutant, the labyrinth was reduced in thickness (black bars in Figure 1F). Even when the embryo was less severely affected, the mutant placenta was often abnormal, with a thinner, less well developed labyrinth. At E9.5, we observed a range of placental defects, which varied in severity from those that were indistinguishable from wild type, to very severe cases (Figure S1A-C, and data not shown). In less severely affected mutants some formation of fetal blood vessels within the labyrinth was visible, and fetal blood cells were seen within them (Figure S1A, B). In more severely affected mutants, there were almost no vessels visible and the entire labyrinth appeared compacted (Figure S1C). Similarly, at E13.5, embryos that were significantly smaller than their littermates were almost always Tgif null, and had placental defects. The severity of the placental defect at E13.5 generally correlated with the degree of growth retardation of the embryo (for example, Figure S1D-F). From our analysis of embryos at E9.5-E13.5, we estimate that severe placental defects affect around 10% of the Tgif null embryos from heterozygous intercrosses.
A maternal contribution to the placental defect
Since the Tgif genotype of the mother affected embryo viability, we analyzed placental defects in Tgif null mothers at E10.5. Although very few mice were weaned from homozygous mutant mothers, relatively normal numbers of embryos were found at E10.5 (Table 1). Sections from E10.5 placentas were stained with H&E, and the central sections identified based on the site of umbilical attachment. To span the range of possible genotypes, we analyzed placentas from heterozygous embryos in wild type, heterozygous and homozygous mutant mothers, as well as completely wild type or Tgif null placentas. H&E stained sections of representative placentas are shown in Figure 2. There is a clear decrease in overall placental size between the two extremes; wild type embryo from wild type mother, compared with Tgif null embryo from Tgif null mother (Figure 2A, E). In addition, the size of the placenta from heterozygous embryos also decreases from wild type to heterozygous, to homozygous Tgif mutant mother (Figure 2B-D). More strikingly, there is an apparent compaction of the labyrinth, which correlates with maternal genotype (Figure 2F-J). This is indicative of a decrease in the number of fetal blood vessels or maternal blood spaces within the labyrinth. Thus in a well-developed wild type there are numerous fetal vessels in close proximity to the maternal blood spaces (Figure 2F), whereas with loss of Tgif from both mother and embryo, there appear to be fewer blood spaces and fetal vessels and they are less closely intermingled (see Figure 6 for quantification). This is also apparent at lower magnification, and there is a clear contribution of the maternal genotype to the development of the labyrinth, which, other than maternal blood, is composed entirely of embryo-derived cells (Figure 2K-M). We also noticed that in defective labyrinths, there often appeared to be fewer, large maternal blood spaces with very few surrounding fetal vessels (compare 2K and M). The giant cell and spongiotrophoblast cell layers were both visible in the fetal regions of the placenta from all genotypes analyzed, although there was often a decrease in the size of these layers in defective placentas (Figure 2K-M).
Fig. 2. Defects in the embryonic placenta from Tgif mutant mothers.
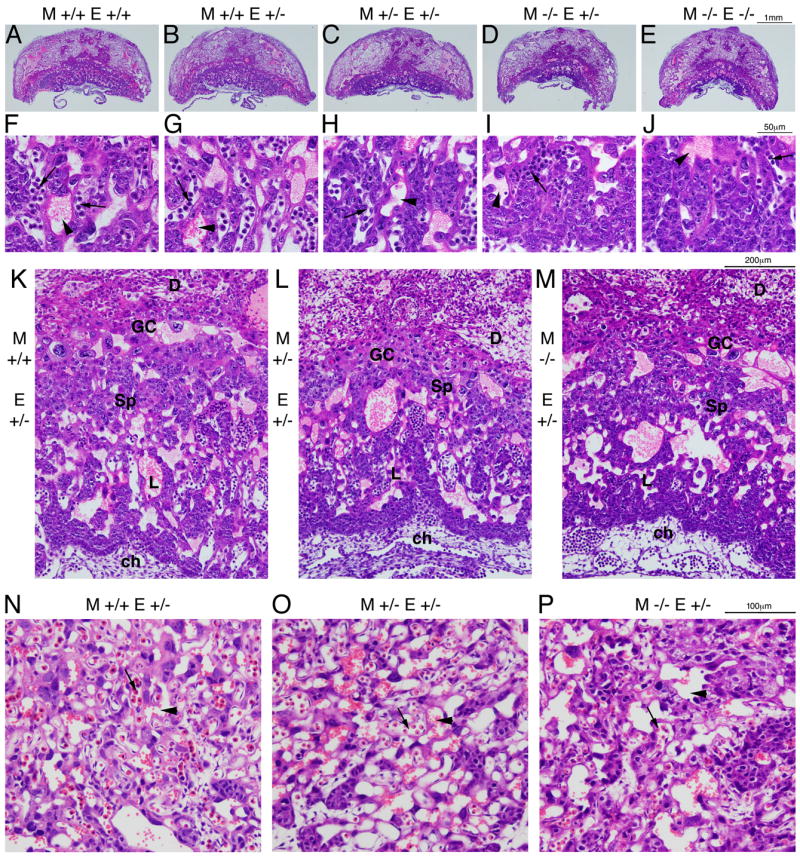
A-E) H&E stained sections through central sections of E10.5 placentas from the indicated Tgif genotypes are shown (2× magnification). F-J) 40× images of the labyrinth of the corresponding placentas shown in A-E. Arrows indicate fetal vessels, arrowheads show maternal blood spaces. K, L, M) 10× images of placentas from heterozygous embryos in each maternal genotype are shown. The decidua (D), giant cell (GC) and spongiotrophoblast (Sp) areas, as well as the labyrinth (L) and chorion (ch) are shown. N, O, P) 20× images of central regions of the labyrinth of E13.5 placentas from heterozygous embryos in each maternal genotype are shown. Arrows and arrowheads indicate fetal and maternal blood spaces respectively. The genotype of both the mother (M) and the embryo (E) are indicated for all sections.
We examined a number of placentas from heterozygous embryos in each maternal genotype at E13.5. It appears that by this stage differences in the overall size of the placenta are less obvious and that those embryos present at this stage in homozygous null mothers have an apparently more normally vascularized labyrinth. Figure 2N-P shows representative central regions of the labyrinths from heterozygous embryos from each maternal genotype. In the mutant mother, the labyrinth appears to be relatively better formed (compared to the wild type) than at E10.5, but is still somewhat less well vascularized. Taken together, these data suggest that there is an important contribution of the maternal genotype to the development of the embryo-derived regions of the placenta.
