

Abstract
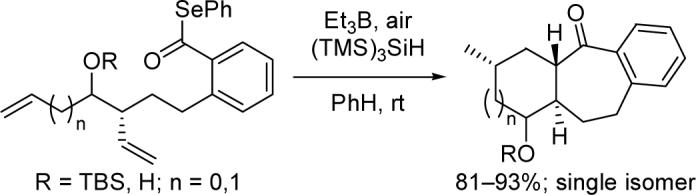
Radical cascades that feature a 7-exo acyl radical cyclization followed by a 6-exo or 5-exo alkyl radical cyclization proceed with very good yields and diastereoselectivities. Two stereocenters are created by the reaction, and a single isomeric product was obtained from each of the five substrates examined. The relative configurations of the products are consistent with cyclizations occurring via chairlike or pseudochairlike transition states.
The synthesis of seven-membered rings via radical cyclization is rare and usually limited to specialized substrates.1 Acyl radical cyclizations represent a large subset of the known 7-exo radical processes. The slower reduction rates of acyl radicals relative to alkyl radicals render them more useful in 7-exo cyclizations.2 In a seminal report, Boger demonstrated that 7-exo-trig acyl radical cyclizations with unactivated alkene acceptors are facile provided an aromatic ring is present in the tether between the radical and the acceptor.3 Evans,4 Bonjoch,5 and Ryu6 have subsequently disclosed various 7-exo ring closures involving acyl radicals. Significantly, Evans found that these reactions can proceed with excellent diastereoselectivity.4 Despite these advances, to the best of our knowledge there are no published reports of cascade reactions that incorporate a 7-exo acyl radical cyclization.7
Lyconadin A (1, Figure 1) is an anticancer alkaloid isolated by Kobayashi from the club moss Lycopodium complanatum.8 Its unique molecular skeleton makes 1 an attractive target for total synthesis. In the course of planning such an endeavor, we recognized that a bicyclo[5.4.0]undecane ring system is embedded within the framework of 1 (depicted in bold in Figure 1). A tandem 7-exo–6-exo radical cyclization would be a particularly efficient means of preparing this structural motif. Accordingly, we decided to study the facility and stereoselectivity of this process with model substrates prior to commencing the total synthesis. Herein, we report that 7-exo–6-exo and 7-exo–5-exo cascade cyclizations which employ an acyl radical in the initial ring closure proceed in good yields with excellent levels of diastereoselectivity.
Figure 1.
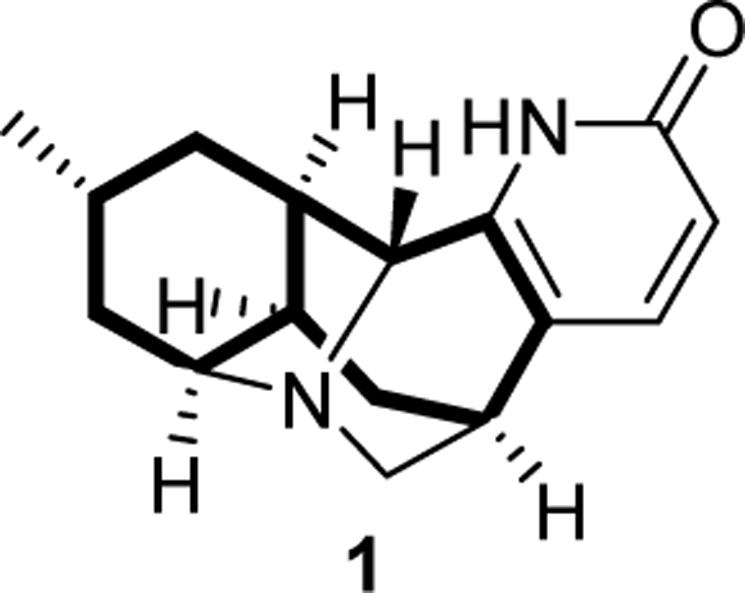
Lyconadin A (bicyclo[5.4.0]undecane shown in bold).
