

Abstract
Water-soluble amyloid-β (wsAβ) is present in cerebral cortex of subjects at risk of Alzheimer's disease (AD) as well as in normal elderly subjects as a mixture of three major amyloid-β (Aβ) species: 1–42, py3–42 and py11–42. The three wsAβ species are nondetectable in brains of young people, free of immunohistochemically detectable amyloid plaques. In the brains of Down's syndrome and APP-mutant transgenic mice, wsAβ appears long time before amyloid deposition, indicating that it represent the first form of Aβ aggregation and accumulation. In normal brain, wsAβ is bound to apolipoprotein E that favours its degradation by proteases. The composition of wsAβ, in terms of the ratio between the full-length 1–42 and the py3–42 peptides, correlates with the severity of clinical and pathological phenotype in familial early onset AD. Water-soluble Aβ is the native counterpart of the Aβ small aggregates (soluble oligomers) that show in vitro an early and high neuronal toxicity.
Keywords: accumulation, Alzheimer's disease, clearance, pathogenesis, soluble amyloid-β
Introduction
Amyloid plaques and neurofibrillary tangles are the classical histological lesions of Alzheimer's disease (AD). The studies carried out in the last 20 years, since the identification of amyloid-β peptide (Aβ) by Glenner & Wong (1984), have clearly indicated three major points that underlie the pathogenesis of AD: 1. In AD brain occurs a progressive accumulation and aggregation of Aβ, a normal product of intracellular proteolysis of a precursor protein (βAPP) (Selkoe 1996;Hardy 1997). 2. Accumulation of Aβ is the final effect of various mechanisms, that influence production, aggregation and degrations of the molecule (Galli et al. 1998; Russo et al. 1998; Paola et al. 2000; Wolozin 2001; Carson & Turner 2002). 3. The aggregates of Aβ consist in a mixture of various peptides, having different N- and C-termini, that dictate the physical properties of Aβ itself (Saido et al. 1995; He & Barrow 1999; Russo et al. 2002).
The mode of Aβ neuronal toxicity is still poorly known, even if a variety of different mechanisms have been proposed (Mattson et al. 1992; Lue et al. 1999; Butterfield et al. 2001; Lashuel et al. 2002; Eckert et al. 2003). Independently from the factors that induce neuronal damage, the recent data showed that the more harmful physical form of Aβ are small, still soluble and diffusible aggregates of low-molecular weight and that Aβ shares this behaviour with other amyloidogenic peptides (Walsh et al. 1999; Dahlgren et al. 2002; Walsh et al. 2002; Kayed et al. 2003; Stefani & Dobson 2003). This early physical state of Aβ aggregation corresponds, in brain tissue, to the so-called water-soluble Aβ (wsAβ), which we identified in 1994 by extraction from the water-soluble fraction of cerebral cortex of AD patients (Tabaton et al. 1994).
Water-soluble Aβ in AD and normal brain
Total wsAβ is extractable from the cytosolic fraction of cerebral cortex (total brain homogenate in saline buffer ultracentrifuged at 100,000 g for 1 h) of AD cases by immunoprecipitation with antibodies recognizing the C-terminal region of the molecule and is revealed by immunoblotting with 4G8 (recognizing Aβ portion 17–24), which shows three bands of 4.5 kDa (B1), 4.2 kDa (B2) and 3.5 kDa (B3) (Figure 1a). The use of monoclonal antibodies recognizing different N- and C-terminal portions of Aβ allowed the initial characterization of wsAβ. The three bands corresponded to the full-length peptide and two N-terminal-truncated species, all ending at residue 42, as showed by the reactivity with an antibody having alanine at position 42 as essential part of the epitope (Figure 1b). Water-soluble Aβ is nondetectable by immunoblotting in the cerebral cortex of normal cases free of Aβ deposits, as ascertained by immunocytochemistry in sections adjacent to those used for biochemical analysis. Immunoblotting has a sensitivity threshold of 5 ng of Aβ 42, and therefore, the amount of wsAβ present in normal brain has to be below this limit (see ELISA). Water-soluble Aβ was further analysed with antibodies specific for the N-termini: B1, B2 and B3 correspond to three major species Aβ 1–42, Aβ py3–42, Aβ py11–42, the last two forms starting with pyroglutamate in place of glutamate (Figure 1c) (Saido et al. 1996). MALDI-TOF mass spectrometry confirmed the identity of the three Aβ species and showed other minor peptides that were nonconstantly present in AD tissue (Aβ 4–42 and Aβ 1–40) (Figure 2). Quantitative analysis of wsAβ with sandwich ELISA specific for Aβ 42 species demonstrated that in frontal cortex of AD cases is present an average amount of 1.5 µg/g of tissue, while in normal brain free of amyloid plaques, only 2 ng/g of tissue are detectable (Figure 3). The low level of wsAβ in normal brain explains the failure to detect Aβ species by immunoblotting. Moreover, most of the wsAβ in normal brain is not in a free state, but it is sequestered by apolipoproteins and other macromolecules of extracellular space (see below).