Correlation between an abnormally small labyrinth and growth retarded embryos
To compare differences in placental size more quantitatively, we analyzed the size of the placenta from seven litters (48 placentas) from heterozygous by homozygous null crosses (three in which the mother was Tgif-/- and four with Tgif+/- mothers), as well as 23 placentas from wild type mothers crossed to heterozygous males. To allow us to make a relatively simple comparison of a large number of samples, we selected central sections and measured the cross-sectional area of the entire placenta, and of the maternal decidua and embryonic region independently. The overall size of the placenta was slightly smaller in both heterozygous and homozygous mutant mothers, compared to wild types (Figure 3A). Comparison of only heterozygous embryos in each of the three maternal genotypes revealed that this decrease in placental cross-sectional area was significant. Interestingly, the areas of the embryo-derived regions of the placenta, from Tgif mutant mothers was dramatically smaller than in wild type mothers (Figure 3B). Again, this difference appeared to track more with the maternal genotype than the embryonic. We observed less difference in the size of the decidua than in the embryonic regions, although there was a significant difference between wild type and homozygous null (Figure 3C). These results suggest that while the most severe defects are in the embryo-derived layers of the placenta, the maternal Tgif genotype plays the major role.
Fig. 3. Decreased placenta size in Tgif null mothers.
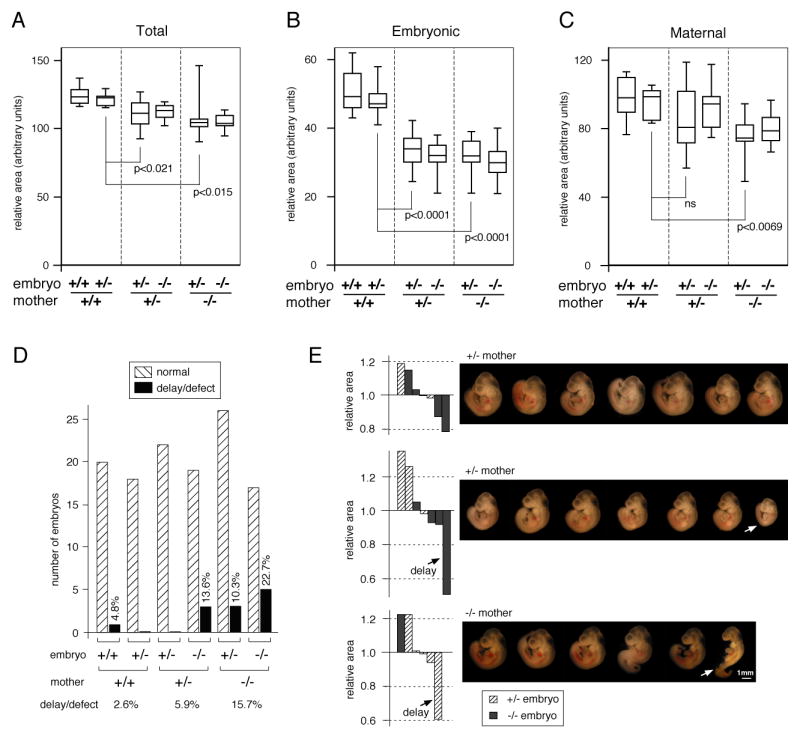
The cross-sectional area of central sections of E10.5 placentas of the indicated genotypes was determined. Data is presented as box-plots (median, upper and lower quartiles and extremes) for total placenta (A), the embryonic region (B) and the decidua (C). The significance levels (by T test) for pairwise comparisons of placentas from heterozygous embryos in each maternal genotype are shown. D) Embryos from the crosses analyzed for placental area were scored for growth delay or defect. The number of normal and defective embryos for each genotype is plotted. The percentage of delayed or defective embryos for each genotype is also shown. E) The relative area of the embryonic placenta for each of three litters is plotted, as a proportion of the litter average. The embryos from each litter are shown from left to right in the same order as the labyrinth area. The maternal genotype is shown, and delayed embryos and the corresponding placental areas are indicated by arrows.
To determine whether a defective placenta, as judged by cross-sectional area of the embryonic region, correlated with embryo defects, we compared embryos from the litters analyzed above. In a heterozygous mother, 13.6% of the mutant embryos had a serious growth delay, whereas none of the heterozygotes did (Figure 3D). This agrees well with our estimate of the frequency of severe placental defects from heterozygous intercrosses. In a mutant mother more than 10% of heterozygous embryos were delayed and almost a quarter of the homozygous mutants showed some delay or defect (Figure 3D). In contrast, we observed only one delayed embryo in a wild type mother in this sample. Comparison of the proportion of defective heterozygous embryos in each maternal genotype again revealed an increase in the severity of defects with decreasing number of Tgif alleles in the mother. The relative cross-sectional area of the embryonic placentas varied considerably within individual litters (Figure 3E). In litters in which the area did not vary by more than about 20% from the litter mean, we did not generally find growth retarded embryos; for example, the first litter from a heterozygous mother shown in Figure 3E (the corresponding embryos are shown in the same order as the placenta size is plotted). When we identified a placenta in which the area was reduced by significantly more than 25% compared to the litter average, the embryo was clearly growth retarded. Two examples are shown in Figure 3E, for a growth retarded homozygous null embryo in a heterozygous mother and a heterozygous embryo in a homozygous mutant mother. Thus it appears that considerable variation in placental size can be tolerated, but if it decreases too far, then embryonic development is compromised.
Decreased expression of trophoblast markers in Tgif null placentas
We next analyzed marker gene expression in placentas pooled from whole litters, of Tgif null or wild type in-crosses. E9.5 placental RNA was analyzed by qRT-PCR from four wild type and four Tgif null litters. We first tested expression of the spongiotrophoblast and giant cell markers, Tpbpα and Hand1. Expression of both genes was reduced in at least two of the Tgif mutant litters, whereas, expression of Actin, Hand2 and Tgif2 was not (Figure 4A-E). We next tested expression of the genes encoding three transcription factors, with roles in the formation of specific placental structures or cell types. Mash2 is required for the development of spongiotrophoblasts, Gcm1 for syncytiotrophoblasts, and both Gcm1 and Tfeb are important for branching of fetal vessels within the labyrinth. Expression of the genes encoding all three of these proteins is generally lower in the Tgif null placentas (Figure 4F-H). Similarly, expression of the giant cell specific genes, PL-I and PL-II, was decreased in Tgif mutant placentas (Figure 4I, J). PL-I and PL-II are members of the prolactin (PRL) family of hormones, which in rodents has undergone considerable gene duplication, resulting in a large family of related genes, many of which are expressed exclusively in trophoblast cells (Soares et al., 2007). However, in mice, two members of this family, PLP-J and decidual PRL-related protein (dPRP), are expressed only in the decidua. We, therefore, tested expression of PLP-J and dPRP to determine whether the decrease in expression of placenta-specific genes was specific to trophoblasts or also affected maternal tissue. We observed no decrease in expression of either dPRP or PLP-J (Figure 4K, L), whereas expression of three trophoblast-specific PRL family members (proliferin [Plf], proliferin 4 [Plf4] and proliferin related protein [Prp]) was decreased in the mutants, as seen for other trophoblast specific markers (Figure 4M-O).
Fig. 4. Defective trophoblast development in Tgif null placentas.

