

Abstract
Myocardin gene has been identified as a master regulator of smooth muscle cell differentiation. Smooth muscle cells play a critical role in the pathogenesis of hypoxia-induced pulmonary hypertension (PH) and pulmonary vascular remodelling (PVR). The purpose of this study was to investigate the change of myocardin gene expression in the pulmonary vessels of hypoxia-induced PH affected by Sildenafil treatment and the involvement of endothelial cells transdifferentiation into smooth muscle cells in the process of hypoxia-induced PH and PVR. Myocardin and relative markers were investigated in animal models and cultured endothelial cells. Mean pulmonary artery pressure (mPAP) was measured. Immunohistochemistry and immunofluorescence were used to show the expression of smooth muscle α-actin (SMA), in situ hybridization (ISH) and reverse transcription polymerase chain reaction (RT-PCR) were performed respectively to detect the myocardin and SMA expression at mRNA levels. Small interfering RNA (siRNA) induced suppression of myocardin in cultured cells. We confirmed that hypoxia induced the PH and PVR in rats. Sildenafil could attenuate the hypoxia-induced PH. We found that myocardin mRNA expression is upregulated significantly in the hypoxic pulmonary vessels and cultured cells but downregulated in PH with Sildenafil treatment. The porcine pulmonary artery endothelial cells (PAECs) transdifferentiate into smooth muscle-like cells in hypoxic culture while the transdifferentiation did not occur when SiRNA of myocardin was applied. Our results suggest that myocardin gene, as a marker of smooth muscle cell differentiation, was expressed in the pulmonary vessels in hypoxia-induced PH rats, which could be downregulated by Sildenafil treatment, as well as in hypoxic cultured endothelial cells. Hypoxia induced the transdifferentiation of endothelial cells of vessels into smooth muscle-like cells which was regulated by myocardin.
Keywords: endothelial cell, hypoxia, myocardin, pulmonary hypertension, smooth muscle cell, transdifferentiation
Pulmonary arterial hypertension (PAH) is a serious clinical problem with significant morbidity and mortality. It is characterized by increased pulmonary vascular resistance caused by vasoconstriction and structural remodelling of pulmonary arterioles, with thickness of medial walls of the vessels (Cheever 2005). Hypoxia-induced pulmonary hypertension (PH) is common in people who live at high altitude and also plays a significant role in the pathogenesis of the PH that accompanies chronic obstructive pulmonary disease (COPD) (Wright et al. 2005). Chronic hypoxia is therefore a well-characterized experimental model of PH and pulmonary vascular remodelling (PVR). Vascular remodelling is characterized by hyperplasia and hypertrophy of smooth muscle cells (SMCs) in small, muscularized pulmonary arteries and by the appearance of new SMCs in distal, previously non-muscular, pulmonary arteries. However, the mechanism governing these architectural changes is largely unknown. A widely accepted viewpoint is that the SMCs, in the media of the pulmonary arteries, would show a proliferative activity while in intact pulmonary arteries, although this phenomenon did not occur during periods of chronic hypoxia exposure (Wohrley et al. 1995). However, in distal non-muscular pulmonary arteries, there are no pre-existing SMCs. Therefore, the proliferation of SMCs does not appear to be the reason for the muscularization of the arteries after exposure to hypoxia.
Recently, it was reported that transdifferentiation of aortic endothelial cells into smooth muscle-like cells was also observed in some conditions (Arciniegas et al. 1992; Frid et al. 2002), and SMCs progenitors in bone marrow and peripheral circulating blood could differentiate into SMCs and participate in the development of embryo and in vessels diseases (Kashiwakura et al. 2003; Hirschi & Majesky 2004; Owens et al. 2004).
Myocardin, which was thought as a pivotal gene in the differentiation of SMC (Wang et al. 2001, 2003; Chen et al. 2002), belongs to a family of transcription factors, and activates smooth and cardiac muscle reporter genes by interacting with serum response factors (SRF). Myocardin is likely to be one of the effectors of the signalling pathways involved in the pathogenesis of many vessel diseases, such as heart failure (Torrado et al. 2003), and arteriosclerosis (Owens et al. 2004).
We hypothesized that myocardin expression could be involved in the process of hypoxia-induced PH and PVR, and the hypoxia could induce the cultured endothelial cells transdifferenatiation into smooth muscle-like cells regulated by myocardin. This could be an alternative source of SMCs in hypoxia-induced PH and PVR.
To determine the possible role of myocardin in the process of PH and PVR, we tested the expression of myocardin in the walls of remodelling pulmonary vessels in a rat model of hypoxia-induced PH and the hypoxic cultured endothelial cells. We also explored the myocardin expression change with drug sildenafil treatment (Della Torre et al. 2005) in rat model and SiRNA of myocardin in cultured cells, respectively.
Methods
Animal models and experimental protocols
Chronic hypoxia and sildenafil treatment in animals
Sixty Adult male Sprague–Dawley (SD) rats (purchased from the Animal Experimental Centre of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China) weighing 260–300 g were divided into six groups in the experiment: hypoxic sildenafil treated for 8 weeks (n = 10), hypoxia for 8 weeks (n = 10), hypoxic sildenafil treated for 4 weeks (n = 10), hypoxia for 4 weeks (n = 10), normoxic sildenafil treated (n = 10) and normoxic controls (n = 10).
