

Abstract
While there is a significant amount of information available on the clinical presentation(s) and pathological changes associated with tendinopathy, the precise aetiopathogenesis of this condition remains a topic of debate. Classically, the aetiology of tendinopathy has been linked to the performance of repetitive activities (so-called overuse injuries). This has led many investigators to suggest that it is the mechanobiologic over-stimulation of tendon cells that is the initial stimulus for the degradative processes which have been shown to accompany tendinopathy. Although several studies have been able to demonstrate that the in vitro over-stimulation of tendon cells in monolayer can result in a pattern(s) of gene expression seen in clinical cases of tendinopathy, the strain magnitudes and durations used in these in vitro studies, as well as the model systems, may not be clinically relevant. Using a rat tail tendon model, we have studied the in vitro mechanobiologic response of tendon cells in situ to various tensile loading regimes. These studies have led to the hypothesis that the aetiopathogenic stimulus for the degenerative cascade which precedes the overt pathologic development of tendinopathy is the catabolic response of tendon cells to mechanobiologic under-stimulation as a result of microscopic damage to the collagen fibres of the tendon. In this review, we examine the rationale for this hypothesis and provide evidence in support of this theory.
Keywords: apoptosis, gene expression, matrix metalloproteinases, mechanobiology, tendinopathy, tendon cell
Introduction
Mechanoreponsiveness is a fundamental feature of all living tissues and tendons are no exception (Banes et al. 1995a; Ingber 1997; Lavagnino & Arnoczky 2005; Wang 2006). The ability of tendon cells to sense and respond to load is central to the concept of mechanotransduction and the subsequent maintenance of tissue homeostasis (Wang & Ingber 1994; Banes et al. 1995a; Ingber 1997; Ruoslahti 1997). Tendon cells sense load through a mechano-electrochemical sensory system(s) which detects mechanical load signals through the deformation of the cellular membrane and/or the cytoskeleton (Ben-Ze’ev 1991; Watson 1991; Adams 1992; Wang et al. 1993; Wang & Ingber 1994; Banes et al. 1995a; Ingber 1997; Brown et al. 1998; Wang 2006). The cellular deformation which occurs with extracellular matrix strain produces tension in the cytoskeleton which can be sensed by the cell nucleus through a mechano-sensory tensegrity system to elicit a metabolic response (Ben-Ze’ev 1991; Watson 1991; Adams 1992; Wang et al. 1993; Wang & Ingber 1994; Banes et al. 1995a; Ingber 1997; Arnoczky et al. 2002; Wang 2006). Cellular deformation in response to tissue strain is thought to occur through the binding of the cell to extracellular matrix proteins such as collagen and fibronectin (Banes et al. 1995a; Rosales et al. 1995; Sung et al. 1996). These connections are mediated by the integrin family of cell surface receptors which link the extracellular matrix to the interior of the cell through the cytoskeleton (Ingber 1991; Wang et al. 1993; Banes et al. 1995; Janmey 1998). While the precise level (magnitude, frequency and duration) of mechanobiological stimulation required to maintain normal tendon homeostasis is not currently known, it is very likely that an abnormal level(s) of stimulation may play a role in the aetiopathogenesis of tendinopathy (Józsa & Kannus 1997; Arnoczky et al. 2002, 2007).
A proposed algorithm for the onset of overuse tendinopathy involves altered cell-matrix interactions in response to repetitive loading (Figure 1) (Archambault et al. 1995). In this scenario, repeated strains below the injury threshold of the tendon induce degenerative changes in the tendon-matrix composition and organization (Järvinen et al. 1997; Józsa & Kannus 1997; Jones et al. 2006). The degeneration of the extracellular matrix leads to a transient weakness of the tissue making it more susceptible to damage from continued loading. This damage then accumulates until the overt pathology of tendinopathy develops (Archambault et al. 1995). While this is a feasible algorithm for the development of overuse tendinopathy, the precise mechanism(s) which lead to altered cell-matrix interactions have not been described.
Figure 1.

Proposed algorithm of the aetiopathogenesis of tendinopathy. Modified from Archambault et al. 1995; Arnoczky et al. 2007.
Several investigators have suggested that it is the tendon cells’ mechanobiologic response to over-stimulation secondary to repetitive loading that initiates the degenerative cascade that leads to tendinopathy (Archambault et al. 2001, 2002; Skutek et al. 2001; Tsuzaki et al. 2003; Wang et al. 2003). Over-stimulation of tendon cells in vitro has been shown to induce increases in inflammatory cytokines and degenerative enzymes (Almekinders et al. 1993; Banes et al. 1995a, 1999; Archambault et al. 2002; Tsuzaki et al. 2003; Wang et al. 2003). However, many of these investigations have utilized non-physiologic strain patterns (Almekinders et al. 1993; Wang et al. 2003) (high strain amplitudes and frequencies, as well as long durations) or the addition of external factors (Archambault et al. 2002; Tsuzaki et al. 2003) to elicit these cellular responses. Thus the clinical relevance of these studies must be called into question (Arnoczky et al. 2007).
Experimental studies from our lab have shown that mechanobiological under-stimulation of tendon cells can also produce a pattern of catabolic gene expression that results in extracellular matrix degradation and subsequent loss of tendon material properties (Lavagnino et al. 2003, 2005a, 2006, 2006a; Arnoczky et al. 2004; Lavagnino & Arnoczky 2005; Egerbacher et al. 2006). We have also shown that at the extremes of physiologic loading, isolated fibril damage can occur in tendons which alters normal cell-matrix interactions in this damaged area (Lavagnino et al. 2006a). The inability of the damaged fibrils to transmit extracellular matrix loads to the tendon cells results in an under-stimulation of these cells which, in turn, initiates a catabolic response that can weaken the tendon making it more susceptible to damage from subsequent loading (Lavagnino et al. 2006a).