RNA was isolated from whole E9.5 placentas, pooled by litter. Genotypes were either wild type or Tgif null (mother and embryos), as indicated. Expression of the indicated genes, analyzed by qRT-PCR, is plotted in arbitrary units, normalized to cyclophilin (mean + sd of triplicates). The dashed lines indicate the average expression level for each gene among the four wild type samples.
Mutations in a number of genes in mice have been shown to affect implantation and decidualization, (Lee et al., 2007; Wang and Dey, 2006). To begin to test potential changes in decidualization in the absence of Tgif, we analyzed the expression of a panel of genes by qRT-PCR. The genes analyzed encode transcriptional regulators, such as CoupTFII, the progesterone receptor and Ncoa2, and signaling molecules such as Bmp2 and IL-11Rα, all of which play a role in decidualization. As shown in Figure S2, we did not observe a significant decrease in the expression of any of them. Furthermore, analysis of the expression patterns of the Ptgs2 and CEBPβ proteins, which are required for normal decidualization, was not changed in the absence of Tgif (data not shown). Taken together, these data demonstrate that in the Tgif null placenta there is decreased expression of numerous trophoblast specific genes, including markers of several trophoblast cell types, such as giant cells and spongiotrophoblasts. In contrast, expression of decidua-specific genes appeared not to be affected.
To confirm the decrease in multiple trophoblast cell types indicated by qRT-PCR, we analyzed the cross sectional areas of placentas from heterozygous embryos isolated from either wild type or Tgif null mothers in greater detail. As shown in Figure 5A (see also Figure 3), placentas from wild type mothers were somewhat larger than those from Tgif null mothers. This decrease in size affected primarily the embryonic regions of the placenta rather than the decidua (compare Figure 5B and 5F), despite the fact that the embryonic genotype was constant. Indeed, as a proportion of the total area, the decidua was slightly greater in the null mother than the wild type. When we quantified specific areas within the embryonic placenta, it was clear that in the absence of Tgif the labyrinth and the giant cell and spongiotrophoblast areas were all significantly smaller (Figure 5C-E). Together with the decreased expression of markers of different trophoblast cell types, this suggests an overall decrease in the differentiation or development of the major trophoblast cell types from TS cells in the absence of Tgif.
Fig. 5. Decreased trophoblast cell areas in placentas from Tgif null mothers.
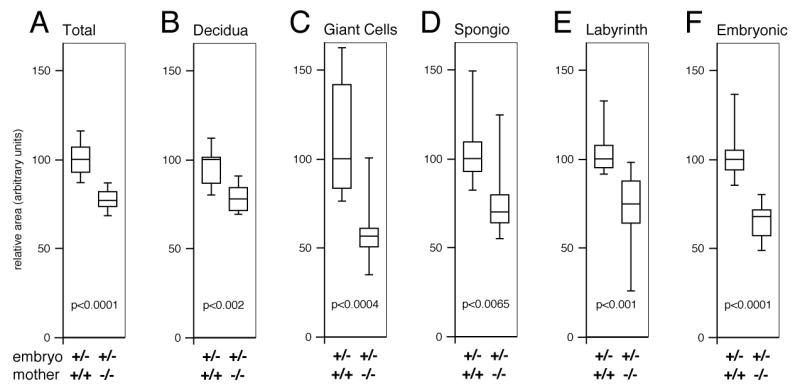
Central sections of E10.5 placentas from heterozygous embryos in either wild type (N=9) or Tgif null mothers (N=10) were analyzed for relative area by cell type. Areas are presented as box-plots (median, upper and lower quartiles and extremes), with the median value for the wild type in each case set to 100. Areas quantified were whole placenta (A), the decidua (B), trophoblast giant cells (C), spongiotrophoblasts (D), and labyrinth (E). In addition the overall area of the embryonic regions was analyzed (F). The significance levels (by T test) for comparisons of placentas from each maternal genotype are shown. The embryonic and maternal genotypes are shown below.
Decreased labyrinth vascularization in placentas lacking Tgif
To identify fetal vessels within the labyrinth, we stained central sections from a number of the placentas isolated from various Tgif mutant crosses with a laminin specific antibody. Laminin is present on the basal lamina of fetal capillaries, but is not detected in maternal blood spaces (Natale et al., 2006). We stained the same sections with Hoechst 33342, to identify the giant cells, and to distinguish between maternal blood and the nucleated fetal blood. In wild type mothers, numerous small fetal vessels form an intricate pattern throughout the labyrinth out to the giant cell layer, and this appears not to be affected by whether the embryo is wild type or heterozygous for Tgif (Figure 6A, B). This is indicative of a highly branched network of fetal vessels, which are in close apposition to maternal blood spaces (arrows [fetal] and arrowheads [maternal] in 6A-E). In a placenta from a heterozygous Tgif embryo from a Tgif null mother, while there is clearly some branching of the fetal vessels close to the chorion (Figure 6D), they have not formed the network of small vessels seen in placentas from wild type mothers (compare 6B and D). In the most severe cases, the formation of branched fetal vessels is even more dramatically reduced in embryos from a homozygous null mother (Figure 6E). There is some branching of fetal vessels from the chorion, but very few of the smaller fetal capillaries can be seen and there are very few fetal blood vessels between the giant cell layer and the major branches from the chorion (Figure 6D). Even in the placenta from a heterozygous embryo in a heterozygous mother, there is some decrease in the intricacy of the network of fetal blood vessels (Figure 6C), compared to placentas from wild type mothers. There also appears to be some disruption of the maternal blood spaces. However, this is more variable, with an apparent decrease in the number of maternal blood spaces in some defective placentas (for example compare 6C and D), and a few, larger maternal blood spaces visible in others.
Fig. 6. Abnormal fetal blood vessels in Tgif null labyrinths.