Pulmonary hypertension was induced by exposure to hypoxia (10% inspired O2 fraction) in an isobaric hypoxic chamber; rats were exposed to hypoxia for between 4 and 8 weeks. The experiment (hypoxic sildenafil treated) group of rats were treated with sildenafil citrate 25 mg·kg−1·d−1 in their drinking water, starting 4 days before exposure to hypoxia this dose was used as per the previous reports (Zhao et al. 2001). The other subgroup of animal exposure to hypoxia was treated with placebo. Normoxic control animals were maintained in a normal oxygen environment with placebo (lactose, the same dose as sildenafil) treatment. For hypoxic exposure, we modified an isobaric hypoxic chamber described by Cryer and Bartley (1974). Our chamber consisted of a hypoxic animal chamber of transparent Perspex and an oxygen analyser (HT-6101 oxygen analyser; Kanda Electrical Co. Ltd, Chengdu, China) was used to monitor the chamber environment. Nitrogen was used in the chamber to replace oxygen. The oxygen content of the chamber was adjusted at 10% ± 0.5% (V/V). Carbon dioxide was absorbed with soda lime, and the condensed water vapour was absorbed by silica gel. All rats had free access to standard rat chow and water during the whole experiment. Rats were exposed to hypoxic conditions for 8 h a day, 6 days a week for 4–8 weeks.
Measurement of pulmonary artery pressure
Rat pulmonary artery pressure (PAP) was measured with an implanted multiconduct physiological recording system (RM-6000; Nihon Konden Co., Tokyo, Japan). The protocol of measurement of PAP was according to the menu of the machine. In brief, after rats were anaesthetized with pentobarbital sodium (40 mg/kg intraperitoneally), a specially designed single-lumen catheter was inserted into the main pulmonary artery through the right jugular vein at which point the position of the catheter was judged by the waveform of the pressure signal. The shape of the pressure tracing displayed on an oscilloscope guided the placement of the catheter.
Tissue processing
After 4–8 weeks of exposure, the rats were killed as per protocol after being anaesthetized with pentobarbital sodium (60 mg/kg i.p.). Lung tissues were removed from the animals for histology and immunohistochemistry (ICH) and fixed in 4% paraformaldehyde (PFA) for 4 h. Paraffin-embedded sections were cut at 4 μm thickness and were stained with haematoxylin and eosin. The sections were also used for elastic fibre staining with the van Gieson method and smooth muscle α-actin (SMA) ICH. Lung tissue for in situ hybridization (ISH) and reverse transcription polymerase chain reaction (RT-PCR) was snap-frozen in liquid nitrogen and stored at −80 °C.
Morphological analysis and immunohistochemistry
Transverse lung sections were stained with haematoxylin and eosin, elastic fibre staining and monoclonal SMA antibody (1:100; Sigma, St. Louis, MO, USA) for assessment of vascular morphology by displaying the percentage of intra-acinar muscularized vessels in the whole pulmonary vasculature and the thickness of the vascular wall (data of H&E, fibre staining not shown). Briefly, serial sections of formalin-fixed paraffin-embedded lung tissues were digested with 3% H2O2 for 20 min at room temperature, and then preincubated with 10% non-immunized serum. Sections were incubated with specific primary antibodies overnight at 4 °C (for negative control studies, the antibodies were substituted by phosphate-buffered saline). After unbound antibodies were washed off, the sections were incubated with biotinylated second antibodies against rat and thereafter incubated with streptavidin peroxidase. Subsequently, peroxidase activity was visualized by a colour reaction with diaminobenzidine as the substrate. Brown and yellow colours indicated positive results. Finally, the sections were counterstained with haematoxylin and mounted. The optic density (OD) value of the hybridization signals was assessed by image analysis system (PIPS-2020; Kangli Co. Ltd, Hangzhou, China).
Peripheral pulmonary arteries associated with alveolar sacs and ducts were classified as non-muscular (0–25% of circumference with actin staining), partially muscular (26–75%), and fully muscular (>75% of circumference). All intra-acinar blood vessels were identified. Muscular blood vessels (a blood vessel with ≥2 layer of elastic lamina for ≥50% of the vessel circumference) were identified and the proportion of intra-acinar blood muscularized vessels was calculated. All sections were assessed by a double-blinded reviewer who was unaware of the experimental condition to which each lung had been exposed. At least 40 vessels were counted per section, one section per rat.
In situ hybridization
Digoxigenin (Dig)-labelled myocardin riboprobes were generated by random-primed labelling as described previously by an other group (Du et al. 2004). In situ hybridization analyses were performed on sections of lung tissues, which were embedded in optimum cutting temperature compound, cryosectioned (6 μm), mounted on slides, and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4, 4 h, 4 °C). The sections were cleaned and rehydrated, all sections were treated with proteinase K (20 mg/ml; Sigma) for 8 min at room temperature and washed twice with PBS in DEPC treated ddH2O. After postfixation in 4% PFA for 1 h, the sections were rinsed with 0.1 M triethanolamine (pH 8.0) and acetylated with 0.25% acetic anhydride for 15 min at room temperature. Slides were rinsed with 2 × SSC and hybridized with Dig-labelled riboprobes in 50% formaminde. 4 × SSC, 1 × Denhardt's, 10 × dextran sulphate, and 0.5% mg/ml. Salmon sperm DNA overnight at 50 °C. After posthybridization washes, sections were blocked with 2% sheep serum in buffer 1 (100 mM tris-HCL, pH 7.5, and 150 mM NaCl) for 1 h at room temperature and incubated with streptavidin–biotin–peroxidase. Peroxidase activity was visualized by a colour reaction with diaminobenzidine (DAB) (Wuhan Boster Biological Technology Co. Ltd, Wuhan City, China). Brown and yellow colours indicated positive results.