In order to mimic the 2-pyridone moiety of 1 and take advantage of the precedent of Boger,3 we targeted phenyltethered phenyl selenoester 12 as our initial cyclization substrate and prepared it as outlined in Scheme 1. Thus, saponification of 1-isochromanone (2)9 and exhaustive PMB protection afforded ester 3, which was reduced with LiAlH4. Treatment of the resultant crude alcohol with TBS-Cl provided differentially protected diol 4. Cleavage of the PMB ether delivered phenethyl alcohol 5, which was converted into the corresponding bromide in excellent yield. Alkylation of diethyl acetone 1,3-dicarboxylate (DEADC) by this bromide provided 6 in good yield. Careful optimization of Johnson's conditions10 was required in order to preclude styrene formation via elimination of the bromide.
Scheme 1.
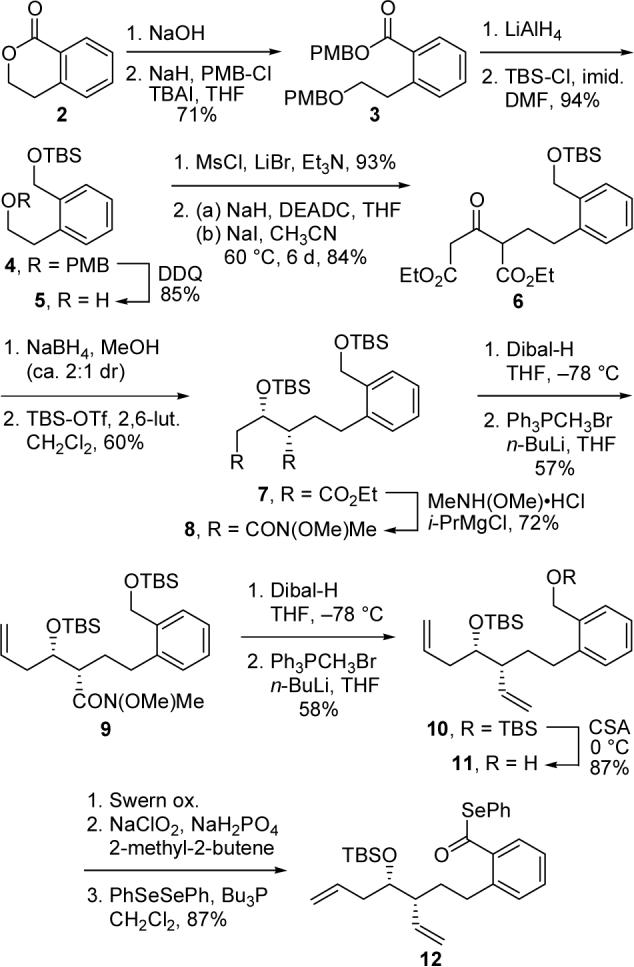
Preparation of Phenyl Selenoester 12
Reduction of 6 with NaBH4 was modestly syn-selective11 and provided 7 as a mixture of diastereomers after TBS protection. The minor anti diastereomer was gradually removed over the course of the next few steps. Conversion of both ester moieties to the corresponding Weinreb amides was followed by attempts to form bisolefin 10 via a reduction–Wittig olefination sequence. Unfortunately, significant differences in the reduction rates of the two amides necessitated the stepwise approach to 10 portrayed in Scheme 1. Then, selective cleavage of the primary benzylic silyl ether mediated by camphorsulfonic acid (CSA) was followed by oxidation and phenyl selenoesterification,12 delivering cyclization substrate 12 in good yield.