In this study, we will forward the hypothesis that it is a mechanobiological under-stimulation resulting from altered cell-matrix interaction and not a repetitive over-stimulation of tendon cells that is the aetiopathogenic stimulus for the degenerative cascade which may eventually lead to tendinopathy.
Mechanobiology and the aetiopathogenesis of tendinopathy
An increasing body of clinical material has suggested that tendons from tendinopathy patients exhibit an increase in degradative enzymes (matrix metalloproteinases) as well as an induction of apoptosis (Ireland et al. 2001; Fu et al. 2002; Riley et al. 2002; Yuan et al. 2002, 2003; Alfredson et al. 2003; Lo et al. 2004; Riley 2004; Hosaka et al. 2005; Magra & Maffulli 2005; Sharma & Maffulli 2005; Tuoheti et al. 2005; Jones et al. 2006). Therefore, many investigators have focused on inducing expression of these molecular ‘markers’ of tendinopathy by exposing tendon cells to various loading regimes (Almekinders et al. 1993; Banes et al. 1995, 1999; Skutek et al. 2001; Archambault et al. 2002; Lavagnino et al. 2003, 2006a; Tsuzaki et al. 2003; Wang et al. 2003; Arnoczky et al. 2004; Lavagnino & Arnoczky 2005).
Numerous studies have suggested that over-stimulation of tendon cells, secondary to repetitive loading, results in a pattern of gene expression that can lead to tendinopathy (Almekinders et al. 1993; Banes et al. 1995, 1999; Skutek et al. 2001; Archambault et al. 2002; Tsuzaki et al. 2003; Wang et al. 2003). The majority of these in vitro studies have been based on the response of tendon cells cultured on artificial substrates to various regimes of mechanical loading (Almekinders et al. 1993; Banes et al. 1995, 1999; Archambault et al. 2002; Arnoczky et al. 2002a; Tsuzaki et al. 2003; Wang et al. 2003). In these culture systems, large numbers of cells are subjected to a uniform loading regime. While this permits analysis of large amounts of cellular material and cellular products, it may not replicate the normal in situ environmental conditions of tendon cells within a three-dimensional collagenous matrix (Figure 2). As mechanotransduction signals are known to be mediated through the pericellular matrix to the nucleus via integrin-based cell-matrix connections (Sachs 1988; Ingber 1991; Watson 1991; Wang et al. 1993; Banes et al. 1995a; Ritty et al. 2003) it is not clear how, or even if, the complex cell-matrix interactions which occur in situ could be maintained or recreated in monolayer cell cultures.
Figure 2.

(a) Phase contrast, microscopic image of rat tail tendon cells in monolayer. The random orientation, density, and number of the cells does not replicate the normal in situ cellular environment. (b) Confocal laser microscopic image of a rat tail tendon fascicle stained with acridine orange. Using this model system, the natural distribution, number, and orientation of the tendon cells within their normal extracellular matrix is maintained.
In addition, the strain magnitudes and durations required to achieve an up-regulation in the expression of these inflammatory and catabolic genes may not be clinically relevant. Some of these studies have used a sustained (>20 h) application of cyclic strains in excess of 8% to elicit catabolic and inflammatory gene expression in tendon and ligament cells cultured on artificial substrates (Almekinders et al. 1993; Wang et al. 2003; Bhargava et al. 2004). Because tendon cell strain in situ has been shown to be appreciably less than whole tendon strain (Arnoczky et al. 2002), it is unlikely that such high levels of repetitive tendon cell strain could be reached and maintained in vivo without significant damage occurring within the extracellular matrix of the tendon (Woo et al. 1982). Also, as tendons are known to exhibit non-homogeneous strain patterns in response to tensile load (Kastelic et al. 1978), it would seem impossible to precisely recreate the complex and varied patterns of strain amplitudes experienced by a population of tendon cells in situ by uniformly straining a large population of isolated tenocytes in monolayer.
Finally, the large number of confluent, or near confluent, cells studied in these monolayer systems (Almekinders et al. 1993; Banes et al. 1995, 1999; Archambault et al. 2002; Arnoczky et al. 2002a; Tsuzaki et al. 2003; Wang et al. 2003) could produce a marked paracrine cellular stimulus that may not occur in the more limited cell populations seen in normal tendons. This could, in turn, provide an artificially enhanced cellular response to the repetitive loading stimulus. Thus, the in vitro application of high magnitudes of cyclic cellular strain for excessively long durations to tendon cells in monolayer may have little bearing on what is actually occurring to tendon cells in situ.
Therefore, to better understand how the mechanotransduction response of tendon cells under tensile load may contribute to the aetiopathogenesis of tendinopathy, our lab has utilized an in situ rat tail tendon model in an effort to maintain the tendon cells’ natural cell-matrix interactions as well as the naturally occurring strain fields that are developed in response to tensile loading (Lavagnino et al. 2003, 2006, 2006a; Arnoczky et al. 2004, 2007).
Previous studies from our lab have demonstrated that mechanobiologic under-stimulation of tendon cells in situ through stress-deprivation results in an immediate and significant up-regulation of interstitial collagenase mRNA expression and protein synthesis in our rat tail tendon model (Lavagnino et al. 2003, 2005a, 2006a; Arnoczky et al. 2004). This has led to the theory that the destructive mechanism(s) that precedes overt pathological development of tendinopathy may, in fact, be a catabolic response of the tendon cells to the local loss of homeostatic strain as a result of isolated, microscopic, collagen fibre damage (Jones et al. 2006; Arnoczky et al. 2007).