In vitro experiments of cultured cells
Cell isolation, culture and experimental groups
Small arteries of the lung were isolated from healthy adult pig under careful sterile conditions. After washed repeatedly with sterile D-Hank's buffer, the arterioles were digested with 0.25% trypsin for 6–8 min. Furthermore, the Porcine pulmonary artery endothelial cells (PAECs) were obtained by collecting the cell suspension in the lumen of the vessels, washed repeatedly with medium M199, supplemented with 20% foetal calf serum (Hyclone, Logan, VT, USA), 10 mmol/l N-2-hydroxyethylpiperazine-N′-2-ethane sulphonic acid (HEPES) and 100 U/ml penicillin and streptomycin (Gibico, Gaithersburg, MD, USA), pH 7.4. The suspension was centrifuged at 150 × g for 10 min, and the supernatant was removed. The cells were resuspended in 5 ml PBS-F (containing 2% newborn calf serum), and 50 μl platelet endothelial cell adhesive molecules-1(PECAM-1) antibody (1:100; Sigma) was added misce bene in the mixer for 30 min. The suspension was centrifuged at 150 × g for 10 min, and the supernatant was removed. The cells were resuspended in 2.5 ml PBS-F, incubated with Dybeads M-280 for 30 min, washed and processed through a magnetic separation column (Miltenyi Biotec, Bergisch-Gladbach, Germany) to obtain purified PAECs. The cells were resuspended and counted, adjusted to 1 × 105 cells/ml in PBS-F. Consequently, the cells were cultured in fibronectin-coated dishes.
Adherent cells were harvested with typsin/EDTA and washed in ice-cold PBS. Cells were fixed with methanol for 5–10 min at −20 °C, and blocked with 5% BSA/0.2% Triton X-100 for 30 min at room temperature. Cells were then incubated with primary antibody (rabbit anti-human VIII related antigen, 1:100; Zhongshan Co., Beijing, China) overnight at 4 °C washed with PBS for 5 min, and incubated with secondary antibody (goat anti-rabbit IgG labelled with Cy3, 1:200; Sigma) for 1 h at room temperature. They were washed twice, incubated with Hoechst 33258 (1 μg/ml; Sigma) for 15 min, and then washed twice, finally they were moved to the fluorescence microscope for PAECs morphological analysis. Primary PAECs were divided into four groups for culture: normoxic group (21% O2, 5% CO2, 74% N2); normoxia + myocardin prohibited group; hypoxic group (1% O2, 5% CO2, 94% N2); hypoxia + myocardin prohibited group. We selected cells cultured after 1 and 7 days as the targets for PAECs analysis.
RNAi of myocardin mRNA analysis
According the sequence of myocardin in Genbank, the corresponding siRNA was designed: sense strand: 5′-TCC CGC TCA AGT ACC ACC AGT ATA TTC AAG AGA TAT ACT GGT GGT ACT TGA GCT T-3′; antisense strand: 5′-CAA AAA GCT CAA GTA CCA CCA GTA TAT CTC TTG AAT ATA CTG GTG GTA CTT GAG C-3′, and synthesized by Boya Biotech Co. (Shanghai, China). The two single-strand oligos were dissolved in annealing temperature buffer in equal volume, water-bathed for 2 min at 80 °C, cooled to 35 °C naturally, and maintained at −20 °C for next step. PsiRNA-hHlne (Invitrogen, Carlsbad, CA, USA) was digested by BbsI enzyme for 12–18 h. The electrophoresis of digested mixture was carried out on the 0.7% agarose gel plate. Purified linear psiRNA was reclaimed with Gel Extraction Kit (Boya Biotech Co., Shanghai, China). The following reagents were combined into mixture: 1 μl siRNA template, 1 μl (100 ng) linear plasmids, 1 μl T4DNA ligase, 2 μl 10 × T4DNA ligase buffer, and also 15 μl water. The mixture was water-bathed overnight at 16 °C; consequently, the recombinant plasmid psimyocardinRNA was obtained. Competent DH5α was produced with Calcium chloride method, recombinant plasmids transformed the competent bacteria, then the converters were cultured overnight at 37 °C on the agarose plate with ampicillin added. The recombinant colonies were obtained after the ampicillin selection, subsequently amplified for culture. A small volume plasmid DNA was obtained by using DNA extraction kit (Boya Biotech Co., Shanghai, China). The inserting fragment was confirmed by DNA sequence analysis. The recombinant plasmid was used to transform into cells with lipofectamine (Invitrogen).
Identification of the smooth muscle-like cell with indirect immunofluorescence and morphology analysis
Cultured cells grown to confluence on coverslips were washed with warmed (37 °C) PBS, and fixed in warmed paraformaldehyde–lysine–sodium periodate (PLP) fixation solution (2% paraformaldehyde, 75 mmol/L lysine, 10 mmol/l sodium periodate, 45 mmol/l sodium phosphate, pH 7.4). Then the cells were treated with 1% sodium dodecyl sulphate (SDS) for 5 min to unmask epitopes. Cells were blocked for 1 h in PBS containing 0.2% bovine serum albumin (BSA). Specific primary antibodies (Rat against human SMA, 1:100; Sigma) were layered onto coverslips and incubated for 1 h at room temperature. Coverslips were washed in PBS. Furthermore, secondary antibody conjugated to FITC (goat against rat IgG, 1:50; Pierce, Rockford, IL, USA) was applied for 1 h at room temperature. Fluorescence was observed by confocal laser scanning microscopy (Leica TCS NT, Germany). The green cytoplasmic fluorescence was positive. The smooth muscle-like cells appeared spindle-shaped or polygonal. The ratio of SMA positive cells on the sections (five sections were selected for calculation each group) was calculated as the transdifferentiation ratio of smooth muscle-like cells (‰).