Figure 1.

Immunoprecipitation and identification of water-soluble amyloid-β peptides from cerebral cortex of Alzheimer's disease (AD) subjects. (a) On immunoblots, antibody 4G8 (Aβ 17–24 epitope) detects three bands of 4.5 kDa (B1), 4.2 kDa (B2) and 3.5 kDa (B3) in six representative AD cases. (b) The three bands recognized by 4G8 are also revealed by an antibody specific for Aβ ending at residue 42 (α-42). (c) B1, B2 and B3, revealed with 4G8 are, respectively, detected with antibodies specific for Aβ residue 1 (α-N1), Aβ residue 3 with pyroglutamate (α-py3) and Aβ residue 11 with pyroglutamate (α-py11).
Figure 2.

Characterization of the water-soluble amyloid-β (wsAβ) peptides in Alzheimer's disease (AD) brain. MALDI-TOF mass spectra of wsAβ immunoprecipitated from AD brain. Peaks that were consistently present in all samples are indicated by lines.
Figure 3.

ELISA quantification of amyloid-β (Aβ) 42 in water-soluble brain fractions of Alzheimer's disease (▪) and control (□) subjects. Values are expressed as ng/g of tissue.
The concept of soluble Aβ has been variously interpreted by investigators. Most of the studies in which a quantification of Aβ was done by ELISA, considered the formic acid-extracted Aβ as soluble Aβ (Roher et al. 1993; Tamaoka et al. 1994). Instead, we used the term ‘water-soluble Aβ’ to define the form of the peptide that derives from poorly aggregated deposits and possibly from nonfibrillar intracellular accumulation and that is partially in equilibrium with the insoluble form (Tabaton et al. 1994).
Timing of wsAβ accumulation
Water-soluble Aβ was supposed to be the earliest form of Aβ accumulation (Tabaton et al. 1994). This hypothesis was confirmed by analysis of brain tissues of cases with Down's syndrome (DS) as well as by studies of APP-mutant transgenic mice (TG). Water-soluble Aβ is already detectable in 50% of foetal brains with trisomy 21 and in all postnatal DS cases examined that ranged from 4 days to 61 years of age (Teller et al. 1996). In the youngest DS cases (4 days–22 years) free of amyloid plaques, wsAβ concentration is low (20 ng/g of net tissue), and it increased by 100 fold (2 ng/g) in the 26-year-old patient in whom diffuse amyloid plaques appeared by immunohistichemistry. The quality of wsAβ peptides in DS brain is comparable to AD. It resolved into three bands, corresponding to the full-length and two N-terminal-truncated species, all ending at residue 42. Interestingly, the 100-fold increase of wsAβ levels paralleled the increase of Aβpy3–42 form, suggesting that this peptide have a nucleation effect, favouring aggregation and accumulation of wsAβ mixture. thus, in DS, wsAβ appears in cerebral cortex 20 years before amyloid deposition and 40 years before neurofibrillary pathology. In DS is the Aβ overproduction due to βAPP overexpression, the primary cause of wsAβ accumulation. Hence, we cannot assume that a similar time progression occurs in sporadic AD in which a primary and single cause of Aβ accumulation does not occur. Independently, from a possible different time course, the observations in DS clearly indicate a precise mechanism of Aβ accumulation that begins with the failure of Aβ clearance and the consequent appearance in brain tissue of Aβ in soluble, poorly aggregated form.