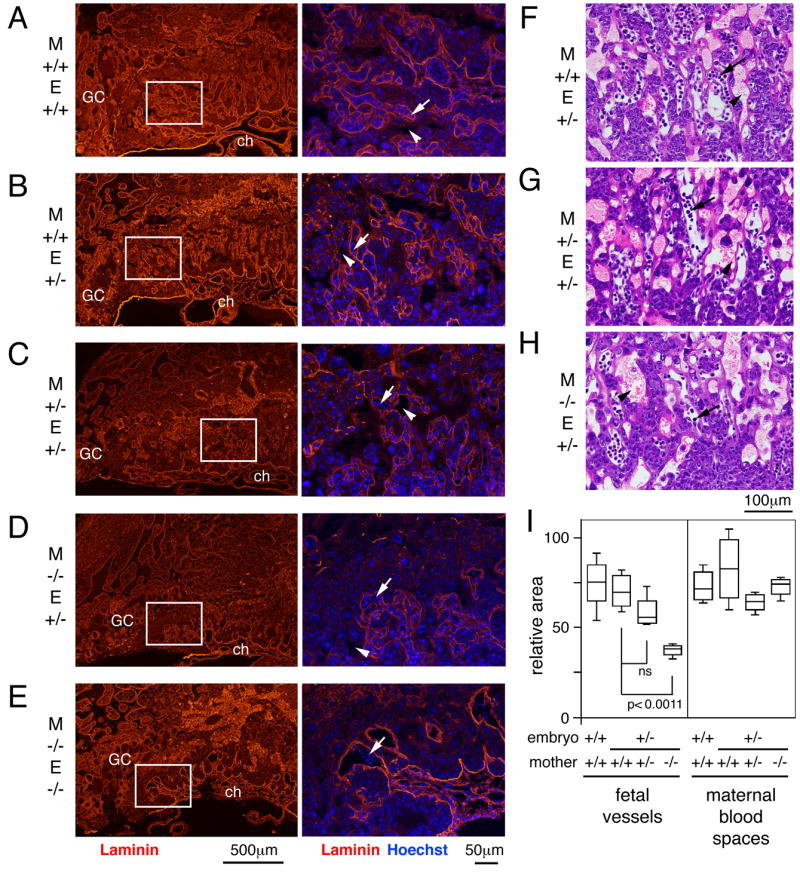
A-E) Central sections of placentas of the indicated maternal (M) and embryonic (E) genotypes were examined by indirect immunofluorescence. Staining with a laminin specific antibody, to visualize fetal vessels, is shown in red and Hoechst stain for DNA is shown overlayed in blue (right hand panels). The regions shown at higher magnification are indicated by a white box in the left hand panels. GC, giant cell; ch, chorion. Arrows and arrowheads indicate fetal and maternal blood spaces respectively. Images were captured at 4× (left) and 20× (right). F, G, H) 20× images of representative areas of H&E stained placental sections used for quantitative analysis of blood spaces. I) Central regions of the labyrinth from central placental sections were analyzed for the relative areas taken up by fetal blood vessels and maternal blood spaces. Data is shown as box plots (as in Figure 3), in arbitrary units.
To more quantitatively compare maternal and fetal blood spaces, we determined the relative areas occupied by each in fixed areas within the labyrinth from multiple central sections of each genotype. Representative H&E stained images of the regions analyzed for placentas from heterozygous embryos in each maternal genotype are shown in Figure 6F-H. There was a significant difference in fetal vessel area between heterozygotes from wild type and Tgif null mothers (Figure 6I). The fetal vessel cross-sectional area in heterozygous mothers was also less than that in wild types, but was not statistically significant with the number of sections analyzed. In contrast, we did not see any consistent changes in the areas occupied by maternal blood spaces (Figure 6I). Together, these data suggest that there is a decrease in vascularization of the labyrinth in Tgif mutants, and that this defect results primarily from loss of Tgif from the mother, with a lesser dependence on embryonic Tgif.
Decreased Cx26 expression in Tgif null mothers
We were interested to know whether the decrease in vascularization might be accompanied by other defects in vessel structure and function. Expression of a number of gap junction proteins (connexins) has been documented within the murine placenta (Malassine and Cronier, 2005). Interestingly, connexin 26 (Cx26, or Gjb2) is required for trans-placental glucose uptake, and mutation of the Cx26 gene results in embryonic growth retardation (Gabriel et al., 1998). We therefore tested expression of Cx26 by qRT-PCR in the panel of E9.5 placental RNAs used before. Expression of Cx26 was dramatically decreased in all four mutant litters, by an average of four-fold compared to the wild type average (Figure 7A). We also observed a smaller decrease in Cx31 expression (Figure 7B), but did not see a dramatic decrease in expression levels of any of several other connexins (Figure 7C-F). Interestingly, Cx43 expression was consistently increased in the mutants by a little more than 50% (Figure 7E).
Fig. 7. Connexin expression in Tgif null placentas.

A-F) The expression of the indicated connexin genes was analyzed at E9.5 by qRT-PCR, and is presented, as in Figure 4. G-L) Sections of E10.5 placentas from heterozygous embryos in either wild type or Tgif null mothers were stained with a Cx26 antibody. Images were captured at 10× (G, J), 40× (H, K) and 100× (I, L). Maternal (m) and fetal (f) blood spaces are indicated. The arrow in L shows a region of disorganized Cx26.
To test whether the decrease in Cx26 expression seen by qRT-PCR reflected a more serious defect in placental structure, we analyzed expression of Cx26 by immunostaining. As shown in Figure 7, Cx26 was observed in a pattern that approximately surrounds the fetal blood vessels. Comparison with adjacent sections stained with laminin shows that although this is generally true, in some places Cx26 expression is seen more towards adjacent maternal blood spaces (Figure S3). However, in severely defective placentas with few fetal vessels, Cx26 expression is dramatically reduced, even when large maternal blood spaces are present (Figure S3E). Interestingly, careful comparison of placentas from heterozygous embryos in wild type and Tgif null mothers revealed decreased Cx26 staining in placentas from null mothers (Figure 7 J, K). As shown in Figure 7L (arrow), gaps in the normally contiguous band of Cx26 were readily visible between fetal vessels and maternal blood spaces in placentas from Tgif null mothers.
We did not see any dramatic differences in Tgif null mothers for Cx31 staining, which is expressed in a punctate pattern throughout the labyrinth, rather than the tight association with fetal vessels seen for Cx26 (data not shown). We also analyzed the expression pattern of Cx43 in sections of E10.5 placentas, since its expression measured by qRT-PCR was consistently increased in the mutants (Figure 7E). In contrast to the labyrinth specific expression of Cx26, Cx43 was found exclusively in the decidua at this stage of gestation (Figure S4). Expression was seen throughout the decidua, up to the giant cell layer of the embryonic placenta, and was particularly intense surrounding maternal blood vessels in the decidua (Figure S4). By E12.5, Cx43 expression is also found in giant cells and spongiotrophoblasts (Plum et al., 2001), but we did not see any increased expression of Cx43 in the embryonic regions of Tgif null placentas at E10.5. Together these data suggest that there is a specific decrease in Cx26 expression in the absence of Tgif, whereas, expression of other connexins is not dramatically altered.
Loss of maternal Tgif causes placental defects
The previous analyses indicate a contribution of the maternal genotype to development of the embryonic placenta. However, where the mother is null for Tgif, the embryo has at least a heterozygous mutation, such that we cannot rule out an effect of a predisposing mutation in the embryo. We, therefore, performed embryo transfer experiments in which wild type ICR embryos at E0.5 were transferred into the oviduct of pseudopregnant wild type or Tgif null C57BL/6J mice, and placentas were isolated at E11. Embryo transfer to wild type recipients was quite successful, with four of five recipients carrying embryos at E11, and 74% of the embryos (34/46) transferred to these four still present at E11 (Figure 8A). In contrast, only one of eight Tgif null recipients still had embryos at E11, and only 6 of the 12 embryos transferred to this individual survived to E11. We did not observe a significant number of resorbing embryos, suggesting that Tgif null females may also have defects in uterine function, resulting in lower rates of implantation. However, we cannot rule out the possibility that the nature of the embryo transfer experiment itself also contributed to the apparent increased failure of implantation. From natural matings, of 17 mutant mothers for which vaginal plugs were observed, 11 (65%) had embryos at E9.5 or E10.5, with an average of 5.6 embryos per female. For comparison, 13 out of 17 wild type mothers (76%) had embryos at E9.5/E10.5, with an average of 6.4 embryos per litter. Thus, it appears that with natural matings there is not a dramatic difference in the implantation rate between wild type and mutant mothers. However, analysis of late stage embryos from matings in which the mother was Tgif null reveals that most litters are lost after mid-gestation, with only two of nine mothers (22%) which plugged having embryos by E18.5.