Reverse transcription polymerase chain reaction
Total RNA was purified from tissue with TRI-ZOL reagent (Invitrogen) according to the manufacture's instructions. For RT-PCR, total RNA was used as a template for reverse transcriptase and random hexamer primers. The following primer pairs were utilized: myocardin (5′-CTC GGA GTC AGC AGA TGG ATG-3′) and (5′-CCT CAC TGT CGG TGG CAT AGT-3′) yielding a 217 bp product; SM α-actin (5′-TCA ATG TCC CTG CCA TGT ATG-3′) and (5′-TAG AGG TCC TTC CTG ATG TCA-3′) yielding a 502 bp product; β-actin as control gene: (5′-GAG CAC CCC GTG CTG CTC ACC CTA GG-3′) and (5′-GTG GTG GTG AAG CTG TAG CCA CGC T-3′) yielding a 310 bp product in animal model and (5′-GGC TAC AGC TTC ACC ACC AC-3′) and (5′-TAC TCC TGC TTG CTG ATC CAC-3′) yielding a 498 bp product in cultured cells. PCR amplifications were performed in 10 μL volumes using a PTC100-machine (MJ Research, Boston, MA, USA). The PCR conditions used were 95 °C for 5 min, followed by 35 cycles of 94 °C for 1 min, 50 °C for 45 s, and 72 °C for 45 s. A final extension of 72 °C for 7 min was done to flush unfinished ends. PCR products were electrophoresed on 2% agarose gels and visualized by ethidium bromide staining.
Statistical analysis
Values are presented as the mean ± SD. Statistical significance was evaluated by SPSS12.0. Comparisons between groups were performed with one-way analysis of variance (anova), followed by Student–Newman–Keuls post hoc test when appropriate. A value of P < 0.01 or P < 0.05 was considered to be significant.
Results
Effects of sildenafil and hypoxia on mean pulmonary arterial pressure
No significant change of mPAP in normal non-hypoxic rats was observed after treatment with sildenafil. mPAP was almost double in the 4 week hypoxic animals compared with normoxic controls (Figure 1) although there was no additional increase between 4 and 8 weeks of hypoxia exposure. Sildenafil treated animals significantly attenuated the increase of mPAP compared with hypoxic animals without sildenafil treatment at both 4 and 8 week periods (Figure 1).
Figure 1.

Mean pulmonary artery pressure (mPAP) among the six groups of rats. n = 10 rats for all the groups. Results are mean ± SD. *P < 0.01 for normoxia vs. all the other groups but normoxia + sildenafil. **P < 0.01 for within hypoxia 4 weeks group (hypoxia vs. hypoxia + sildenafil) and for within hypoxia 8 weeks group (hypoxia vs. hypoxia + sildenafil), respectively.
Effects of sildenafil and hypoxia on PVR
As shown in the Table 1 and Figure 2, pulmonary arterioles in normoxic animals were thin walled, whereas after 4 weeks of hypoxic exposure they developed an increased percentage of muscular vessels in the whole pulmonary vasculature and medial thickness characteristic of PH. In hypoxia-treated animals, there were significantly increased numbers of muscularized vessels and the vascular walls were thicker than those of normoxic ones. Treatment with sildenafil was associated with a marked attenuation in the musclularization and the thickness of the walls of these arteries induced by hypoxia. The present medial wall thickness of arteries 10–150 μm in external diameter was significantly attenuated by sildenafil treatment compared with hypoxic subgroup without sildenafil treatment. We selected 4–8 weeks as the most appropriate exposure periods for hypoxia in this experiment, while the previous reports from other groups (Hoshikawa et al. 2003) chose <4 weeks because we found that during periods shorter than 4 weeks not all of the hypoxic group had developed the characteristic medial thickness and musclularization of the vessels. After 4 weeks exposure to hypoxic conditions, all animals had developed the hypoxic PH.
Table 1.
Quantification of degree of muscularization of rat alveolar arteries
| Vessel type | Normoxia % | Normoxia + sildenafil % | Hypoxia 4ws % | Hypoxia 4ws + sildenafil % | Hypoxia 8ws % | Hypoxia 8ws + sildenafil % |
|---|---|---|---|---|---|---|
| Non-muscular arteries | 78.91 ± 8.71 | 81.87 ± 8.88 | 50.98 ± 6.34a | 68.02 ± 6.28b | 28.83 ± 6.79c | 55.01 ± 5.44d |
| Partially muscular arteries | 12.24 ± 5.24 | 10.7 ± 6.05 | 24.0 ± 2.80 | 20.0 ± 3.09 | 18.1 ± 5.03 | 22.9 ± 4.61 |
| Fully muscular arteries | 8.85 ± 4.45 | 7.33 ± 4.06 | 25.0 ± 4.17e | 11.9 ± 4.14f | 53.0 ± 4.81g | 22.0 ± 3.19h |
| Total number of vessels counted | 410 | 411 | 425 | 454 | 463 | 490 |
Values (mean ± SD) reflect the percentage of vessels associated with alveolar spaces and ducts categorized by vessel type in each group; n = 20 rats for hypoxia group and n = 10 for all other groups.
P < 0.01 for hypoxia 4 weeks vs. normoxia
P < 0.01 for hypoxia 4 weeks + sildenafil vs. hypoxia 4 weeks
P < 0.01 for hypoxia 8 weeks vs. hypoxia 4 weeks
P < 0.01 for hypoxia 8 weeks + sildenafil vs. hypoxia 8 weeks
P < 0.01 for hypoxia 4 weeks vs. normoxia
P < 0.01 for hypoxia 4 weeks + sildenafil vs. hypoxia 4 weeks
P < 0.01 for hypoxia 8 weeks vs. hypoxia 4 weeks
P < 0.01 for hypoxia 8 weeks + sildenafil vs. hypoxia 8 weeks.
Figure 2.

Representative photomicrographs of rat lung after 4 and 8 weeks of normoxia, hyoxia, hyoxia + sildenafil treatment. SMA immunohistochemistry-stained sections (a, d and g) show a change in hypoxia-induced wall thickening and (j, k and l) in sildenafil treated ones; in situ hybridization analysis of myocardin mRNA expression (b, e and h) when exposed to hypoxia; myocardin mRNA expression is shown by sildenafil treatment (c, f and i). Bars represent a length of 50 μm.