This sequence of events was confirmed by the analysis of brains of TG overexpressing mutant APP, in which wsAβ precedes by 3–4 months the appearance of amyloid plaques (Johnson-Wood et al. 1997). Moreover, in TG APP mice brain, the levels of wsAβ correlates with memory deficit more than amyloid plaques burden (Koistinaho et al. 2001).
Accumulation and clearance of wsAβ
The progression of Aβ accumulation from the early, water-soluble oligomers to the deposition of amyloid plaques is the effect of various factors that favour either its production and aggregation or its dismission. Increased production of Aβ mostly ending at residue 42, is induced by several agents, such as gene mutations, high cholesterol, oxidative stress, drop of oestrogens, trauma and ischaemia, although various mechanisms (Hardy 1997; Jaffe et al. 1994; Jendroska et al. 1997; Paola et al. 2000; Wolozin 2001; Olsson et al. 2004). Macromolecules, apolipoproteins and metal ions bind Aβ, facilitating its aggregation (Yanagisawa et al. 1995; Atwood et al. 1998; Russo et al. 1998; Cotman et al. 2000). Then, several extracellular proteases are involved in the degradation of Aβ, mainly in the form of monomers and oligomers (Saido 1998; Carson & Turner 2002). Most of these findings were derived from studies carried out in vitro or in mutant APP TG mice. Very few studies indeed investigated the role of Aβ-linked molecules directly in AD brain tissue.
We showed (Russo et al. 1998) that wsAβ 42 and apolipoprotein E (apoE) form SDS-insoluble complexes in normal brain free of amyloid deposits and that the bindings between the two molecules is weaker in AD, leading to the release of unbound Aβ in denaturating conditions (Figure 4). The Aβ42/apoE complexes are detectable in normal and AD brain independently from various apoE isoforms, although in less amount in the presence of apoE ε4 (Figure 4). When complexed with apoE, wsAβ is more prone to proteases degradation, as demonstrated by the lower concentration of proteinase K needed to digest apoE-bound wsAβ, in comparison with free wsAβ (Russo et al. 1998).
Figure 4.

Release of amyloid-β (Aβ) from the aggregate with apolipoprotein E (apoE) in Alzheimer's disease (AD) brains. Immunoprecipitation with the apoE antibody AB947 and immunodetection with the monoclonal anti-apoE Mab1062 show in the range of 34–40 kDa broad band corresponding to an apoE/Aβ42 complex that is under-represented in AD subjects in comparison with controls (CTR). The band corresponding to apoE/Aβ42 is visible in the subject homozygous ε4 only after a longer exposure. The lower portion of the gel, immunostained with the monoclonal anti-Aβ antibody 6E10, reveals the typical Aβ pattern for AD cases, indicating that a portion of Aβ peptides is released from the complex with apoE after SDS/PAGE, only in AD samples.
Because unbound wsAβ is nondetactable in amyloid-free normal brain (Figure 4), these findings clearly indicate that Aβ is physiologically sequestered by apoE that can favour its degradation by extracellular proteases. The lower affinity of apoE isoform 4 for Aβ (LaDu et al. 1994), as well as the less expression in the brain of apoE ε4 (Bray et al. 2004), makes less efficient this specific activity of apoE, leading to progressive accumulation of free wsAβ. At this point, when free wsAβ reaches the oligomeric state, the binding with apoE facilitates its aggregation and accumulation. This sequence of events conciliates the positive effect of apoE on Aβ degradation, as shown in apoE-null mice (Holtzman et al. 1999), with that on Aβ aggregation, that is independent from apoE isoforms and is in common with other apolipoproteins expressed in brain tissue (Matsubara et al. 1995).