Previous biomechanical studies have suggested that isolated collagen fibril damage occurs near the end of the linear portion of the load deformation curves of ligaments and tendons (Figure 3) (Viidik & Ekholm 1968; Viidik 1972, 1980; Woo et al. 1982; Józsa & Kannus 1997). The ability to produce isolated fibril failure within an otherwise intact tendon is likely attributable to the multicomposite structure of the tissue (Kastelic et al. 1978, 1980; Viidik 1980). The sequential straightening and loading of crimped collagen fibrils, as well as interfibrillar sliding and shear between fibres and/or fibrils, produce a non-linear, load-deformation behaviour of tendons that may put certain fibrils ‘at risk’ for damage before others (Viidik 1980). Using confocal laser microscopy and a previously described in vitro tensile loading apparatus (Arnoczky et al. 2002), our lab has been able to demonstrate that isolated collagen fibril damage does occur in tendons in response to increasing tensile loads (Figure 4). As previously predicted, this damage occurs well in advance of complete tendon rupture (Viidik & Ekholm 1968; Viidik 1972, 1980; Woo et al. 1982; Józsa & Kannus 1997). While such damage may not affect the ultimate tensile strength of the tissues (Panjabi et al. 1996) it could alter the cell-matrix interactions within the damaged portion of the tendon. A previous study has demonstrated that following isolated fibrillar damage in tendons the damaged fibrils relax (Knorzer et al. 1986). This would suggest an inability of these damaged fibrils to transmit load and therefore maintain a homeostatic mechanobiological stimulus to those cells associated with the damaged fibrils.
Figure 3.

Schematic drawing of a load deformation curve illustrating the mechanical response of a tendon to tensile loading. Modified from Józsa & Kannus 1997; Arnoczky et al. 2007.
Figure 4.

Time-lapse confocal images of a rat tail tendon being strained at a rate of 20 μm/s. The cell nuclei have been stained with acridine orange and the pairs of short and long arrows identify cell nuclei used as fiduciary markers to demonstrate fibril sliding. At 7% strain (image 3) the lower (long) pair of arrows can be seen separating indicating fibre slippage. This separation continues to increase with increasing strain (images 4 and 5). After 8% strain (image 4) the upper (short) pair of arrows begin to get closer and pass over one another at 9% strain (image 5). This slippage continues to increase at 10% strain (image 6). In both instances, fibril slippage occurred in advance of complete tendon rupture (Arnoczky et al. 2007).
Based on the above findings, our laboratory has forwarded the hypothesis that an alteration of cell-matrix interaction secondary to isolated fibrillar damage could result in a mechanobiological under-stimulation of tendon cells which has been shown to result in an upregulation of collagenase mRNA expression and protein synthesis (Lavagnino et al. 2003, 2005a; Arnoczky et al. 2004; Lavagnino & Arnoczky 2005). This, in turn, causes an initial degeneration of the pericellular matrix that may further compromise cell-matrix interactions and mechanobiological signalling (Egerbacher et al. 2006). The degenerative process then progresses throughout the extracellular matrix resulting in a decrease in the material properties of the tendon (Figure 5). These changes could then put more of the extracellular matrix at risk for further damage with subsequent loading (Lavagnino & Arnoczky 2005). Then, when a critical level of damage has been reached the clinical and histological signs of tendinopathy may become evident.
Figure 5.

Confocal images of a fresh and 21-day stress deprived rat tail tendon. Parallel registration lines have been photobleached onto the surface of the tendons. When strained to 3% (grip-to-grip strain) the registration lines on the fresh tendon remain parallel. This is in contrast to the 21-day stress deprived tendon that demonstrated an altered strain pattern due to breakdown of the extracellular matrix by collagenase which is upregulated in these tendons following stress-deprivation (Arnoczky et al. 2007).
Recently, our laboratory has demonstrated that creation of isolated tendon fibrillar damage within an otherwise intact tendon fascicle results in an up-regulation of collagenase mRNA expression and protein synthesis by only those tendon cells associated with the damaged fibrils (Figures 6 and 7) (Lavagnino et al. 2006a). This would suggest a loss of load-transmitting function in the damaged fibril(s) and a subsequent altered cell-matrix interaction within the affected area. The presence of increased levels of collagenase protein in these injured tendons is similar to what has been reported in clinical cases of tendinopathy (Fu et al. 2002; Riley et al. 2002; Lo et al. 2004; Magra & Maffulli 2005; Sharma & Maffulli 2005).
Figure 6.

Images of a rat tail tendon fascicle at various points throughout the testing protocol: (a) prior to loading (the crimp pattern is clearly visible), (b) during loading in the linear portion of the stress–strain curve demonstrating the elimination of the crimp pattern, (c) onset of fibrillar damage as manifested by a change in the reflectivity of the damaged fibrils (arrows), and (d) unloading of the tendon to 100 g and the reoccurrence of the crimp pattern within the damaged fibrils (arrows). (bar = 200 μm). (Reprinted from Lavagnino et al. (2006a) Isolated fibrillar damage in tendons stimulates local collagenase mRNA expression and protein synthesis. J. Biomech. 39, 2355–2362, with permission from Elsevier.)
Figure 7.

Representative images of a rat tail tendon fascicle following fibrillar damage. (a) Presence of the crimp pattern on the bottom of the tendon fascicle (arrows) indicates the site of isolated fibrillar damage. (b) In situ hybridization of the tendon fascicle reveals interstitial collagenase mRNA expression in those cells associated with the damaged fibril(s). The borders of the tendon fascicle are delineated by lines. (bar = 100 μm). Reprinted from Lavagnino et al. (2006a). Isolated fibrillar damage in tendons stimulates local collagenase mRNA expression and protein synthesis. J. Biomech. 39, 2355–2362, with permission from Elsevier.)
A clinical study examining matrix metalloproteinase (MMP) activity in ruptured human tendons demonstrated an altered expression and activity of several members of the MMP family (Riley et al. 2002). MMP-1 levels were significantly higher in ruptured tendons compared with normal controls, whereas MMP-2 and 3 levels were reduced, possibly representing a failure in the normal remodeling process (Riley et al. 2002; Riley 2004). This increase in collagenase activity was associated with a deterioration in the quantity of the collagen network. Increased expression of MMP-1 was also found in human patellar tendinosis tissue (Fu et al. 2002).
