

Abstract
Docosahexaenoic acid (DHA, 22:6n-3), an n-3 fatty acid highly concentrated in the central nervous system, is essential for proper neuronal and retinal function. While a high level of DHA is generally maintained in neuronal membranes, inadequate supply of n-3 fatty acid or ethanol exposure leads to a significant loss of DHA in neuronal cells. The roles DHA in neuronal signaling have been emerging. In this review, biological, biochemical and molecular mechanisms supporting the essential function of DHA in neuronal survival and development are described in relation to n-3 fatty acid depleting conditions.
Keywords: Docosahexaenoic acid, neuronal cells, survival, development, ethanol, n-3 fatty acid deficiency, phosphatidylserine, Akt, hippocampal neurons
I. Introduction
Polyunsaturated fatty acids, especially arachidonic acid (20:4n-6, AA) and docosahexaenoic acid (22:6n-3, DHA), are highly concentrated in neuronal membranes within the brain and retina. Numerous studies have indicated that DHA is essential for proper neuronal and retinal functions (Salem et al., 2001). It has been shown that inadequate supply of n-3 fatty acids during prenatal and postnatal development decreases DHA in the brain and reciprocally increases docosapentaenoic acid (DPAn6, 22:5n-6), resulting in a variety of cognitive and/or behavioral deficits in animal models (Moriguchi et al., 2000). In contrast, dietary supplementation of DHA during infancy has been shown to improve mental development in humans (Willatts et al., 1998). In addition, it has been indicated that low levels of DHA in the brain are associated with neuro-degenerative diseases, such as generalized peroxisomal disorders (Martinez, 1990) and Alzheimer's disease (Soderberg, 1991). Furthermore, DHA supplementation has been shown to alleviate symptoms in some patients with peroxisomal disorders (Martinez et al., 2000), prevent dendritic pathology observed in a mouse model of Alzheimer's disease (Calon et al., 2004), and reduce neuronal injury in experimental brain ischemia (Okada et al., 1996). Although all this evidence indicated a beneficial effect of DHA and suggested a requirement of DHA accretion in neural cells, underlying mechanisms have not been well-understood.
The research aim of our laboratory has been to elucidate the biochemical and biological mechanisms supporting the need for this particular fatty acid in the central nervous system. The polyunsaturated fatty acids are mostly esterified to membrane phospholipids and mobilized upon activation of phospholipases. As the hydrolysis of AA and the production of its oxygenated metabolite (eicosanoids) have been implicated in signal transduction, DHA or eicosanoid-like DHA products may also play an important role in neuronal signaling. However, we found that DHA is neither easily released nor enzymatically oxygenated in neuronal cells under a normal condition, but tenaciously retained in membrane phospholipids (Kim et al., 1991; Sawazaki et al., 1994; Kim et al, 1996; Garcia and Kim, 1997; Kim et al, 1999; Kim and Edsall 1999). In contrast, astroglia cells, which support neurons by providing trophic factors, have been shown to release this fatty acid readily (Garcia and Kim, 1997; Kim et al, 1999). These findings suggested that DHA may be trophic and enrichment of this fatty acid in neuronal membranes may be an important aspect of neuronal development and survival. Long-term ethanol exposure has been shown to decrease the astroglial release of DHA (Garcia et al, 1997), indicating effects of ethanol on the neuronal supply of DHA.
DHA, delivered from the blood stream or biosynthesized in the brain, is uniquely processed in neural cells and incorporated into neuronal membranes (Kim, 2007). DHA accumulates particularly in phosphatidylserine (PS) primarily as 1-stearoyl-2-docosahexaenoyl-PS (18:0,22:6-PS) in the brain. This lipid profile appears to be rigorously maintained and may be important for normal brain function in view of the fact that the lipid environment can affect functions of membrane proteins (Niu et al., 2004). Generally, it is difficult to alter fatty acyl composition in the neuronal membranes of adult mammals. However, ethanol can lower the DHA fatty acyl content in brain as has been indicated by several reports (Alling et al., 1982; Harris et al., 1984; Pawlosky et al., 1995), particularly from PS. Considering the fact that PS is the major negatively charged phospholipid class in many mammalian cell membranes and many of the signaling proteins such as protein kinases are influenced by PS, the accumulation of DHA in PS of neural membranes and its depletion by ethanol may have a significant modulatory role in signaling pathways in the nervous system. With this background, following hypotheses have been developed.