Pleasingly, treatment of 12 with Et3B, air, and tris(trimethylsilyl)silane according to a slight modification of the conditions disclosed by Evans4c provided tricycle 13 in excellent yield as a single diastereomer (Scheme 2). The trans ring fusion and relative configuration of the methyl-bearing stereocenter of 13 are consistent with data obtained from both 1D and 2D NMR experiments.13 Notably, (TMS)3SiH proved critical to the success of the cyclization, as use of the more rapid hydrogen atom donor Bu3SnH resulted in a complex mixture from which prematurely reduced byproducts could be tentatively identified. Interestingly, we found that vigorous stirring of the reaction mixture was crucial to the reproducibility of the reaction. We propose that fast stirring leads to an increase in the surface area of the solution that is exposed to air, thereby increasing the concentration of oxygen in solution and resulting in more efficient initiation of the radical cascade process.
Scheme 2.
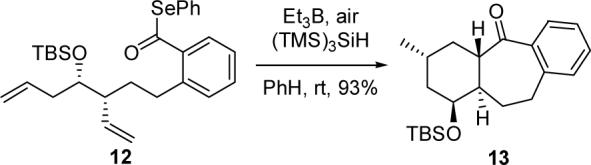
Tandem 7-exo–6-exo Cyclization of 12
Intrigued by this result, we next examined cascade cyclizations of substrates related to 12 in order to ascertain the origins of the excellent diastereoselectivity. To probe the role of the OTBS group, we generated free alcohol 14 from 12 via treatment with CSA as illustrated in Scheme 3. Interestingly, cyclization of 14 provided alcohol 15 as a single diastereomer which was identical in structure to material obtained from deprotection of 13. Accordingly, the bulky silyl ether moiety is not required to achieve stereoselectivity in this process.
Scheme 3.
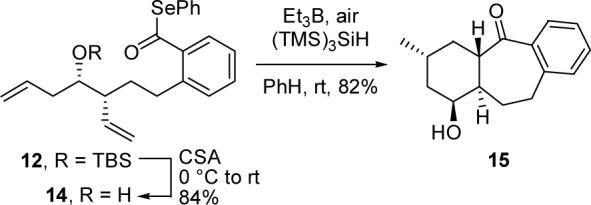
Tandem Cyclization of Alcohol 14
By simply changing the conditions for reduction of ketone 6, we were able to access phenyl selenoester 21, the diastereomer of our original cyclization substrate 12. Thus, exposure of 6 to freshly prepared Zn(BH4)214 and silylation of the resultant alcohol delivered anti isomer 16 in 4:1 dr (Scheme 4). Conversion of 16 into 21 proceeded without incident via the route employed for the synthesis of 12. Gratifyingly, 21 produced a single tricyclic product upon treatment with Et3B–(TMS)3SiH. The structure of this compound was formulated as 22 based on NMR experiments.13 Therefore, it appears that this 7-exo–6-exo cascade radical cyclization occurs with outstanding diastereoselectivity regardless of the relative configuration of the OTBS substituent.
Scheme 4.