The result of this MMP-mediated degradation of the extracellular matrix is reflected in the histopathological findings in tendinosis that reveal irregular orientation of collagen, fibre disruption, change in fibre diameter, a decrease in the overall density of collagen and an upregulation of collagen type III production (Józsa et al. 1990; Kannus & Józsa 1991; Järvinen et al. 1997). Another study that examined the histopathology of ruptured and tendinopathic Achilles tendons suggested that while the ruptured tendons were significantly more degenerated than the tendinopathic tendons, the general pattern of degeneration was common to both groups (Tallon et al. 2001).
In addition to the documented increase in collagenase activity seen in clinical cases of tendinopathy, other studies have suggested that apoptosis may play a role in the pathogenesis of tendinopathy (Yuan et al. 2002, 2003; Hosaka et al. 2005). Studies on the pathogenesis of rotator cuff disorders demonstrate a significant increase in the number of apoptotic cells detected in degenerative supraspinatus tendons compared with normal control tendons (Yuan et al. 2002; Tuoheti et al. 2005). It is theorized that the increased number of apoptotic cells seen in the degenerative tissues of tendinopathy patients could adversely affect the rate of collagen synthesis and the potential for repair (Yuan et al. 2003). Apoptosis was also detected in samples of inflamed superficial digital flexor tendon in the horse possibly resulting in tendon weakness and increased risk of tendinopathy (Hosaka et al. 2005). However, at present, it is still unknown whether apoptosis is the result or the cause of tendon degeneration.
Experimental studies from our laboratory have documented an increase in Caspase-3 mRNA expression and protein synthesis as well as an increase in the number of apoptotic cells (demonstrated by detection of single stranded DNA) following 24 h of stress-deprivation in our rat tail tendon model (Egerbacher et al. 2007). While another ex vivo study was able to induce apoptosis in tendon cells following exposure to high strains (20% strain for 6 h at the rate of 1 Hz), it is probable that the high strains utilized in the experiment likely damaged tendon fibres or fibrils (Scott et al. 2005). This could result in the under-stimulation of the tendon cells associated with the damaged fibres or fibrils and the subsequent induction of apoptosis secondary to the release of cellular tension (Grinnell et al. 1999). Thus, these experimental and clinical studies point to a possible effect of mechanical stimulus, or lack thereof, on the induction of apoptosis in tendon cells. However, the precise mechanism(s) that trigger programmed cell death under these conditions must still be defined.
In addition to the increase in MMP expression and apoptosis reported in clinical cases of tendinopathy, classic histological changes have been described. These include collagen disruption and disarray along with an alteration in the crimping pattern (Järvinen et al. 1997). There can also be great variation in the density and appearance of the tendon cells ranging from hypercellularity to hypocellularity (Järvinen et al. 1997). In the latter case, many of the remaining cells may appear necrotic, or as noted above, apoptotic. This loss of metabolically active cells can lead to the classically described changes in the extracellular matrix of the tendons such as mucoid degeneration, hyaline degeneration and fibrocartilagenous metaplasia (Galliani et al. 2002; Józsa & Kannus 1997). An in vitro experimental study from our laboratory has demonstrated that under-stimulation of tendon cells can, in fact, produce a histological picture commensurate with that seen in tendinopathy (Hannafin et al. 1995). In this study, stress-deprived tendons underwent a progressive, cell-mediated degeneration of their extracellular matrix (Hannafin et al. 1995). These changes were manifested by widespread hypocellularity, alterations (rounding up) in the morphology of the tenocytes, and a loss of collagen orientation and packing (Figure 8). The histological changes induced by stress-deprivation provide further support for the theory that mechanobiological under-stimulation of tendons cells is responsible for initiating the degenerative cascade that is associated with tendinopathy.
Figure 8.
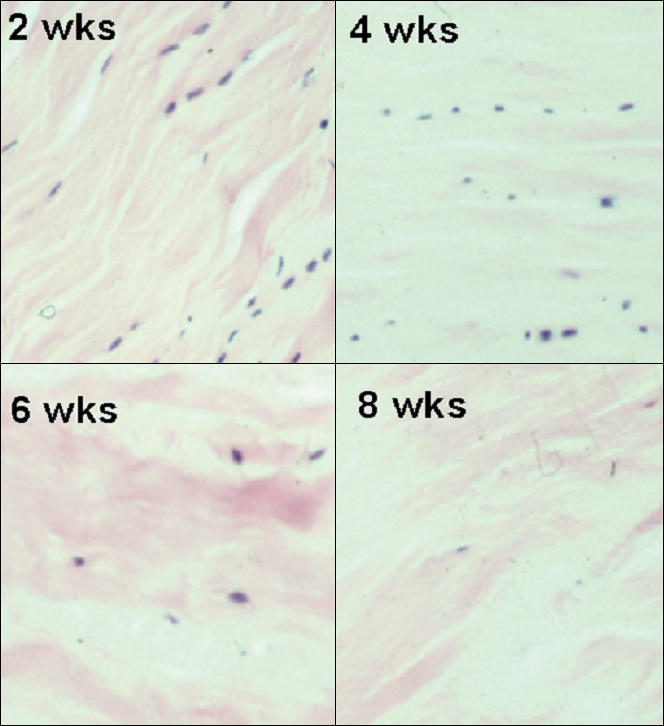
Photomicrographs of the histological changes seen in canine flexor digitorum profundus tendons following stress-deprivation for 2, 4, 6 and 8 weeks. Note how the tendon cells change morphology and ‘round up’ in response to the loss of normal homeostatic tension. Also note the progressive decrease in cell number and the progressive disruption of the collagen architecture with time of stress-deprivation. (H & E ×100)
Clinical relevance and significance
The aetiology of tendinopathy remains unclear, and many causes have been theorized (Józsa & Kannus 1997; Sharma & Maffulli 2005). Central to these theories is the concept that excessive loading of tendons during vigorous physical activity is the main pathological stimulation for degeneration of the extracellular matrix (Selvanetti et al. 1997). Some investigators have suggested that it is the tendon cells’ mechanobiologic response to excessive loading that initiates the degenerative cascade of events that leads to tendinopathy (Skutek et al. 2001; Archambault et al. 2002; Tsuzaki et al. 2003; Wang et al. 2003). The idea being that prolonged mechanical stimuli of tendon cells induce production of degradative cytokines and inflammatory prostaglandins which are thought to be mediators of tendinopathy (Sharma & Maffulli 2005). However, as noted above, the level of stimuli required to elicit these cellular responses are not clinically relevant and have, to date, only been demonstrated in cultured cells on artificial substrates (Almekinders et al. 1993; Wang et al. 2003; Bhargava et al. 2004).