II. Hypotheses/Propositions
DHA is neurotrophic.
The incorporation of DHA in membrane phospholipids, especially accumulation in PS is an important aspect of its biological function in the nervous system.
Ethanol exerts its effect, at least in part, through alteration of the DHA supply and phospholipid remodeling processes modulated by DHA.
To test these hypotheses we asked questions concerning the effect of DHA and ethanol on PS regulation, neuronal survival and differentiation as well as underlying biochemical and biological mechanisms. This review will summarize our findings for those questions, which furthered our understanding for the mechanistic role of DHA and ethanol in neuronal function and the signaling processes involved.
III. Findings
1. Does DHA have a specific biochemical function in PS biosynthesis and accumulation?
It is well known that DHA is highly concentrated in aminophospholipids, PE and PS, in neuronal cells. Compositional studies have indicated that DHA comprises 35-40% of total fatty acid in synaptic membrane PS (Hitzemann 1981), implying that probably more than at least 60% of the PS molecular species contain DHA, based on the fact that di-22:6-PS species is rather a minor species in neuronal membranes except the retina. Since PS biosynthesis in animal cells occurs exclusively through the serine base exchange reaction using PC or PE as substrates, the high compositional profile of DHA in PS may be due to the specificity in PS biosynthesis and degradation although the specificity in deacylation/reacylation reactions may also contribute. In such case, the PS content may in turn be modulated according to the DHA status in substrate phospholipids.
The effect of the DHA compositional status on the synthesis of PS was investigated using two approaches: an n-3 fatty acid deficiency animal model and a DHA-supplemented cell system (Garcia et al., 1998). Fatty acid composition of brain microsomes from the offspring of rats artificially reared on an n-3 deficient diet showed a dramatic reduction of DHA (1.7±0.1%) when compared to control animals (15.0±0.2%). Instead, docosapentaenoic acid (DPAn-6, 22:5n-6) quantitatively replaced DHA. The molecular species analysis by HPLC/electrospray mass spectrometry revealed that 18;0,22:6 was the major PS species which was replaced by 18:0,22:5-PS by n-3 fatty acid deficiency. It was also revealed that the total polyunsaturated PS level decreased after depleting n-3 fatty acids although the total amount of polyunsaturated phospholipids in brain microsomes remained unchanged. The decrease in PS was mainly due to the incomplete replacement of 18:0:22:6 with 18:0:22:5 species. This reduction of PS was also accompanied by an accumulation of 18:0:22:5-PC, suggesting a preferential PS synthesis from 18:0:22:6-PC in comparison to 18:0:22:5-PC. The serine base exchange activity evaluated by the incorporation of [3H]serine into the microsomal membranes was also decreased significantly by n-3 fatty acid deficiency. These data suggested that the presence of DHA phospholipid species in the membrane may promote the serine base exchange reaction, either as a preferred substrate or as an enhancer of enzymatic activity. Similarly, Neuro 2A or C6 glioma cells cultured for 24-48h in DHA-supplemented media (10-40μM) significantly increased their PS content as well as the synthesis of [3H]PS when compared with non-supplemented or AA-supplemented cells. The increase of total PS was primarily due to a dramatic increase in 18:0,22:6-PS in DHA supplemented cells. These data indicated that neuronal and glial PS synthesis was sensitive to the changes in DHA levels in phospholipids and suggested that DHA may be a positive modulator of PS synthesis.