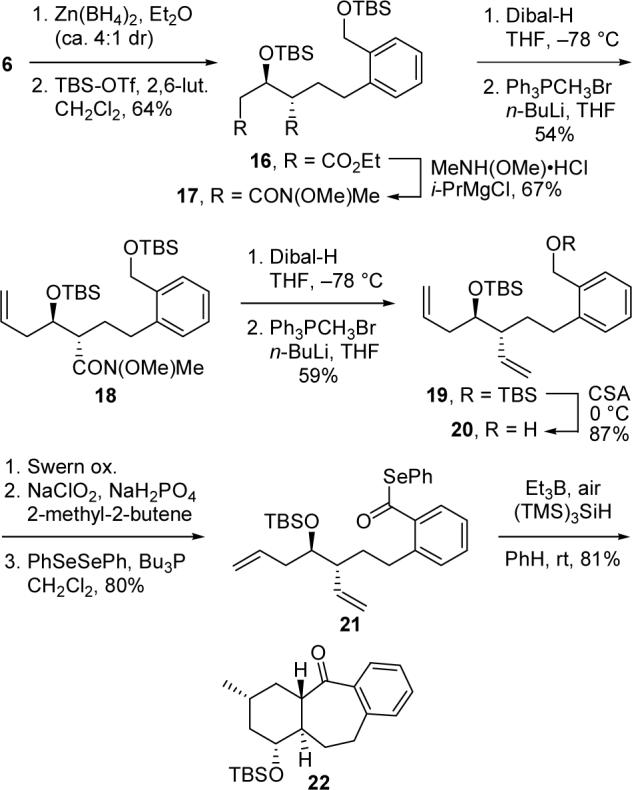
Synthesis and Cyclization of Phenyl Selenoester 21
Although not directly relevant to our targeted natural product 1, we also examined the corresponding 7-exo–5-exo tandem radical cyclizations in order to learn more about the scope of the cascade reaction. The synthesis of requisite phenyl selenoester 31 is detailed in Scheme 5. Alkylation of diethyl malonate with the bromide derived from 5 and reduction of the resultant diester afforded 1,3-diol 24 in excellent yield. Monoprotection of this diol as a PMB ether followed by oxidation and vinyl Grignard addition provided allylic alcohol 26 as a ca. 1:1 mixture of diastereomers. This mixture was carried on as such through the remainder of the synthesis. Then, TBS protection of the secondary alcohol and PMB ether cleavage delivered primary alcohol 28. This compound was transformed into diene 29 via an oxidation–Wittig methylenation sequence. Conversion of 29 into phenyl selenoester 31 mirrors the routes shown in Schemes 1 and 4 used to prepare the 7-exo–6-exo cyclization substrates.
Scheme 5.
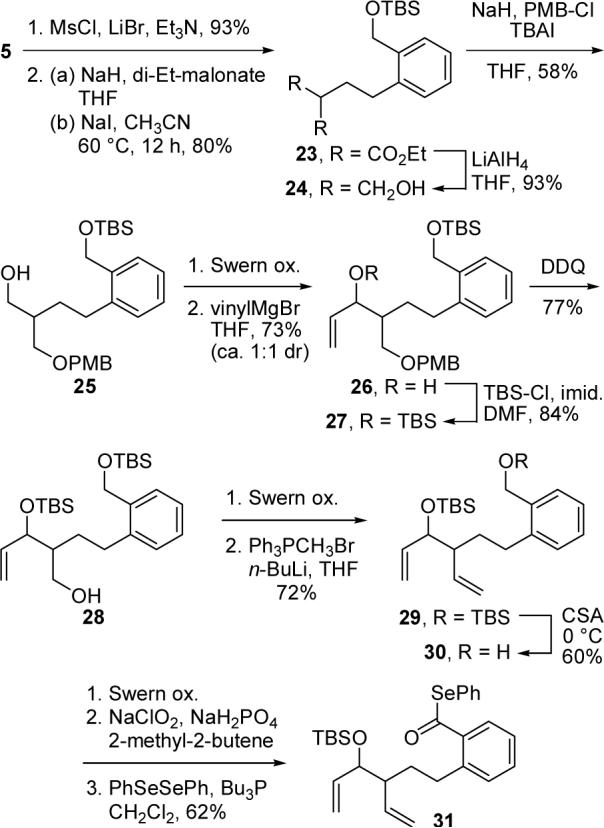
Preparation of Phenyl Selenoester 31
Exposure of 31 to the previously developed tandem cyclization conditions resulted in a separable 1:1 mixture of tricyclic compounds 32 and 33, whose structures were elucidated via 1D and 2D NMR spectroscopy (Scheme 6).13 Since 32 and 33 are epimeric only at the OTBS-bearing carbon, we believe that the 7-exo–5-exo radical cyclization cascade was highly stereoselective, with each diastereomer of the starting material delivering a single product.
Scheme 6.