Fig. 8. Defective wild type placentas in Tgif null mothers.
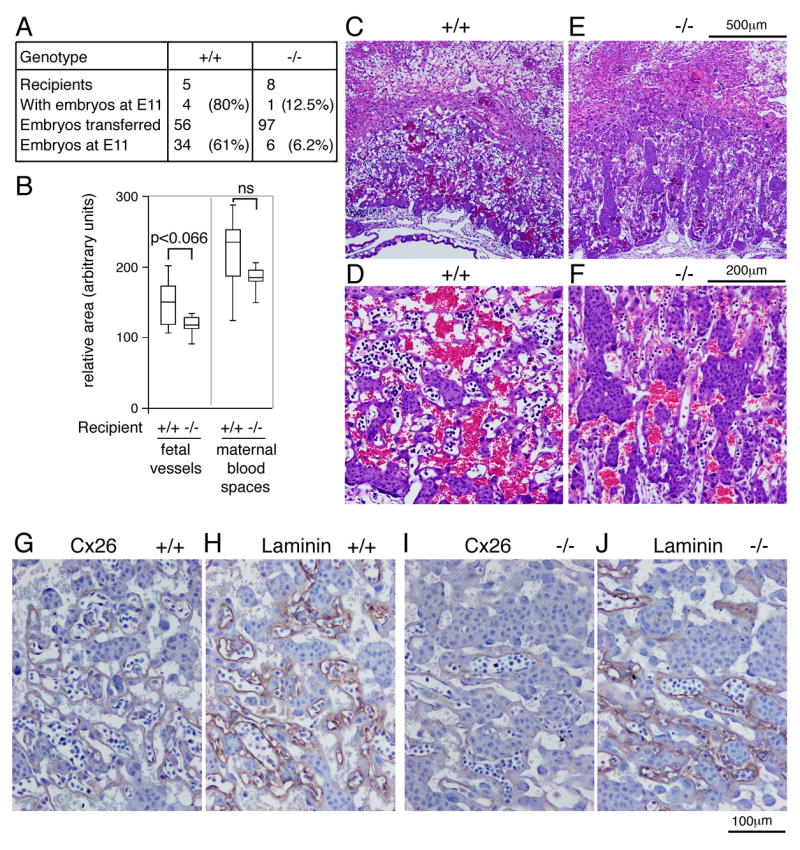
A) A summary of the embryo transfer experiments is shown. Wild type ICR embryos at E0.5 were transferred to the uteri of pseudopregnant wild type or Tgif null C57BL6/J mice, and placentas were isolated at E11. B) Quantitative analysis of the relative areas of maternal and fetal blood spaces in the labyrinths of central sections of placentas from the embryo transfer experiments. C-F) Representative H&E stained images of the labyrinth of placentas from embryo transfer experiments. Images were captured at 4× (C, E) and 10× (D, F). G-J) Immunostaining for laminin or Cx26 is shown (captured at 20×) in placentas from embryo transfer experiments. The genotype of the recipient mothers is shown in all panels.
To test whether placental development is impaired in a wild type embryo in a Tgif null mother, we sectioned placentas from the Tgif null and from three wild type embryo transfer recipients. We did not observe a decrease in overall placental cross-sectional area, or in the embryonic region specifically, in the Tgif null recipient. However, analysis of central sections of the labyrinth from 10 placentas from wild type mothers and five from the Tgif null revealed a decrease in the amount of fetal blood vessels (Figure 8B). Although this did not quite reach significance, it clearly points to a contribution of the maternally expressed Tgif to development of the fetal vessels. Examination of the H&E stained sections indicated that the labyrinth appeared to be less well vascularized, with less intimate contacts between fetal vessels and maternal blood spaces (Figure 8C-F). We next examined laminin and Cx26 staining in sections from either wild type or Tgif null recipients. As shown in Figure 8 (G-J), there was an overall decrease in the number of fetal vessels outlined by laminin, and a similar decrease in the amount of Cx26 staining, suggesting that even when maternal blood spaces are present, Cx26 expression is decreased if the number of fetal vessels is reduced. Together, these data clearly point to a major contribution of maternally expressed Tgif in the development and function of the embryonic placenta.
Discussion
Here we show that loss of Tgif function in the mother has severe consequences for the development of the embryonic regions of the placenta. In contrast to the minimal effects of Tgif loss of function on a mixed C57BL/6J x 129Sv/J strain background, we observed embryonic or neonatal lethality with about 50% penetrance in a relatively pure C57BL/6J background. In the mixed strain, this lethality is presumably compensated for by genetic determinants from the 129Sv/J strain background. We do not know what these strain specific differences are, and their identification is complicated by the partially penetrant and variable nature of the defects. We do not yet know what causes the majority of the lethality of Tgif null animals in heterozygous intercrosses, and this will be the subject of future work. However, we have clearly demonstrated that Tgif null mothers have severe placental defects, which are also present sporadically in heterozygous mothers, particularly if the embryo is homozygous null for Tgif.
In heterozygous intercrosses, severe placental defects were seen in only a small proportion of the mutant embryos. We also observed some post-natal growth delay in Tgif null mice. This may, at least initially, be a consequence of placental defects, but could also be due to independent effects of loss of Tgif on the adult mice. In homozygous Tgif null mothers the embryonic and placental defects were both more severe and more frequent, and defects were clearly seen even in placentas from heterozygous embryos in this maternal background. This suggests that there is an important function of maternally expressed Tgif that affects the normal development of the embryonic placenta. Comparison of the fetal vasculature suggests that even a heterozygous mutation in Tgif in the mother can cause defects in development of the embryonic regions of the placenta. However, in most cases this is not enough to cause severe embryonic defects. That a proportion of the placentas from homozygous Tgif null embryos in this background have severe defects clearly suggests that loss of expression of Tgif in the placenta itself can contribute to the phenotype. Thus the function of both maternally expressed and embryonic Tgif appear to be required for normal placental development.