Effects of sildenafil and hypoxia on expression of myocardin and SMA mRNA
In situ hybridization showed the myocardin mRNA as a brown–yellow and granular deposit observed mainly within the cytoplasm of SMCs in tunica media of pulmonary arterial walls. The expression levels were stronger after 4 and 8 weeks hypoxia exposure than that in normoxic controls and correspondingly weaker in hypoxic sildenafil treated groups (Figure 3). Consequently, the change of SMA expression (Figure 4, ICH result) was associated with that of myocardin expression.
Figure 3.

In situ hybridization analysis of myocardin mRNA expression (OD value) among the six groups of rats as in Figure 1. *P < 0.01 for normoxia vs. all the other groups but normoxia + sildenafil. **P < 0.01 for within hypoxia 4 weeks group (hypoxia vs. hypoxia + sildenafil) and for within hypoxia 8 weeks group (hypoxia vs. hypoxia + sildenafil), respectively.
Figure 4.

Immunohistochemistry (ICH) analysis of SMA protein expression (OD value) among the six groups of rats as in Figure 1. *P < 0.01 for normoxia vs. all the other groups but normoxia + sildenafil. **P < 0.01 for within hypoxia 4 weeks group (hypoxia vs. hypoxia + sildenafil) and for within hypoxia 8 weeks group (hypoxia vs. hypoxia + sildenafil), respectively.
Reverse transcription polymerase chain reaction results showed overexpression of myocardin mRNA in hypoxic animals with or without sildenafil treatment compared with the nomorxic control; and with the sildenafil treatment, it was decreased compared with the hypoxic group without sildenafil treatment (Figure 5), as well as SMA mRNA expression (Figure 6).
Figure 5.

Reverse transcription polymerase chain reaction (RT-PCR) analysis of myocardin among the six groups of rats. (a) RT-PCR analysis of myocardin and β-actin gene expression in representative rat lungs from six groups. (b) Quantitation of myocardin mRNA levels, results are means ± SD; n = 10 rats in each group. *P < 0.01 for normoxia vs. all the other groups but normoxia + sildenafil. **P < 0.01 for within hypoxia 4 weeks group (hypoxia vs. hypoxia + sildenafil) and for within hypoxia 8 weeks group (hypoxia vs. hypoxia + sildenafil) respectively.
Figure 6.

RT-PCR analysis of SMA among the six groups of rats. (a) RT-PCR analysis of SMA and β-actin gene expression in representative rat lungs from six groups. (b) Quantitation of SMA mRNA levels, results are mean ± SD; n = 10 rats in each group. *P < 0.01 for normoxia vs. all the other groups but normoxia + sildenafil. **P < 0.01 for within hypoxia 4 weeks group (hypoxia vs. hypoxia + sildenafil) and for within hypoxia 8 weeks group (hypoxia vs. hypoxia + sildenafil), respectively.
There is difference between myocardin mRNA expression in 4 and 8 weeks hypoxic animals, but it is not associated with PAPs change accordingly. We suggested that it could be related with the change of hypoxic pulmonary vasoconstriction (HPV) after a long period of hypoxia exposure (Stenmark & McMurtry 2005; Wright et al. 2005).
PAECs transdifferentiate into smooth muscle-like cells in hypoxic culture
The isolated PAECs exhibited typical ‘cobblestone’ morphology after purified culture (Figure 7). The indirect immunofluorescene of VIII-related antigen in the cells showed red fluorescence as positive results (Figure 8). After hypoxia exposure for 7 days, the cells changed from their ‘cobblestone’ morphology to a more elongated, spindle-shaped morphology characteristic of SMCs in culture, and the expression of SMA was positive green fluorescence in cytoplasm (Figure 9). The ratio of transdifferentiation in each group was obtained (Table 2): in N group and 1 day group, there were no SMA positive cells; while the ratio of SMA positive cells was significantly higher (2.07‰ ± 0.06) in H group after 7 days.
Figure 7.

The isolated endothelial cells exhibited typical ‘cobblestone’ morphology after purified culture.
Figure 8.

Double-label immunofluorescence staining in isolated endothelial cells (in the same field) Hoechst fluorescence showing cell nuclei (left); VIII relative antigen expression is detected in cytoplasm (right).
Figure 9.

Double-label immunofluorescence staining in cultured endothelial cells of hypoxic group (in the same field) Hoechst fluorescence showing cell nuclei (left), SMA expression is negative in the cytoplasm by day 1 (top, right); SMA is expressed in a cytoplasmic pattern by days 7, and the SMA-positive cells alter their morphological appearance toward an elongated, spindle-shaped form (bottom, right).
Table 2.
The ratio of smooth muscle-like cells transdifferentiation in each group of cultured endothelial cells(‰)
| Group | Day 1 | Day 7 |
|---|---|---|
| N | 0 | 0 |
| N + pSi | 0 | 0 |
| H | 0 | 2.07 ± 0.06* |
| H + pSi | 0 | 0.21 ± 0.04 |
The ratio of α-SM-actin positive cells on the sections (five sections were selected for calculation each group) was calculated as the transdifferentiation ratio of smooth muscle-like cells (‰).
P <0.01 for day 7 H vs. day 7 H + pSi.
Hypoxia induced the expression of myocardin mRNA in the PAECs
The RT-PCR results showed that there was no expression of myocardin mRNA in N group and hypoxic group by day 1, but myocardin mRNA expression was detected in hypoxic group by days 7 (0.23 ± 0.03) (Figure 11).
Figure 11.