Clinical and pathological phenotypes of wsAβ composition
It is well known that the conformation of pathologic prion protein dictates the clinical and pathological phenotype of transmissible spongiform encephalopaties (Parchi et al. 1999). This behaviour has been discovered to be a general rule for other proteins involved in the pathogenesis of several neurodegenerative diseases (Kirkitadze et al. 2002).
We showed (Russo et al. 2000) that in the soluble fraction of brains from early-onset familial AD cases with mutations of Presenilin 1 gene, in comparison with sporadic AD, prevail the N-terminal-truncated Aβ species (py3–42 and py11–42) on the full-length 1–42 form. Most importantly, the increase of the N-terminal-truncated species is proportional to the severity of the disease, in terms of course duration and early onset (Russo et al. 2000). Hence, we predicted that the composition of wsAβ can influence the physical properties of Aβ aggregates, which in turn regulates the toxicity of the molecule (Russo et al. 2000). Indeed, Aβ py3–42 has a more quick rate of aggregation and shows a higher toxicity than the full-length molecule (He & Barrow 1999; Russo et al. 2002). Accordingly, with our hypothesis, in the brain of cognitively normal elderly subjects showing abundant amyloid deposition and scarce neurofibrillary pathology, there is a mixture of wsAβ species different from AD, with the prevalence of the full-length Aβ 1–42 form on the Aβ py3–42 form (Figure 5). Consequently, the intensity of neuronal degeneration and the severity of the clinical phenotype seem to be directly proportional to the predominance of Aβ py3–42 peptide. The generation of this N-terminal-truncated form is still unknown. It may be created from APP through an alternative β-secretase cleavage, although Aβ 3–40/42 species were not observed as products of constitutive APP processing (Shirotani et al. 2002). Alternatively, Aβ py3–42 may be produced by extracellular peptidases and then modified by glutaminyl cyclase to obtain pyroglutamate (Shirotani et al. 2002). Interestingly, the proteolysis of N-terminal cyclized parts of Aβ requires neprylisin, a metallopeptidase that is reduced in AD brains (Yasojima et al. 2001).
Figure 5.
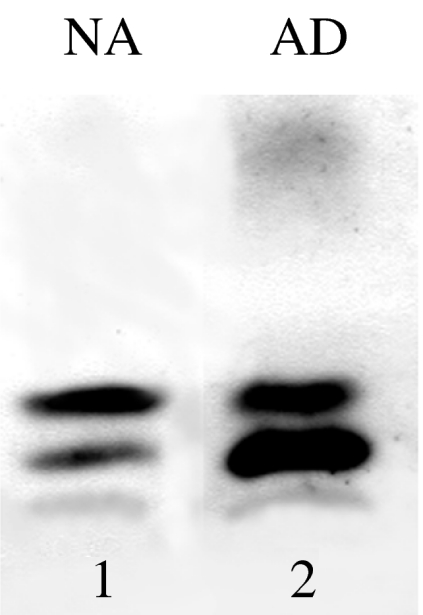
Phenotypic characterization of water-soluble amyloid-β peptides in cerebral cortex from normal elderly cases with abundant amyloid deposits (NA) and Alzheimer's disease (AD) subjects. On immunoblots, cerebral wsAβ is prevalently composed by the Aβ1–42 (B1) in NA and by the Aβpy3–42 (B2) in AD.