From the analysis of the severity of placental defects, and associated embryonic growth retardation in embryos and mothers of different Tgif genotypes, it appears that loss of Tgif function from the mother is the major contributing factor. The best test for this is to transfer wild type embryos to Tgif null mothers, to completely rule out the possible requirement for a predisposing mutation in the embryo. Our attempts to do this clearly point to a problem with homozygous Tgif null mothers, since only one of eight Tgif null recipients was successful, compared to four out of five wild types. We did not observe a significant number of resorbing embryos at E11, suggesting that the mutants may have a problem with implantation. Although this may be pointing to a genuine defect, one success out of eight likely over-estimates the severity of potential implantation defects in Tgif null animals, and we cannot rule out a contribution of the embryo transfer procedure to this. Further work will be required to carefully test possible implantation defects. Interestingly, analysis of placental sections from the mutant recipient demonstrated that there were defects in the fetal blood vessels within the labyrinth. Since the transferred embryos were wild type, and of a different strain background (ICR), this result strongly suggests that loss of Tgif function from the mother is enough to cause defects in the development of the embryo-derived portions of the placenta. The embryos obtained from the Tgif null recipient in this transfer experiment were not obviously defective, suggesting that the changes in placental structure were not severe enough to result in embryonic growth retardation of a wild type embryo. However, since only one mutant recipient had embryos at E11 and the phenotypes seen in the Tgif null C57BL/6J strain are variable in severity, it is possible that this one represents the mild end of the spectrum.
Despite the significant contribution of the maternal genotype, the major defects in the placenta appear to be in embryo-derived structures. The majority of other targeted mutations in mice that affect the development of the embryonic placenta are due to the lack of a particular gene in the embryonic tissue itself. In many cases such defects are severe enough that the additional effect of homozygous loss of gene function in the mother has not been, or cannot easily be tested. Like the Tgif mutation described here, mutation of the IL-11Rα gene results in a failure of null females to carry embryos to term, while mutant mice were born at normal mendelian ratios to heterozygous mothers (Bilinski et al., 1998). Although defects in development of the embryonic placenta were observed, they were accompanied by a failure of normal decidualization, suggesting that the primary defect in the absence of IL-11Rα may be in the maternal tissue, followed by degeneration of the embryonic placenta. Interestingly, it was recently demonstrated that an ovary and uterus-specific deletion of the gene encoding the COUP-TFII nuclear receptor results in defects in both the uterus and placenta, suggesting that loss of a maternal transcription factor can affect placental development (Petit et al., 2007). Maternal loss of COUP-TFII also resulted in altered trophoblast differentiation and decreased vascularization due to a defective labyrinth. The mutations in Tgif and COUP-TFII may represent a paradigm for placental development in which the maternal function of regulators of placental development are required. In the case of Tgif, it appears that both maternal and embryonic functions are required for normal placental development. This suggests that in the absence of Tgif the maternal environment can predispose to placental defects, which are enhanced by lack of Tgif in the embryo. The maternal effect of loss of Tgif could be due to defects in decidualization or other uterine functions. Our attempts so far to identify defects in the decidua in the absence of Tgif have not been successful. Thus at least at a gross level, the predominant defect is in formation of the embryonic regions of the placenta. However, we are unable to rule out more subtle changes in the decidua in the absence of Tgif, and further work will be required to fully explain the mechanism of the maternal effect.
Many mutations in mice cause what has been termed a small labyrinth phenotype, with reduced vascularization (Watson and Cross, 2005). In addition, a number of mutations in specific placental regulators result in an alteration of the balance between giant cells and spongiotrophoblasts, with trophoblast cells preferentially differentiating to one cell type versus another. Our analysis suggests that either trophoblast differentiation is decreased in general, or the development of mature spongiotrophoblasts and giant cells is delayed. There is evidence for involvement of both Nodal and RA signaling in the differentiation of trophoblast stem cells. Loss of Nodal expression in the mouse placenta results in an absence of spongiotrophoblasts, a decrease in the labyrinth, and an expansion of the number of giant cells (Ma et al., 2001). In contrast to the role of Nodal, RA signaling increases giant cell formation at the expense of spongiotrophoblasts (Yan et al., 2001). Our analysis of both gene expression and of specific areas within the placenta suggests that the formation of both spongiotrophoblasts and giant cells is compromised in the mutants, making it less likely that the underlying defect is simply due to deregulation of either the Nodal or RA pathway. Additionally, we have not detected any dramatic deregulation of either pathway in the Tgif null placentas. Interpretation of the phenotype within the context of a single pathway is also complicated by the contributions of both maternal and embryonic Tgif.
In addition to altered trophoblast differentiation, there is a clear defect in the formation of the intricate pattern of small, highly branched fetal vessels in the labyrinth, with only a few larger, less branched vessels extending from the chorion in the severe mutants. Since we did not observe defects in chorio-allantoic fusion, this may point towards a defect in vessel branching. Many signaling pathways are implicated in branching morphogenesis, including Wnt and FGF signaling (Cross et al., 2006). We have no evidence that Tgif regulates these pathways directly, and it is possible that the defects are due to indirect effects via TGFβ/Nodal and/or RA signaling. In addition to the overall decrease in vascularization in Tgif null placentas, we observed a specific and consistent decrease in expression of Cx26, which is known to be required for transplacental glucose uptake (Gabriel et al., 1998). Since expression of Cx26 decreased in the absence of Tgif, it is unlikely that Tgif itself directly regulates Cx26 gene expression, as Tgif is an obligate repressor. Indeed, since there is a delay in trophoblast development in the absence of Tgif, it may be that defects in the Cx26 gap junctions are secondary to defective or delayed formation of the normal trilaminar trophoblast layer which separates maternal and fetal blood (Watson and Cross, 2005).
In summary, loss of Tgif causes defects in the embryonic placenta, which appear to be primarily dependent on loss of Tgif function from the mother. That the maternal effect is primary may have implications for recurrent spontaneous abortion and other complications of human pregnancies that cannot be explained by embryonic defects. Our results with Tgif may represent a paradigm for the role of maternal factors in regulating the development of the embryonic placenta.
Supplementary Material
H&E stained sections through a wild type (A) and two Tgif null (B, C) E9.5 placentas are shown. The labyrinth is indicated by a black bar. Arrows in A and B indicate fetal blood vessels. D and E show H&E stained sections through a normal E13.5 heterozygous and defective Tgif null placenta. Black bars indicate the depth of the labyrinth and arrows and arrowheads indicate fetal and maternal blood spaces respectively. The corresponding embryos are shown in F. Images were captured at 10× (A-C, left), 40× (A-C, right), 4× (D, E, left) and 20× (D, E, right). G) Tgif expression in placental tissue was analyzed by RT-PCR from E9.5 placentas from embryos of the indicated Tgif genotypes from a homozygous Tgif null mother. The same RNAs were tested for expression of GFP and Gapdh.
RNA was isolated from whole E9.5 placentas, pooled by litter. Genotypes were either wild type or Tgif null (mother and embryos), as indicated. Expression of the indicated genes, analyzed by qRT-PCR, is plotted in arbitrary units, normalized to cyclophilin (mean + sd of triplicates). The dashed lines indicate the average expression level for each gene among the four wild type samples. The genes analyzed were selected for the known contribution of the encoded protein products to decidual development (reviewed in (Lee et al., 2007a; Wang and Dey, 2006)). Mutation of Il11ra1 results in decidual failure and associated degeneration of the embryonic placenta (Bilinski et al., 1998). Deletion of the transcription factor, CoupTFII, from the uterus results in defects in decidualization and formation of the embryonic placenta (Petit et al., 2007). Mutations in the genes encoding the transcriptional regulators Pgr (progesterone receptor) and Ncoa2 (nuclear receptor coactivator 2) result in both failure of implantation and decidualization, and loss of the CCAAT/enhancer binding protein beta (C/EBPβ) causes defects in ovulation and decidualization. Similarly, loss of Ptgs2 (prostaglandin-endoperoxidase synthase 2), which is required for prostaglandin synthesis, causes defects in ovulation, implantation and decidualization (Lim et al., 1997). Interestingly, like our results with Tgif, these effects are to some degree dependent of the strain background of the mice (Wang et al., 2004). No significant decrease in expression of any of the genes tested in this panel was observed. Indeed, the only change was a small increase in BMP2 expression. However, it is loss of BMP2 from the uterus, which results in defects in decidualization (Lee et al., 2007b).