We contend that it is actually an absence of mechanical stimuli, secondary to microtrauma that is the mechanobiological stimulus for the degradative casade that leads to tendinopathy. Numerous investigators have postulated that during excessive, repetitive loading, microtrauma occurs within the tendon matrix (Archambault et al. 1995; Józsa & Kannus 1997; Sharma & Maffulli 2005; Wang et al. 2006). If this microtrauma is not balanced by an active repair response from the tenocytes, it will result in cumulative damage and ultimately, degradation of the tendon (Ker 2002). Our research supports the concept that isolated fibril damage can occur during the extremes of physiologic loading (Lavagnino et al. 2006a). This damage alters cell-matrix interactions (mechanobiologic signalling) in the area causing an upregulation in collagenase and a weakening of the collagen structure (Lavagnino & Arnoczky 2005; Lavagnino et al. 2006a). This could make the tendon more susceptible to damage from additional loading at lower strains. We have also shown that mechanobiological under-stimulation can induce apoptosis in tendon cells (Egerbacher et al. 2007). The loss of cells could further compromise the tendon’s ability to repair itself or even maintain its local extracellular matrix.
While mechanobiological under-stimulation of tendon cells is a feasible explanation for the increase in apoptosis and collagenase expression reported in clinical cases of tendinopathy (Ireland et al. 2001; Fu et al. 2002; Riley et al. 2002; Yuan et al. 2002, 2003; Alfredson et al. 2003; Lo et al. 2004; Riley 2004; Hosaka et al. 2005; Magra & Maffulli 2005; Sharma & Maffulli 2005; Tuoheti et al. 2005), the question remains as to the role of repetitive strain in the aetiopathogenesis of tendinopathy. As we have demonstrated in our model system, a single high load event was able to cause sufficient fibril damage to initiate a cell-mediated response as a result of mechanobiological under-stimulation (Lavagnino et al. 2006a). Tendon microtrauma can also result from a non-uniform stress occurring within a tendon producing abnormal loading concentrations and localized fibre damage (Ker 2002). Therefore, it is possible that during a series of repetitive loading cycles a single abnormal loading cycle could produce strains sufficient enough to induce isolated fibril damage but not cause clinical injury. This abnormal loading cycle could be a result of muscle fatigue and/or altered kinematics that can occur with the performance of repetitive activities (Józsa & Kannus 1997). It has long been suggested that mental fatigue (and altered neuromuscular responses) may also play a role in tendinopathy (Darling 1899a, 1899b).
Thus, while repetitive loading, per se, may not be responsible for initiating the cascade of events that lead to tendinopathy, it is likely that continued loading of the compromised tissue plays a significant role in the progression of the pathological process. Additional research is needed to determine the magnitude of tendon forces experienced in activities that are often associated with the development of tendinopathy (jumping, running and throwing). In addition, the effect of muscle fatigue and/or altered kinematics on these tendon forces must be determined to gain insight into the mechanobiological mechanism(s) which may play a role in the aetiopathogenesis of tendinopathy.
The response of tendon cells to changing loading conditions has significant implications in unraveling the aetiopathogenesis of tendinopathy. While the knowledge base regarding the potential role(s) of tendon cell mechanobiology in tendon health, injury, and repair is continuing to expand (Wang 2006), additional research is required to determine how changes (mechanical, chemical and structural) in the in situ extracellular environment affect the mechanotransduction response(s) of tendon cells. In addition, we must determine how (or if) tendon cells can adapt to these changing loading conditions and/or changes in extracellular matrix composition.
Finally, we must assure that these in vitro investigations into tendon cell mechanobiology are clinically relevant so that the basic science data gleaned from these studies can be appropriately translated into the clinical situation. To do this, we must have a comprehensive understanding of what is happening to tendons (on both a structural and material level) during actual in vivo activities.
Summary
The role of the mechanobiological response of tendon cells in the aetiopathogenesis of tendinopathy remains a point of controversy and debate. In this study, we have presented an argument for mechanobiological under-stimulation of tendon cells, secondary to microtrauma and isolated collagen fibril damage, as a predisposing factor for the pathological changes (collagen disruption, increased MMP levels and apoptosis) reported in clinical cases of tendinopathy. While our basic science data appears to support this hypothesis, additional translational research is needed to determine how, or even if, these proposed mechanobiological mechanisms are involved in the aetiopathogenesis of clinical tendinopathy.