Discussion
In 1993, two research groups discovered that Aβ in soluble nonaggregated form is recovered in cell media as normal product of APP proteolytic processing (Golde et al. 1993; Haass et al. 1993). This finding immediately suggested that the physiologically produced Aβ could be detectable in soluble form at low concentration in normal tissues. Indeed, soluble Aβ was shown to be present in cerebrospinal fluid (CSF) and plasma of normal subjects (Vigo-Pelfrey et al. 1993; Tabaton et al. 1994) but was surprisingly nondetectable in normal brain, indicating that existing mechanisms that rapidly dismisses soluble Aβ and that the failure of such mechanisms causes its progressive accumulation in AD (Tabaton et al. 1994). This prediction was confirmed to be true by a series of evidence that emerged in the following years. The clearance of Aβ is regulated by the rate of production by the binding with extracellular molecules (Yanagisawa et al. 1995; Atwood et al. 1998; Russo et al. 1998; Cotman et al. 2000) as well as by the activity of various extracellular proteases (Saido 1998; Carson & Turner 2002). Accumulation of soluble Aβ is the final result of the three factors that are altered in sporadic AD mainly because of age-related causes (Galli et al. 1998; Hardy & Selkoe 2002).
Several findings indicate that soluble, small and diffusible Aβ aggregates are the earliest and most toxic agents of AD (Walsh et al. 1999; Kayed et al. 2003; Stefani & Dobson 2003). In oligomeric form, Aβ produces functional and structural neuronal damage (Lue et al. 1999; Dahlgren et al. 2002; Walsh et al. 2002). Converging evidence shows that cerebral wsAβ represents the native counterpart of the nonfibrillar conformational state of the peptide. Futher studies must be focused on understanding how wsAβ interferes with neuronal functioning to define precise therapeutic strategies for AD.
Acknowledgments
The authors studies have been supported by the Alzheimer's Association, CARIGE Foundation and CARISA Foundation. We thank Dr Claudio Russo for technical assistance.
References
- Atwood CS, Moir RD, Huang X, et al. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- Bray NJ, Jehu L, Moskvina V, et al. Allelic expression of APOE in human brain: effects of epsilon status and promoter haplotypes. Hum Mol Genet. 2004;13:2885–2892. doi: 10.1093/hmg/ddh299. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Carson JA, Turner AJ. Beta-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J Neurochem. 2002;81:1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- Cotman SL, Halfter W, Cole GJ. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Mol Cell Neurosci. 2000;15:183–198. doi: 10.1006/mcne.1999.0816. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Eckert A, Keil U, Marques CA, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol. 2003;66:1627–1634. doi: 10.1016/s0006-2952(03)00534-3. 10.1016/S0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]
- Galli C, Piccini A, Ciotti MT, et al. Increased amyloidogenic secretion in cerebellar granule cells undergoing apoptosis. Proc Natl Acad Sci USA. 1998;95:1247–1252. doi: 10.1073/pnas.95.3.1247. 10.1073/pnas.95.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Golde TE, Cai XD, Shoji M, Younkin SG. Production of amyloid beta protein from normal amyloid beta-protein precursor (beta APP) and the mutated beta APPS linked to familial Alzheimer's disease. Ann NY Acad Sci. 1993;695:103–108. doi: 10.1111/j.1749-6632.1993.tb23036.x. [DOI] [PubMed] [Google Scholar]
- Haass C, Hung AY, Schlossmacher MG, Oltersdorf T, Teplow DB, Selkoe DJ. Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann NY Acad Sci. 1993;695:109–116. doi: 10.1111/j.1749-6632.1993.tb23037.x. [DOI] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. 10.1016/S0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- He W, Barrow CJ. The A beta 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater beta-sheet forming and aggregation propensities in vitro than full-length A beta. Biochemistry. 1999;38:10871–10877. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, et al. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. Estrogen regulates metabolism of Alzheimer amyloid β precursor protein. J Biol Chem. 1994;269:13065–13068. [PubMed] [Google Scholar]