Adjacent E10.5 sections were stained for either laminin or Cx26 expression and images were captured at 20×. The genotypes of both mother (M) and embryo (E) are indicated. Note that as the amount of fetal vessels (outlined by laminin immunorecativity) decreases, so does the amount of Cx26 expression, even in cases where relatively large maternal blood spaces are still present.
Expression of Cx43 was examined by immunohistochemistry, in sections of E10.5 placentas from the indicated maternal (M) and embryonic (E) genotypes. Images were captured at 4× (A-D) and 20× (E, F). Note that Cx43 expression is exclusive to the decidua at this stage, and no aberrant expression is seen in the embryonic regions, even in the placenta from a Tgif null embryo in a Tgif null mother. In contrast, Cx26 expression is found exclusively in the labyrinth at E10.5. D, decidua; L, labyrinth; GC, giant cells; mv, maternal blood vessel.
Acknowledgments
We thank L.F. Pemberton for critical reading of the manuscript, and the UVA Research Histology Core and Gene Targeting and Transgenic Facility for invaluable assistance. This work was supported in part by research Grant 6-FY04-77 from the March of Dimes (D.W.), and by NIH grants to D.W. (HD52707) and to A.E.S. (HD034807).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anson-Cartwright L, Dawson K, Holmyard D, Fisher SJ, Lazzarini RA, Cross JC. The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat Genet. 2000;25:311–314. doi: 10.1038/77076. [DOI] [PubMed] [Google Scholar]
- Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M, Wotton D. TGIF inhibits retinoid signaling. Mol Cell Biol. 2006;26:990–1001. doi: 10.1128/MCB.26.3.990-1001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basyuk E, Cross JC, Corbin J, Nakayama H, Hunter P, Nait-Oumesmar B, Lazzarini RA. Murine Gcm1 gene is expressed in a subset of placental trophoblast cells. Dev Dyn. 1999;214:303–11. doi: 10.1002/(SICI)1097-0177(199904)214:4<303::AID-AJA3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Bertolino E, Reimund B, Wildt-Perinic D, Clerc R. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- Bilinski P, Roopenian D, Gossler A. Maternal IL-11Ralpha function is required for normal decidua and fetoplacental development in mice. Genes Dev. 1998;12:2234–43. doi: 10.1101/gad.12.14.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross JC, Baczyk D, Dobric N, Hemberger M, Hughes M, Simmons DG, Yamamoto H, Kingdom JC. Genes, development and evolution of the placenta. Placenta. 2003;24:123–30. doi: 10.1053/plac.2002.0887. [DOI] [PubMed] [Google Scholar]
- Cross JC, Nakano H, Natale DRC, Simmons DG, Watson ED. Branching morphogenesis during development of placental villi. Differentiation. 2006;74:393–401. doi: 10.1111/j.1432-0436.2006.00103.x. [DOI] [PubMed] [Google Scholar]
- Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–18. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- Gabriel HD, Jung D, Butzler C, Temme A, Traub O, Winterhager E, Willecke K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J Cell Biol. 1998;140:1453–61. doi: 10.1083/jcb.140.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner AL. Essential role of Mash-2 in extraembryonic development. Nature. 1994;371:333–6. doi: 10.1038/371333a0. [DOI] [PubMed] [Google Scholar]
- Hayhurst M, McConnell SK. Mouse models of holoprosencephaly. Curr Opin Neurol. 2003;16:135–41. doi: 10.1097/01.wco.0000063761.15877.40. [DOI] [PubMed] [Google Scholar]
- Imoto I, Pimkhaokham A, Watanabe T, Saito-Ohara F, Soeda E, Inazawa J. Amplification and overexpression of TGIF2, a novel homeobox gene of the TALE superclass, in ovarian cancer cell lines. Biochem Biophys Res Commun. 2000;276:264–70. doi: 10.1006/bbrc.2000.3449. [DOI] [PubMed] [Google Scholar]
- Jin JZ, Gu S, McKinney P, Ding J. Expression and functional analysis of Tgif during mouse midline development. Dev Dyn. 2006;235:547–53. doi: 10.1002/dvdy.20642. [DOI] [PubMed] [Google Scholar]
- Jin L, Zhou Y, Kuang C, Lin L, Chen Y. Expression pattern of TG-interacting factor 2 during mouse development. Gene Expr Patterns. 2005;5:457–62. doi: 10.1016/j.modgep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Kingdom J, Huppertz B, Seaward G, Kaufman P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol. 2000;92:35–43. doi: 10.1016/s0301-2115(00)00423-1. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Dev Biol. 1997;191:297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- Lee KY, Jeong JW, Tsai SY, Lydon JP, DeMayo FJ. Mouse models of implantation. Trends Endocrinol Metab. 2007;18:234–9. doi: 10.1016/j.tem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Ma GT, Soloveva V, Tzeng SJ, Lowe LA, Pfendler KC, Iannaccone PM, Kuehn MR, Linzer DI. Nodal regulates trophoblast differentiation and placental development. Dev Biol. 2001;236:124–35. doi: 10.1006/dbio.2001.0334. [DOI] [PubMed] [Google Scholar]
- Malassine A, Cronier L. Involvement of gap junctions in placental functions and development. Biochim Biophys Acta. 2005;1719:117–24. doi: 10.1016/j.bbamem.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Mar L, Hoodless PA. Embryonic fibroblasts from mice lacking Tgif were defective in cell cycling. Mol Cell Biol. 2006;26:4302–10. doi: 10.1128/MCB.02156-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Melhuish TA, Gallo CM, Wotton D. TGIF2 interacts with histone deacetylase 1 and represses transcription. J Biol Chem. 2001;26:26. doi: 10.1074/jbc.M103377200. [DOI] [PubMed] [Google Scholar]
- Melhuish TA, Wotton D. The interaction of C-terminal binding protein with the Smad corepressor TG-interacting factor is disrupted by a holoprosencephaly mutation in TGIF. J Biol Chem. 2000 doi: 10.1074/jbc.C000416200. [DOI] [PubMed] [Google Scholar]
- Melhuish TA, Wotton D. The Tgif2 gene contains a retained intron within the coding sequence. BMC Mol Biol. 2006;7:2. doi: 10.1186/1471-2199-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 2000;10:262–9. doi: 10.1016/s0959-437x(00)00084-8. [DOI] [PubMed] [Google Scholar]
- Natale DR, Starovic M, Cross JC. Phenotypic analysis of the mouse placenta. Methods Mol Med. 2006;121:275–93. doi: 10.1385/1-59259-983-4:273. [DOI] [PubMed] [Google Scholar]