References
- Adams DS. Mechanisms of cell shape change: the cytomechanics of cellular response to chemical environment and mechanical loading. J. Cell Biol. 1992;117:83–93. doi: 10.1083/jcb.117.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfredson H, Lorentzon M, Bäckman S, Bäckman A, Lerner UH. cDNA arrays and real time qualitative PCR techniques in the investigation of chronic Achilles tendinosis. J. Orthop. Res. 2003;21:970–975. doi: 10.1016/S0736-0266(03)00107-4. [DOI] [PubMed] [Google Scholar]
- Almekinders LC, Banes AJ, Ballenger CA. Effects of repetitive motion on human fibroblasts. Med. Sci. Sports Exerc. 1993;25:603–607. [PubMed] [Google Scholar]
- Archambault JM, Wiley JP, Bray RC. Exercise loading of tendons and the development of overuse injuries. Sports Med. 1995;20:77–89. doi: 10.2165/00007256-199520020-00003. [DOI] [PubMed] [Google Scholar]
- Archambault JM, Hart DA, Herzog W. Response of rabbit Achilles tendon to chronic repetitive loading. Connect. Tissue Res. 2001;42:13–23. doi: 10.3109/03008200109014245. [DOI] [PubMed] [Google Scholar]
- Archambault J, Tsuzaki M, Herzog W, Banes AJ. Stretch and interleukin-1beta induce matrix metalloproteinases in rabbit tendon cells in vitro. J. Orthop. Res. 2002;20:36–39. doi: 10.1016/S0736-0266(01)00075-4. [DOI] [PubMed] [Google Scholar]
- Arnoczky SP, Lavagnino M, Whallon JH, Hoonjan A. In situ cell nucleus deformation under tensile load: a morphologic analysis using confocal laser microscopy. J. Orthop. Res. 2002a;20:29–35. doi: 10.1016/S0736-0266(01)00080-8. [DOI] [PubMed] [Google Scholar]
- Arnoczky SP, Tian T, Lavagnino M, Gardner K, Schuler P, Morse P. Activation of stress-activated protein kinases (SAPK) in tendon cells following cyclic strain: the effects of strain frequency, strain magnitude, and cytosolic calcium. J. Orthop. Res. 2002b;20:947–952. doi: 10.1016/S0736-0266(02)00038-4. [DOI] [PubMed] [Google Scholar]
- Arnoczky SP, Tian T, Lavagnino M, Gardner K. Ex vivo static tensile loading inhibits MMP-1 expression in rat-tail tendon cells through a cytoskeletally based mechanotransduction mechanism. J. Orthop. Res. 2004;22:328–333. doi: 10.1016/S0736-0266(03)00185-2. [DOI] [PubMed] [Google Scholar]
- Arnoczky SP, Lavagnino M, Egerbacher M. The response of tendon cells to changing loads: Implications in the etiopathogenesis of tendinopathy. In: Woo SLY, Renstrom P, Arnoczky SP, editors. Tendinopathy in athletes, Encyclopedia of Sports Medicine. Oxford, UK: Blackwell Publishing; 2007. pp. 46–59. [Google Scholar]
- Banes AJ, Tsuzaki M, Hu P, et al. Cyclic mechanical load and growth factors stimulate DNA synthesis in avian tendon cells. J. Biomech. 1995a;28:1505–1513. doi: 10.1016/0021-9290(95)00098-4. [DOI] [PubMed] [Google Scholar]
- Banes AJ, Tsuzaki M, Yamamoto J, et al. Mechanoreception at the cellular level: the detection, interpretation, and diversity of responses to mechanical signals. Biochem. Cell Biol. 1995b;73:349–365. doi: 10.1139/o95-043. [DOI] [PubMed] [Google Scholar]
- Banes AJ, Horesovsky G, Larson C, et al. Mechanical load stimulates expression of novel genes in vivo and in vitro in avian flexor tendon cells. Osteoarthr. Cartil. 1999;7:141–153. doi: 10.1053/joca.1998.0169. [DOI] [PubMed] [Google Scholar]
- Ben-Ze’ev A. Animal cell shape changes and gene expression. Bioessays. 1991;13:207–212. doi: 10.1002/bies.950130502. [DOI] [PubMed] [Google Scholar]
- Bhargava M, Attia E, Hannafin JA. The effect of cyclic tensile strain on MMPs, collagen, and casein degrading activities of fibroblasts isolated from anterior cruciate and medial collateral ligaments. Trans. Orthop. Res. Soc. 2004;29:110. [Google Scholar]
- Brown RA, Prajapati R, McGrouther DA, Yannus IV, Eastwood M. Tensional homeostasis in dermal fibroblasts: mechanical responses to mechanical loading in three dimensional substrates. J. Cell. Physiol. 1998;175:323–332. doi: 10.1002/(SICI)1097-4652(199806)175:3<323::AID-JCP10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Darling EA. The effects of training. A study of the Harvard crews. Boston Medical and Surgical Journal 141. 1899a;9:205–209. [Google Scholar]
- Darling EA. The effects of training. A study of the Harvard crews. Boston Medical and Surgical Journal 141. 1899b;10:229–233. [Google Scholar]
- Egerbacher M, Arnoczky SP, Gardner K, Caballero O, Gartner J. Stress-deprivation of tendons results in alterations in the integrin profile and peri-cellular matrix of tendon cells. Trans. Orthop. Res. Soc. 2006;31:1100. [Google Scholar]
- Egerbacher M, Arnoczky SP, Caballero O, Gardner KL. Stress-deprivation induces apoptosis in tendon cells: a mechanistic explanation for the histologic changes seen in tendinopathy. Trans. Orthop. Res. Soc. 2007;32:862. [Google Scholar]
- Fu SC, Chan BP, Wang W, Pau HM, Chan KM, Rolf CG. Increased expression of matrix metalloproteinase 1 (MMP-1) in 11 patients with patellar tendinosis. Acta Orthop. Scand. 2002;73:658–662. doi: 10.1080/000164702321039624. [DOI] [PubMed] [Google Scholar]