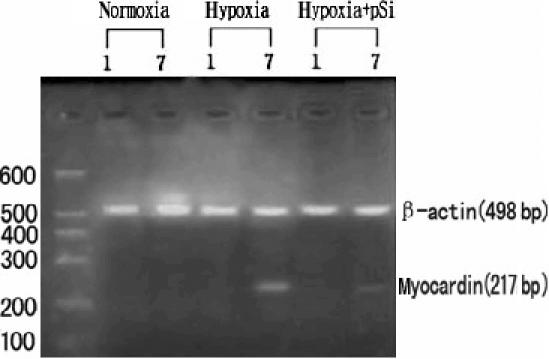
The results of RT-PCR in cultured endothelial cells of all groups there was no expression of myocardin mRNA in N group, H group by day 1, and H + pSi group, but myocardin mRNA expression was detected in H group by days 7, and the level of myocardin mRNA is much weaker in H + pSi group.
The transdifferentiation of PAECs into smooth muscle-like cells was regulated by myocardin
After the transfection of psimyocardinRNA, there were no expressions of myocardin mRNA and no morphological changes of smooth muscle-like cells transdifferentiation in the cells of N group and hypoxic group by day 1 (Figures 10 and 11). The level of myocardin mRNA was lower in H + pSi group by days 7 (Figure 11) and the green cytoplasmic fluorescence in SMA positive cells could be observed (Figure 10). The ratio of smooth muscle-like cells transdifferentiation was lower in H + pSi group by days 7 (Table 2).
Figure 10.

Double-label immunofluorescence staining in cultured endothelial cells of hypoxia + pSi group (in the same field) Hoechst fluorescence showing cell nuclei (left), SMA is not expressed in cytoplasm by day1 (top, right); SMA expression is detected in a cytoplasmic pattern by days 7 (bottom, right), but weaker than that in H group by days 7, and the SMA-positive cells are fewer than those in H group by days 7.
Discussion
After exposure to hypoxia for a period of time, rat can develop changes that mimic those of PH, a condition seen in people who live at high altitudes (Cogo et al. 2004) and in COPD patients (Wright et al. 2005). A widely accepted viewpoint is that the characteristic changes of hypoxia-induced PH are the increased thickness of the medial compartment of the muscular pulmonary arteries due to hypertrophy and proliferation of the individual SMCs and increased synthesis and deposition of extracellular matrix proteins, elastin and collagen (Badesch et al. 1989). In the distal vasculature, in particular in-acinar pulmonary vessels, the musclularization of previously non-musclular arteries is brought about by differentiation and hypertrophy of cells (intermediate cells and pericytes) present in the wall (Durmowicz & Stenmark 1999). In addition, some investigators believe that interstitial fibroblasts are recruited locally and differentiate into cells that exhibit muscle-specific proteins and functions (Das et al. 2002; Rose et al. 2002).
The novel SRF-associated cofactor, myocardin, has provided a new way of identifying another potential source of the SMCs in PH and PVR. As a component of a molecular switch for SMC differentiation, myocardin can potently activate multiple CArG (CC(A/T) 6GG)-containing SMC marker genes including SMA, smooth muscle-myosin heavy chain (SM-MHC) and SM22α (Yoshida et al. 2003). Short interfering RNA (siRNA)-induced suppression of myocardin in cultured SMCs decreased transcription of SMC marker genes and knockout of myocardin in mice resulted in embryonic lethality at embryonic day 10.5 and was associated with failed SMC investment and differentiation(Du et al. 2003). As a hallmark of SMC differentiation, upregulation of myocardin expression in the thickened wall of hypoxic pulmonary arteries indicated that SMC differentiation plays an important role in the process of development of the hypoxia-induced remodelling of pulmonary arteries and the resulting hypertension. Previous studies suggested that SMC proliferation is the key cause of the thickening of hypoxic pulmonary vessels (Mecham et al. 1987; Stenmark et al. 1988; Wohrley et al. 1995). However, for the muscularization of previously intra-acinar, non-muscular vessels, SMC proliferation alone cannot account for the increased thickness of the vascular wall. Although recruited interstitial fibroblast and pericyte hypertrophy could exhibit muscle-specific proteins and functions in response to hypoxia, SMCs progenitor differentiation and relocation into the pulmonary vessels, especially intra-acinar non-muscular arterioles could be an alternative source for the appearance of SMCs. Arciniegas reported that the mesenchymal cells originated from embryonic PAECs could differentiate into SMCs (Arciniegas et al. 2003). In our current report, we found that the mature PAECs, in hypoxic culture, could express the SMA as the reliable marker of SMCs, which exhibited the spindle-shaped or polygonal morphology, different from the shape of endothelial cells. We named them smooth muscle-like cells. It was suggested that there could be smooth muscle-like cell progenitor cells in the mature PAECs. In hypoxic culture, the progenitor cells could transdifferentiate into smooth muscle-like cells. Furukawa et al. (1999) and Hayashi et al. (2000) reported that block of the monocyte chemotactic protein-1 (MCP-1) or p-selectin effect could induce the inhibition of the hyperplasia and migration of SMCs; however, these factors could not induce the hyperplasia and migration of SMCs but could recruit the cells in the peripheral circling blood. It was demonstrated that bone marrow and peripheral circulating blood could be a source of SMCs progenitors and in some pathological conditions including mechanical injury and arteriosclerosis (Saiura et al. 2001; Tanaka et al. 2003; Liu et al. 2004; Xu 2005). Responding to hypoxia, the SMCs progenitors in bone marrow and peripheral circulating blood differentiated into adult pulmonary vascular SMCs (Davie et al. 2004; Hayashida et al. 2005). For instance, several studies suggested that bone marrow-derived SMCs contribute to transplant arteriopathy and neointima formation after vascular injury (Hu et al. 2002). We therefore hypothesized that there would be cells with potential transdifferentiation capability in PAECs, which originated from progenitor cells in the bone marrow (Yeh et al. 2003). The cells could integrate with endothelial cells of the vessel, and the local hypoxia could induce the transdifferentiation of the cells into smooth muscle-like cells. This could be an alternative explanation for the thickening of muscular vessels and muscularization of non-muscular vessels in the process of hypoxia-induced PH. The expression of myocardin in hypoxia-induced PH and remodelling implies that the SMCs progenitor differentiation might participate in the process.