- Jendroska K, Hoffmann OM, Patt S. Amyloid beta peptide and precursor protein (APP) in mild and severe brain ischemia. Ann NY Acad Sci. 1997;826:401–405. doi: 10.1111/j.1749-6632.1997.tb48492.x. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Ort M, Cimadevilla JM, et al. Specific spatial learning deficits become severe with age in beta-amyloid precursor protein transgenic mice that harbor diffuse beta-amyloid deposits but do not form plaques. Proc Natl Acad Sci USA. 2001;98:14675–14680. doi: 10.1073/pnas.261562998. 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, GetZ GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara E, Frangione B, Ghiso J. Characterization of apolipoprotein J-Alzheimer's A beta interaction. J Biol Chem. 1995;270:7563–7567. doi: 10.1074/jbc.270.13.7563. 10.1074/jbc.270.13.7563. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. Beta-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Csajbok L, Ost M, et al. Marked increase of beta-amyloid (1–42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol. 2004;251:870–876. doi: 10.1007/s00415-004-0451-y. [DOI] [PubMed] [Google Scholar]
- Paola D, Domenicotti C, Nitti M, et al. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun. 2000;268:642–646. doi: 10.1006/bbrc.2000.2164. 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W. [PubMed] [Google Scholar]
- Roher AE, Lowenson JD, Clarke S, et al. Beta-amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Angelini G, Dapino D, et al. Opposite roles of apolipoprotein E in normal brains and in Alzheimer's disease. Proc Natl Acad Sci USA. 1998;95:15598–15602. doi: 10.1073/pnas.95.26.15598. 10.1073/pnas.95.26.15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Schettini G, Saido TC, et al. Presenilin-1 mutations in Alzheimer's disease. Nature. 2000;405:531–532. doi: 10.1038/35014735. 10.1038/35014735. [DOI] [PubMed] [Google Scholar]
- Russo C, Violani E, Salis S, et al. Pyroglutamate-modified amyloid beta-peptides – AbetaN3(pE) – strongly affect cultured neuron and astrocyte survival. J Neurochem. 2002;82:1480–1489. doi: 10.1046/j.1471-4159.2002.01107.x. 10.1046/j.1471-4159.2002.01107.x. [DOI] [PubMed] [Google Scholar]
- Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3 (pE), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett. 1996;215:173. doi: 10.1016/0304-3940(96)12970-0. [DOI] [PubMed] [Google Scholar]
- Saido TC. Alzheimer's disease as proteolytic disorders: anabolism and catabolism of beta-amyloid. Neurobiol Aging. 1998;19:S69–S75. doi: 10.1016/s0197-4580(98)00033-5. 10.1016/S0197-4580(98)00033-5. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- Shirotani K, Tsubuki S, Lee HJ, Maruyama K, Saido TC. Generation of amyloid beta peptide with pyroglutamate at position 3 in primary cortical neurons. Neurosci Lett. 2002;327:25–28. doi: 10.1016/s0304-3940(02)00351-8. 10.1016/S0304-3940(02)00351-8. [DOI] [PubMed] [Google Scholar]
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Tabaton M, Nunzi MG, Xue R, Usiak M, Autilio-Gambetti L, Gambetti P. Soluble amyloid beta-protein is a marker of Alzheimer amyloid in brain but not in cerebrospinal fluid. Biochem Biophys ResCommun. 1994;200:1598–1603. doi: 10.1006/bbrc.1994.1634. 10.1006/bbrc.1994.1634. [DOI] [PubMed] [Google Scholar]
- Tamaoka A, Kondo T, Odaka A, et al. Biochemical evidence for the long-tail form (A beta 1-42/43) of amyloid beta protein as a seed molecule in cerebral deposits of Alzheimer's disease. Biochem Biophys Res Commun. 1994;205:834–842. doi: 10.1006/bbrc.1994.2740. 10.1006/bbrc.1994.2740. [DOI] [PubMed] [Google Scholar]
- Teller JK, Russo C, DeBusk LM, et al. Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down's syndrome. Nat Med. 1996;2:93–95. doi: 10.1038/nm0196-93. 10.1038/nm0196-93. [DOI] [PubMed] [Google Scholar]
- Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, et al. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wolozin B. A fluid connection: cholesterol and Abeta. Proc Natl Acad Sci USA. 2001;98:5371–5373. doi: 10.1073/pnas.101123198. 10.1073/pnas.101123198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer's disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaques areas of Alzheimer brain: a possible relationship to deficient degradation of b-amyloid peptide. Neurosci Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. 10.1016/S0304-3940(00)01675-X. [DOI] [PubMed] [Google Scholar]