- Galliani I, Burattini S, Mariani AR, Riccio M, Cassiani G, Falcieri E. Morphofunctional changes in human tendon tissue. Eur. J. Histochem. 2002;46:3–12. doi: 10.4081/1649. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Zhu M, Carlson M, Abrams J. Release of mechanical tension triggers apoptosis of human fibroblasts in a model of regressing granulation tissue. Exp. Cell Res. 1999;248:608–619. doi: 10.1006/excr.1999.4440. [DOI] [PubMed] [Google Scholar]
- Hannafin JA, Arnoczky SP, Hoonjan A, Torzilli PA. Effect of stress deprivation and cyclic tensile loading on the material and morphologic properties of canine flexor digitorum profundus tendon: an in vitro study. J. Ortho. Res. 1995;13:907–914. doi: 10.1002/jor.1100130615. [DOI] [PubMed] [Google Scholar]
- Hosaka Y, Teraoka H, Yamamoto E, Ueda H, Takehana K. Mechanism of cell death in inflamed superficial digital flexor tendon in the horse. J. Comp. Pathol. 2005;132:51–58. doi: 10.1016/j.jcpa.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Integrins as mechanochemical transducers. Curr. Opin. Cell Biol. 1991;3:841–848. doi: 10.1016/0955-0674(91)90058-7. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu. Rev. Physiol. 1997;59:575–599. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- Ireland D, Harrall R, Curry V, et al. Changes in gene expression in chronic human Achilles tendinopathy. Matrix Biol. 2001;20:159–169. doi: 10.1016/s0945-053x(01)00128-7. [DOI] [PubMed] [Google Scholar]
- Janmey PA. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiol. Rev. 1998;78:763–781. doi: 10.1152/physrev.1998.78.3.763. [DOI] [PubMed] [Google Scholar]
- Järvinen M, Józsa L, Kannus P, Järvinen TL, Kvist M, Leadbetter W. Histopathological findings in chronic tendon disorders. Scand. J. Med. Sci. Sports. 1997;7:86–95. doi: 10.1111/j.1600-0838.1997.tb00124.x. [DOI] [PubMed] [Google Scholar]
- Jones GC, Corps AN, Penninton CJ, et al. Expression profiling of metalloproteinases and tissue inhibitors of metalloproteinases in normal and degenerative human Achilles tendon. Arthritis Rheum. 2006;54:832–842. doi: 10.1002/art.21672. [DOI] [PubMed] [Google Scholar]
- Józsa LG, Kannus P. Human tendons: anatomy, physiology, and pathology. Champaign, IL: Human Kinetics; 1997. Overuse injuries of tendons; pp. 164–253. [Google Scholar]
- Józsa LG, Reffy A, Kannus P, Demel S, Elek E. Pathological alterations in human tendons. Arch. Orthop. Trauma Surg. 1990;110:15–21. doi: 10.1007/BF00431359. [DOI] [PubMed] [Google Scholar]
- Kannus P, Józsa LG. Histopathological changes preceding spontaneous rupture of a tendon. A controlled study of 891 patients. J. Bone Joint Surg. Am. 1991;73:1507–1525. [PubMed] [Google Scholar]
- Kastelic J, Galeski A, Baer E. The multicomposite structure of tendon. Connect. Tissue Res. 1978;6:11–23. doi: 10.3109/03008207809152283. [DOI] [PubMed] [Google Scholar]
- Kastelic J, Palley I, Baer E. A structural mechanical model for tendon crimping. J. Biomech. 1980;13:887–893. doi: 10.1016/0021-9290(80)90177-3. [DOI] [PubMed] [Google Scholar]
- Ker RF. The implications of the adaptable fatigue quality of tendons for their construction, repair, and function. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002;133:987–1000. doi: 10.1016/s1095-6433(02)00171-x. [DOI] [PubMed] [Google Scholar]
- Knorzer E, Folkhard W, Geercken W, et al. New aspects of the etiology of tendon rupture. An analysis of time-resolved dynamic-mechanical measurements using synchrotron radiation. Arch. Orthop. Trauma Surg. 1986;105:113–120. doi: 10.1007/BF00455845. [DOI] [PubMed] [Google Scholar]
- Lavagnino M, Arnoczky SP. In vitro alterations in cytoskeletal tensional homeostasis control gene expression in tendon cells. J. Orthop. Res. 2005;23:1211–1218. doi: 10.1016/j.orthres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Lavagnino M, Arnoczky SP, Tian T, Vaupel Z. Effect of amplitude and frequency of cyclic tensile strain on the inhibition of MMP-1 mRNA expression in tendon cells: in vitro study. Connect. Tissue Res. 2003;44:181–187. doi: 10.1080/03008200390215881. [DOI] [PubMed] [Google Scholar]
- Lavagnino M, Arnoczky SP, Frank K, Tian T. Collagen fibril diameter distribution does not reflect changes in the mechanical properties of in vitro stress-deprived tendons. J. Biomech. 2005a;38:69–75. doi: 10.1016/j.jbiomech.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Lavagnino M, Arnoczky SP, Caballero O, Robertson EM, Nashi SM. In vitro stress-deprivation alters the mechanostat set point of tendon cells. Trans. Orthop. Res. Soc. 2006a;31:329. [Google Scholar]
- Lavagnino M, Arnoczky SP, Egerbacher M, Gardner K, Burns ME. Isolated fibrillar damage in tendons stimulates local collagenase mRNA expression and protein synthesis. J. Biomech. 2006b;39:2355–2362. doi: 10.1016/j.jbiomech.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Lo IKY, Marchuk L, Hollinshead R. Matrix metalloproteinases and tissue inhibitors of metalloproteinases mRNA are specifically altered in torn rotator cuff tendons. Am. J. Sports Med. 2004;32:1223–1229. doi: 10.1177/0363546503262200. [DOI] [PubMed] [Google Scholar]
- Magra M, Maffulli N. Matrix metalloproteases: a role in overuse tendinopathies. Br. J. Sports Med. 2005;39:789–791. doi: 10.1136/bjsm.2005.017855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjabi MM, Yoldas E, Oxland TR, Crisco JJ., III Subfailure injury of the rabbit anterior cruciate ligament. J. Orthop. Res. 1996;14:24–31. doi: 10.1002/jor.1100140208. [DOI] [PubMed] [Google Scholar]