It is known that phosphodiesterase type 5 (PDE5) is the most abundant cGMP-metabolizing enzyme in the smooth muscle of the lung and is thought to inhibit the vasodilator and antiproliferative effects of cGMP-mediated vasoactive factors including nitric oxide and the natriuretic peptides on the pulmonary vasculature (Haning et al. 2003; Corbin et al. 2005; Wharton et al. 2005). PDE5 inhibitors markedly attenuate hypoxia-induced PH in animal models and humans (Sebkhi et al. 2003; Hoeper 2005)suggesting that these drugs, in particular sildenafil can attenuate the PH without affecting systemic blood pressure (Zhao et al. 2001; Cubillos-Garzon et al. 2003; Lee et al. 2005; Richalet et al. 2005). In this study, we have also demonstrated that sildenafil could attenuate the hypoxia-induced PH and the muscularization of non-muscular vessels, and we have further shown that sildenafil treated animals also showed downregulation of myocardin expression. The exact relationship between sildenafil treatment and the SMC differentiation as shown by myocardin expression (Wharton et al. 2005) is currently unknown.
In summary, we have shown that the myocardin gene, as a marker of SMC differentiation, was expressed in the pulmonary vessels in hypoxia-induced PH in rats, which could be attenuated by sildenafil treatment, as well as in hypoxic cultured PEACs. Hypoxia could induce transdifferentiation of endothelial cells into smooth muscle-like cells which was regulated by myocardin. Further studies on homing of SMC progenitor cells to the pulmonary arterioles in hypoxia-induced PVR will be required to confirm.
Acknowledgments
This work was supported by grants from National Nature Science Foundations of China (No. 30400192) and Nature Science Foundation of Hubei Province (No. 2003ABA149). We also acknowledge Dr Xinjiang Wu for critical reading of the manuscript and helpful discussions.
Supported by the National nature Sciences Foundation of China (no. 30400192) and nature Science Foundation of Hubei Province (no. 2003ABA149).
References
- Arciniegas E, Sutton AB, Allen TD, Schor AM. Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J. Cell. Sci. 1992;103(Pt 2):521–529. doi: 10.1242/jcs.103.2.521. [DOI] [PubMed] [Google Scholar]
- Arciniegas E, Becerra A, De Sanctis JB, Graterol A, Ramirez R. CD40 and CD40L expression in the chicken embryo aorta: possible role in the endothelial-mesenchymal transdifferentiation process. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2003;274:942–951. doi: 10.1002/ar.a.10105. [DOI] [PubMed] [Google Scholar]
- Badesch DB, Orton EC, Zapp LM, et al. Decreased arterial wall prostaglandin production in neonatal calves with severe chronic pulmonary hypertension. Am. J. Respir. Cell. Mol. Biol. 1989;1:489–498. doi: 10.1165/ajrcmb/1.6.489. [DOI] [PubMed] [Google Scholar]
- Cheever KH. An overview of pulmonary arterial hypertension: risks, pathogenesis, clinical manifestations, and management. J. Cardiovasc. Nurs. 2005;20:108–116. doi: 10.1097/00005082-200503000-00005. quiz 117–118. [DOI] [PubMed] [Google Scholar]
- Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J. Mol. Cell. Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- Cogo A, Fischer R, Schoene R. Respiratory diseases and high altitude. High. Alt. Med. Biol. 2004;5:435–444. doi: 10.1089/ham.2004.5.435. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Beasley A, Blount MA, Francis SH. High lung PDE5: a strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem. Biophys. Res. Commun. 2005;334:930–938. doi: 10.1016/j.bbrc.2005.06.183. [DOI] [PubMed] [Google Scholar]
- Cryer A, Bartley W. The design and use of a hypoxic chamber for small animals. Lab. Pract. 1974;23:713–715. [PubMed] [Google Scholar]
- Cubillos-Garzon LA, Casas JP, Morillo CA. Sildenafil in secondary pulmonary hypertension. Int. J. Cardiol. 2003;89:101–102. doi: 10.1016/s0167-5273(02)00456-4. [DOI] [PubMed] [Google Scholar]
- Das M, Dempsey EC, Reeves JT, Stenmark KR. Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;282:L976–L986. doi: 10.1152/ajplung.00382.2001. [DOI] [PubMed] [Google Scholar]
- Davie NJ, Crossno JT, Jr, Frid MG, et al. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:L668–L678. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- Della Torre F, Della Torre E, Di Berardino F. Sildenafil in pulmonary hypertension. Sarcoidosis Vasc. Diffuse Lung Dis. 2005;22:78–79. [PubMed] [Google Scholar]
- Du KL, Ip HS, Li J, et al. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol. Cell. Biol. 2003;23:2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du KL, Chen M, Li J, Lepore JJ, Mericko P, Parmacek MS. Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J. Biol. Chem. 2004;279:17578–17586. doi: 10.1074/jbc.M400961200. [DOI] [PubMed] [Google Scholar]
- Durmowicz AG, Stenmark KR. Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr. Rev. 1999;20:e91–e102. [PubMed] [Google Scholar]
- Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 2002;90:1189–1196. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Matsumori A, Ohashi N, et al. Anti-monocyte chemoattractant protein-1/monocyte chemotactic and activating factor antibody inhibits neointimal hyperplasia in injured rat carotid arteries. Circ. Res. 1999;84:306–314. doi: 10.1161/01.res.84.3.306. [DOI] [PubMed] [Google Scholar]