- Petit FG, Jamin SP, Kurihara I, Behringer RR, DeMayo FJ, Tsai MJ, Tsai SY. Deletion of the orphan nuclear receptor COUP-TFII in uterus leads to placental deficiency. Proc Natl Acad Sci U S A. 2007;104:6293–8. doi: 10.1073/pnas.0702039104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum A, Winterhager E, Pesch J, Lautermann J, Hallas G, Rosentreter B, Traub O, Herberhold C, Willecke K. Connexin31-deficiency in mice causes transient placental dysmorphogenesis but does not impair hearing and skin differentiation. Dev Biol. 2001;231:334–47. doi: 10.1006/dbio.2000.0148. [DOI] [PubMed] [Google Scholar]
- Sharma M, Sun Z. 5′TG3′ interacting factor interacts with Sin3A and represses AR- mediated transcription. Mol Endocrinol. 2001;15:1918–28. doi: 10.1210/mend.15.11.0732. [DOI] [PubMed] [Google Scholar]
- Shen J, Walsh CA. Targeted disruption of Tgif, the mouse ortholog of a human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol Cell Biol. 2005;25:3639–47. doi: 10.1128/MCB.25.9.3639-3647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MJ, Konno T, Alam SM. The prolactin family: effectors of pregnancy-dependent adaptations. Trends Endocrinol Metab. 2007;18:114–21. doi: 10.1016/j.tem.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Carpino N, Cross JC, Torres M, Parganas E, Ihle JN. SOCS3: an essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. Embo J. 2003;22:372–84. doi: 10.1093/emboj/cdg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Gertsenstein M, Rossant J, Nagy A. Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev Biol. 1997;190:55–65. doi: 10.1006/dbio.1997.8685. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282:2072–5. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:52–54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Varmuza S, Prideaux V, Kothary R, Rossant J. Polytene chromosomes in mouse trophoblast giant cells. Development. 1988;102:127–34. doi: 10.1242/dev.102.1.127. [DOI] [PubMed] [Google Scholar]
- Wang H, Dey SK. Roadmap to embryo implantation: clues from mouse models. Nat Rev Genet. 2006;7:185–99. doi: 10.1038/nrg1808. [DOI] [PubMed] [Google Scholar]
- Watson ED, Cross JC. Development of structures and transport functions in the mouse placenta. Physiology. 2005;20:180–193. doi: 10.1152/physiol.00001.2005. [DOI] [PubMed] [Google Scholar]
- Wotton D, Knoepfler PS, Laherty CD, Eisenman RN, Massague J. The Smad Transcriptional Corepressor TGIF Recruits mSin3. Cell Growth Differ. 2001;12:457–63. [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massague J. A Smad transcriptional corepressor. Cell. 1999a;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Swaby LA, Massague J. Multiple modes of repression by the smad transcriptional corepressor TGIF. J Biol Chem. 1999b;274:37105–10. doi: 10.1074/jbc.274.52.37105. [DOI] [PubMed] [Google Scholar]
- Yan J, Tanaka S, Oda M, Makino T, Ohgane J, Shiota K. Retinoic acid promotes differentiation of trophoblast stem cells to a giant cell fate. Dev Biol. 2001;235:422–32. doi: 10.1006/dbio.2001.0300. [DOI] [PubMed] [Google Scholar]
- Zybina EV, Zybina TG. Polytene chromosomes in mammalian cells. Int Rev Cytol. 1996;165:53–119. doi: 10.1016/s0074-7696(08)62220-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
H&E stained sections through a wild type (A) and two Tgif null (B, C) E9.5 placentas are shown. The labyrinth is indicated by a black bar. Arrows in A and B indicate fetal blood vessels. D and E show H&E stained sections through a normal E13.5 heterozygous and defective Tgif null placenta. Black bars indicate the depth of the labyrinth and arrows and arrowheads indicate fetal and maternal blood spaces respectively. The corresponding embryos are shown in F. Images were captured at 10× (A-C, left), 40× (A-C, right), 4× (D, E, left) and 20× (D, E, right). G) Tgif expression in placental tissue was analyzed by RT-PCR from E9.5 placentas from embryos of the indicated Tgif genotypes from a homozygous Tgif null mother. The same RNAs were tested for expression of GFP and Gapdh.
RNA was isolated from whole E9.5 placentas, pooled by litter. Genotypes were either wild type or Tgif null (mother and embryos), as indicated. Expression of the indicated genes, analyzed by qRT-PCR, is plotted in arbitrary units, normalized to cyclophilin (mean + sd of triplicates). The dashed lines indicate the average expression level for each gene among the four wild type samples. The genes analyzed were selected for the known contribution of the encoded protein products to decidual development (reviewed in (Lee et al., 2007a; Wang and Dey, 2006)). Mutation of Il11ra1 results in decidual failure and associated degeneration of the embryonic placenta (Bilinski et al., 1998). Deletion of the transcription factor, CoupTFII, from the uterus results in defects in decidualization and formation of the embryonic placenta (Petit et al., 2007). Mutations in the genes encoding the transcriptional regulators Pgr (progesterone receptor) and Ncoa2 (nuclear receptor coactivator 2) result in both failure of implantation and decidualization, and loss of the CCAAT/enhancer binding protein beta (C/EBPβ) causes defects in ovulation and decidualization. Similarly, loss of Ptgs2 (prostaglandin-endoperoxidase synthase 2), which is required for prostaglandin synthesis, causes defects in ovulation, implantation and decidualization (Lim et al., 1997). Interestingly, like our results with Tgif, these effects are to some degree dependent of the strain background of the mice (Wang et al., 2004). No significant decrease in expression of any of the genes tested in this panel was observed. Indeed, the only change was a small increase in BMP2 expression. However, it is loss of BMP2 from the uterus, which results in defects in decidualization (Lee et al., 2007b).
Adjacent E10.5 sections were stained for either laminin or Cx26 expression and images were captured at 20×. The genotypes of both mother (M) and embryo (E) are indicated. Note that as the amount of fetal vessels (outlined by laminin immunorecativity) decreases, so does the amount of Cx26 expression, even in cases where relatively large maternal blood spaces are still present.
Expression of Cx43 was examined by immunohistochemistry, in sections of E10.5 placentas from the indicated maternal (M) and embryonic (E) genotypes. Images were captured at 4× (A-D) and 20× (E, F). Note that Cx43 expression is exclusive to the decidua at this stage, and no aberrant expression is seen in the embryonic regions, even in the placenta from a Tgif null embryo in a Tgif null mother. In contrast, Cx26 expression is found exclusively in the labyrinth at E10.5. D, decidua; L, labyrinth; GC, giant cells; mv, maternal blood vessel.