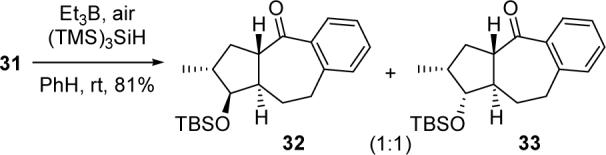
Tandem 7-exo–5-exo Cyclization of 31
The stereochemical course of the reactions is consistent with cyclization via chairlike or pseudochairlike transition states in which the number of equatorial or pseudoequatorial substituents is maximized (Figure 2). Evans has postulated similar transition states for other stereoselective 7-exo-trig acyl radical cyclizations.4a It is noteworthy that the cyclization of 21 provided a single diastereomer. Apparently, the presence of an axial hydrogen in the transition state of the second cyclization (see Figure 2, R1 = H) is sufficient to force the alkene to reside in an equatorial orientation due to the strain inherent in the alternative axial conformer.
Figure 2.
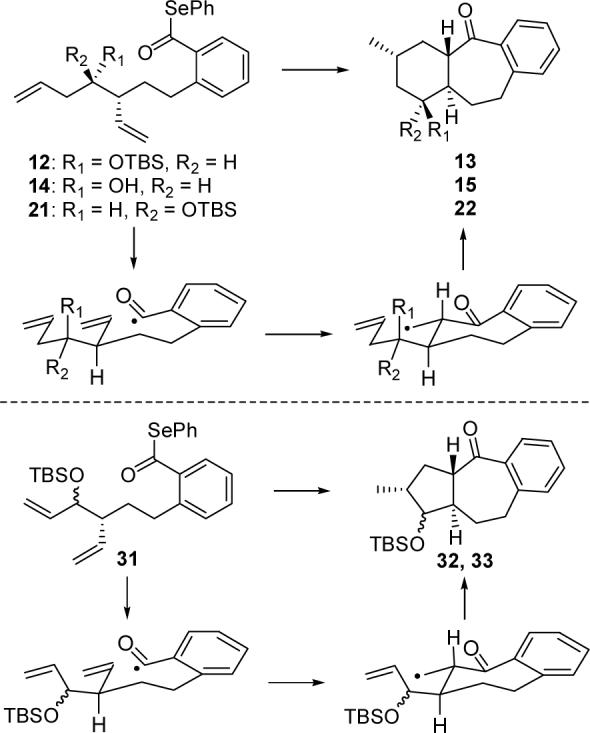
Proposed Pathways of Tandem Cyclizations.
Comparison of the structures of tricycles 13, 15, and 22 with that of lyconadin A reveals that our tandem cyclizations create the proper stereochemistry at the methyl-bearing carbon but the improper stereochemistry at the α-carbonyl ring junction. Fortunately, it should be possible to invert the configuration at the latter carbon. We are currently investigating this transformation.
In conclusion, we have found that 7-exo acyl radical cyclizations followed by 6-exo or 5-exo alkyl radical cyclizations can occur with excellent yields and diastereoselectivities. In fact, of the five cyclization substrates examined,15 all were transformed into a single detectable diastereomer. The stereochemistry of the adducts is consistent with the cyclizations proceeding via chairlike or pseudochairlike transition states. To the best of our knowledge, these reactions represent the first examples of radical cascades that incorporate a 7-exo acyl radical cyclization. Studies to fully define the scope and limitations of this process and apply it to the total synthesis of natural products are in progress.