- Haning H, Niewohner U, Bischoff E. Phosphodiesterase type 5 (PDE5) inhibitors. Prog. Med. Chem. 2003;41:249–306. doi: 10.1016/s0079-6468(02)41007-7. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Watanabe N, Nakazawa K, et al. Roles of P-selectin in inflammation, neointimal formation, and vascular remodeling in balloon-injured rat carotid arteries. Circulation. 2000;102:1710–1717. doi: 10.1161/01.cir.102.14.1710. [DOI] [PubMed] [Google Scholar]
- Hayashida K, Fujita J, Miyake Y, et al. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest. 2005;127:1793–1798. doi: 10.1378/chest.127.5.1793. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, Majesky MW. Smooth muscle stem cells. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004;276:22–33. doi: 10.1002/ar.a.10128. [DOI] [PubMed] [Google Scholar]
- Hoeper MM. Drug treatment of pulmonary arterial hypertension : current and future agents. Drugs. 2005;65:1337–1354. doi: 10.2165/00003495-200565100-00003. [DOI] [PubMed] [Google Scholar]
- Hoshikawa Y, Nana-Sinkam P, Moore MD, et al. Hypoxia induces different genes in the lungs of rats compared with mice. Physiol. Genomics. 2003;12:209–219. doi: 10.1152/physiolgenomics.00081.2001. [DOI] [PubMed] [Google Scholar]
- Hu Y, Davison F, Ludewig B, et al. Smooth muscle cells in transplant atherosclerotic lesions are originated from recipients, but not bone marrow progenitor cells. Circulation. 2002;106:1834–1839. doi: 10.1161/01.cir.0000031333.86845.dd. [DOI] [PubMed] [Google Scholar]
- Kashiwakura Y, Katoh Y, Tamayose K, et al. Isolation of bone marrow stromal cell-derived smooth muscle cells by a human SM22alpha promoter: in vitro differentiation of putative smooth muscle progenitor cells of bone marrow. Circulation. 2003;107:2078–2081. doi: 10.1161/01.CIR.0000070082.64414.B5. [DOI] [PubMed] [Google Scholar]
- Lee AJ, Chiao TB, Tsang MP. Sildenafil for pulmonary hypertension. Ann. Pharmacother. 2005;39:869–884. doi: 10.1345/aph.1E426. [DOI] [PubMed] [Google Scholar]
- Liu C, Nath KA, Katusic ZS, Caplice NM. Smooth muscle progenitor cells in vascular disease. Trends Cardiovasc. Med. 2004;14:288–293. doi: 10.1016/j.tcm.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Mecham RP, Whitehouse LA, Wrenn DS, et al. Smooth muscle-mediated connective tissue remodeling in pulmonary hypertension. Science. 1987;237:423–426. doi: 10.1126/science.3603030. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Richalet JP, Gratadour P, Robach P, et al. Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension. Am. J. Respir. Crit. Care. Med. 2005;171:275–281. doi: 10.1164/rccm.200406-804OC. [DOI] [PubMed] [Google Scholar]
- Rose F, Grimminger F, Appel J, et al. Hypoxic pulmonary artery fibroblasts trigger proliferation of vascular smooth muscle cells: role of hypoxia-inducible transcription factors. FASEB. J. 2002;16:1660–1661. doi: 10.1096/fj.02-0420fje. [DOI] [PubMed] [Google Scholar]
- Saiura A, Sata M, Hirata Y, Nagai R, Makuuchi M. Circulating smooth muscle progenitor cells contribute to atherosclerosis. Nat. Med. 2001;7:382–383. doi: 10.1038/86394. [DOI] [PubMed] [Google Scholar]
- Sebkhi A, Strange JW, Phillips SC, Wharton J, Wilkins MR. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation. 2003;107:3230–3235. doi: 10.1161/01.CIR.0000074226.20466.B1. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal. Circ. Res. 2005;97:95–98. doi: 10.1161/01.RES.00000175934.68087.29. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Orton EC, Reeves JT, et al. Vascular remodeling in neonatal pulmonary hypertension. Role of the smooth muscle cell. Chest. 1988;93:127S–133S. [PubMed] [Google Scholar]
- Tanaka K, Sata M, Hirata Y, Nagai R. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ. Res. 2003;93:783–790. doi: 10.1161/01.RES.0000096651.13001.B4. [DOI] [PubMed] [Google Scholar]
- Torrado M, Lopez E, Centeno A, Medrano C, Castro-Beiras A, Mikhailov AT. Myocardin mRNA is augmented in the failing myocardium: expression profiling in the porcine model and human dilated cardiomyopathy. J. Mol. Med. 2003;81:566–577. doi: 10.1007/s00109-003-0470-7. [DOI] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl Acad. Sci. U. S. A. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton J, Strange JW, Moller GM, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am. J. Respir. Crit. Care. Med. 2005;172:105–113. doi: 10.1164/rccm.200411-1587OC. [DOI] [PubMed] [Google Scholar]
- Wohrley JD, Frid MG, Moiseeva EP, Orton EC, Belknap JK, Stenmark KR. Hypoxia selectively induces proliferation in a specific subpopulation of smooth muscle cells in the bovine neonatal pulmonary arterial media. J. Clin. Invest. 1995;96:273–281. doi: 10.1172/JCI118031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax. 2005;60:605–609. doi: 10.1136/thx.2005.042994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q. The role of stem cells in atherosclerosis. Arch. Mal. Coeur. Vaiss. 2005;98:672–676. [PubMed] [Google Scholar]
- Yeh ET, Zhang S, Wu HD, Korbling M, Willerson JT, Estrov Z. Transdifferentiation of human peripheral blood CD34+-enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo. Circulation. 2003;108:2070–2073. doi: 10.1161/01.CIR.0000099501.52718.70. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Sinha S, Dandre F, et al. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 2003;92:856–864. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- Zhao L, Mason NA, Morrell NW, et al. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. 2001;104:424–428. doi: 10.1161/hc2901.093117. [DOI] [PubMed] [Google Scholar]