- Riley GP. The pathogenesis of tendinopathy. A molecular perspective. Rheumatology. 2004;43:131–142. doi: 10.1093/rheumatology/keg448. [DOI] [PubMed] [Google Scholar]
- Riley GP, Curry V, DeGroot J, et al. Matrix metalloproteinase activities and their relationship with collagen remodeling in tendon pathology. Matrix Biol. 2002;21:185–195. doi: 10.1016/s0945-053x(01)00196-2. [DOI] [PubMed] [Google Scholar]
- Ritty TM, Roth R, Heuser JE. Tendon cell array isolation reveals a previously unknown fibrillin-2-containing macromolecular assembly. Structure. 2003;11:1179–1188. doi: 10.1016/s0969-2126(03)00181-3. [DOI] [PubMed] [Google Scholar]
- Rosales C, O’Brien V, Kornberg L, Juliano R. Signal transduction by cell adhesion receptors. Biochim. Biophys. Acta. 1995;1242:77–98. doi: 10.1016/0304-419x(95)00005-z. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Stretching is good for a cell. Science. 1997;276:1345–1346. doi: 10.1126/science.276.5317.1345. [DOI] [PubMed] [Google Scholar]
- Sachs F. Mechanical transduction in biological systems. Crit. Rev. Biomed. Eng. 1988;16:141–169. [PubMed] [Google Scholar]
- Scott A, Khan KM, Heer J, Cook JL, Lian O, Duronio V. High strain mechanical loading rapidly induces tendon apoptosis: an ex vivo rat tibialis anterior model. Br. J. Sports Med. 2005;39:e25. doi: 10.1136/bjsm.2004.015164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvanetti A, Cipolla M, Puddu G. Overuse tendon injuries: basic science and classification. Oper. Tech. Sports Med. 1997;5:110–117. [Google Scholar]
- Sharma P, Maffulli N. Tendon injury and tendinopathy: healing and repair. J. Bone Joint Surg. 2005;87A:187–202. doi: 10.2106/JBJS.D.01850. [DOI] [PubMed] [Google Scholar]
- Skutek M, van Griensven M, Zeichen J, Brauer N, Bosch U. Cyclic mechanical stretching enhances secretion of interleukin 6 in human tendon fibroblasts. Knee Surg. Sports Traumatol. Arthrosc. 2001;9:322–326. doi: 10.1007/s001670100217. [DOI] [PubMed] [Google Scholar]
- Sung K-LP, Whittemore DE, Yang L, Amiel D, Akeson WH. Signal pathways and ligament cell adhesiveness. J. Orthop. Res. 1996;14:729–735. doi: 10.1002/jor.1100140508. [DOI] [PubMed] [Google Scholar]
- Tallon C, Maffulli N, Ewen SW. Ruptured Achilles tendons are significantly more degenerated than tendinopathic tendons. Med. Sci. Sports Exerc. 2001;33:1983–1990. doi: 10.1097/00005768-200112000-00002. [DOI] [PubMed] [Google Scholar]
- Tsuzaki M, Bynum D, Almekinders L, Yang X, Faber J, Banes AJ. ATP modulates load-inducible IL-1beta, COX 2, and MMP3 gene expression in human tendon cells. J. Cell. Biochem. 2003;89:556–562. doi: 10.1002/jcb.10534. [DOI] [PubMed] [Google Scholar]
- Tuoheti Y, Itoi E, Pradhan RL, et al. Apoptosis in the supraspinatus tendon with stage II subacromial impingement. J. Shoulder Elbow Surg. 2005;14:535–541. doi: 10.1016/j.jse.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Viidik A. Interdependence between structure and function in collagenous tissues. In: Viidik A, Vaust J, editors. Biology of Collagen. New York: Academic Press; 1972. pp. 257–280. [Google Scholar]
- Viidik A. Mechanical properties of parallel-fibered collagenous tissues. In: Viidik A, Vuust J, editors. Biology of Collagen. London: Academic Press; 1980. pp. 237–255. [Google Scholar]
- Viidik A, Ekholm R. Light and electron microscopic studies of collagen fibers under strain. Zeitschrift für Anatomie und Entwicklungsgeschichte. 1968;127:154–164. doi: 10.1007/BF00521981. [DOI] [PubMed] [Google Scholar]
- Wang JHC. Mechanobiology of tendon. J. Biomech. 2006;39:1563–1582. doi: 10.1016/j.jbiomech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Wang N, Ingber DE. Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys. J. 1994;66:2181–2189. doi: 10.1016/S0006-3495(94)81014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Wang JH-C, Jia F, Yang G, et al. Cyclic mechanical stretching of human tendon fibroblasts increases the production of prostaglandin E2 and levels of cyclooxygenase expression: a novel in vitro model study. Connect. Tissue Res. 2003;44:128–133. doi: 10.1080/03008200390223909. [DOI] [PubMed] [Google Scholar]
- Wang JH-C, Iosifidis MI, Fu FH. Biomechanical basis for tendinopathy. Clin. Orthop. Relat. Res. 2006;443:320–332. doi: 10.1097/01.blo.0000195927.81845.46. [DOI] [PubMed] [Google Scholar]
- Watson PA. Function follows form: generation of intracellular signals by cell deformation. FASEB J. 1991;5:2013–2019. doi: 10.1096/fasebj.5.7.1707019. [DOI] [PubMed] [Google Scholar]
- Woo S L-Y, Gomez MA, Woo YK, Akeson WH. Mechanical properties of tendons and ligaments. I. Quasistatic and nonlinear viscoelastic properties. Biorheology. 1982;19:385–396. doi: 10.3233/bir-1982-19301. [DOI] [PubMed] [Google Scholar]
- Yuan J, Murrell GA, Wei AQ, Wang MX. Apoptosis in rotator cuff tendinopathy. J. Orthop. Res. 2002;20:1372–1379. doi: 10.1016/S0736-0266(02)00075-X. [DOI] [PubMed] [Google Scholar]
- Yuan J, Wang MX, Murrell GA. Cell death and tendinopathy. Clin. Sports Med. 2003;22:693–701. doi: 10.1016/s0278-5919(03)00049-8. [DOI] [PubMed] [Google Scholar]