Supplementary Material
Acknowledgment
We thank Brigham Young University and the National Institutes of Health (GM70483) for support of this work. S.W.G. thanks the BYU Cancer Research Center for a Summer Research Fellowship.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for all new compounds, as well as 1H and 13C NMR spectra for selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Justicia J, Oller-López JL, Campaña AG, Oltra JE, Cuerva JM, Buñuel E, Cárdenas DJ. J. Am. Chem. Soc. 2005;127:14911. doi: 10.1021/ja054316o. [DOI] [PubMed] [Google Scholar]; b Lang S, Corr M, Muir N, Khan TA, Schönebeck F, Murphy JA, Payne AH, Williams AC. Tetrahedron Lett. 2005;46:4027. [Google Scholar]; c Srikrishna A. In: Radicals in Organic Synthesis. Renaud P, Sibi MP, editors. Vol. 2. Wiley-VCH; Weinheim: 2001. pp. 163–187. [Google Scholar]
- 2.a Chatgilialoglu C, Crich D, Komatsu M, Ryu I. Chem. Rev. 1999;99:1991. doi: 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]; b Boger DL. Israel J. Chem. 1997;37:119. [Google Scholar]
- 3.a Boger DL, Mathvink RJ. J. Org. Chem. 1988;53:3377. [Google Scholar]; b Boger DL, Mathvink RJ. J. Org. Chem. 1992;57:1429. [Google Scholar]
- 4.a Evans PA, Roseman JD. J. Org. Chem. 1996;61:2252. [Google Scholar]; b Evans PA, Raina S, Ahsan K. Chem. Commun. 2001:2504. [PubMed] [Google Scholar]; c Evans PA, Manangan T, Rheingold AL. J. Am. Chem. Soc. 2000;122:11009. [Google Scholar]; d Evans PA, Manangan T. Tetrahedron Lett. 1997;38:8165. [Google Scholar]; e Evans PA, Manangan T. J. Org. Chem. 2000;65:4523. doi: 10.1021/jo991970b. [DOI] [PubMed] [Google Scholar]
- 5.a Quirante J, Escolano C, Bonjoch J. Synlett. 1997:179. [Google Scholar]; b Quirante J, Vila X, Escolano C, Bonjoch J. J. Org. Chem. 2002;67:2323. doi: 10.1021/jo0163849. [DOI] [PubMed] [Google Scholar]
- 6.a Ryu I, Miyazato H, Kuriyama H, Matsu K, Tojino M, Fukuyama T, Minakata S, Komatsu M. J. Am. Chem. Soc. 2003;125:5632. doi: 10.1021/ja034896u. [DOI] [PubMed] [Google Scholar]; b Tojino M, Otsuka M, Fukuyama T, Matsubara H, Schiesser CH, Kuriyama H, Miyazato H, Minakata S, Komatsu M, Ryu I. Org. Biomol. Chem. 2003;1:4262. doi: 10.1039/b309944j. [DOI] [PubMed] [Google Scholar]
- 7.For a review on radical cascade reactions, see: McCarroll AJ, Walton JC. Angew. Chem., Int. Ed. 2001;40:2224. doi: 10.1002/1521-3773(20010618)40:12<2224::aid-anie2224>3.0.co;2-f. For examples of cascades employing acyl radicals, see ref. 2a.
- 8.Kobayashi J, Hirasawa Y, Yoshida N, Morita H. J. Org. Chem. 2001;66:5901. doi: 10.1021/jo0103874. [DOI] [PubMed] [Google Scholar]
- 9.Kim SS, Sar SK, Tamrakar P. Bull. Korean Chem. Soc. 2002;23:937. [Google Scholar]
- 10.Johnson WS, Plummer MS, Reddy SP, Bartlett WR. J. Am. Chem. Soc. 1993;115:515. [Google Scholar]
- 11.Williams RM, Lee BH, Miller MM, Anderson OP. J. Am. Chem. Soc. 1989;111:1073. [Google Scholar]
- 12.Singh U, Ghosh SK, Chadha MS, Mamdapur VR. Tetrahedron Lett. 1991;32:255. [Google Scholar]
- 13.See Supporting Information for details on structural assignments.
- 14.Overman LE, Angle SR. J. Org. Chem. 1985;50:4021. [Google Scholar]
- 15.This analysis counts each diastereomer of 31 as a separate substrate.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.