2. How is PS accumulation regulated by DHA?
To elucidate the biochemical mechanisms underlying the concentration of PS in neuronal membranes, where the DHA level is high, we investigated substrate preference in PS biosynthesis and degradation in vitro. We also extended our investigation on the effect of the DHA status on PS levels to various neuronal and non-neuronal tissues and cell lines, to gain an insight on neuronal specificity of DHA function. In addition, we searched for other means to alter PS levels in biomembranes by manipulating the expression of PS synthetic enzymes. Our investigations provided the following information; DHA increases the PS content specifically in neuronal cells, primarily due to the accumulation of 18:0,22:6-PS. The DHA-containing PS accumulation was not due to a decrease in degradation, but due to the fact that PS synthesis is most favored for DHA-containing substrates. Depletion of DHA by n-3 fatty acid deficiency has a profound effect on the PS accumulation specifically in neuronal tissues where DHA is highly enriched, despite the compensatory replacement with DPAn6. Over-expressing or silencing genes responsible for PS synthesis did not alter PS accumulation by DHA. The modulation of the PS pool in neuronal membranes due to the DHA status appeared to be a unique mechanism allowing the disruption of membrane homeostasis in living animal cells.
A. Selective decreases in neuronal accumulation of phosphatidylserine by n-3 fatty acid deficiency
The data compiled over the course of our study indicated that n-3 fatty acid deficiency decreases PS selectively from neuronal membranes (Garcia et al., 1998; Hamilton et al., 2000; Murthy et al., 2002). We observed that rat brain cortex, hippocampus, retina and olfactory bulb, where DHA is highly concentrated, contain significantly higher levels of PS in comparison to liver and adrenal where DHA is a rather minor component. Phospholipid molecular species analysis revealed that in brain cortex, hippocampus and olfactory bulb, 18:0,22:6-PS was the most abundant PS species representing 45-65% of the total PS. Dietary depletion of n-3 fatty acids during development decreased the brain DHA content by more than 80% with a concomitant increase in DPAn6 in all tissues. Under these conditions, an approximately 25-35% reduction in total phosphatidylserine in all neuronal tissues was observed, while PS levels in liver and adrenal were unchanged (Figure 1). The observed reduction of PS content in neuronal membranes was due to a dramatic reduction of 18:0,22:6-PS which was not fully compensated with 18:0,22:5n-6-PS. We also observed similar reduction of PS in the pineal gland (Zhang et al., 1998) and retina which are also of a neuronal origin. Collectively, it was concluded that DHA depletion led to a selective decrease in PS accumulation in neuronal tissues where this fatty acid is highly concentrated.
Figure 1.

Neuronal tissue-specific PS reduction by n-3 fatty acid deficiency. Garcia et al., 1998; Hamilton et al., 2000; Murthy et al., 2002
B. Substrate specificity in phosphatidylserine synthesis and degradation
As it became clear that PS content is particularly elevated in neuronal cells due to the specific concentration of 18:0,22:6-PS, the question concerning the biochemical mechanism underlying the observed effect of DHA on PS accumulation naturally followed. In animal cells, PS is synthesized in mitochondria associated membranes (MAM) and microsomes from pre-existing PC or PE by PS synthase1 (PSS1) and PSS2, respectively, which can then be converted to PE by the PS decarboxylation (PSD) enzyme in mitochondria (Vance and Vance, 2004). Therefore, substrate preference in PS synthesis or decarboxylation can contribute to the accumulation of 18:0,22:6-PS species in neuronal membranes. To this end, an in vitro method has been developed to directly determine the substrate preference in PS synthesis and PSD, utilizing liposomes containing deuterium labeled phospholipid substrate species and mass spectrometry. The conversion of PC (PE) or PS substrates to the corresponding PS or PE species was monitored for PSS and PSD activity, respectively, by using reversed-phase liquid chromatography by electrospray ionization mass spectrometry (Figure 2).
Figure 2.

In vitro assay for PS biosynthesis using HPLC/MS and liposomes containing labeled substrates. Kim et al., Biochemistry, 43, 1030-1036, 2004
With this approach we were able to establish a tissue specific substrate preference in PSD in liver (18:0,18:1 ≥ 18:0,22:6 > 18:0,20:4-PS) and brain cortex (18:0,22:6 > 18:0,18:1 > 18:0,20:4-PS) as shown in Figure 3 (Kevala and Kim, 2001). Similarly, PS synthesis by brain (18:0,22:6 > 18:0,22:5 >18:0,18:1 ≥ 18:0,20:4-PS) (Figure 4) and liver microsomes (18:0,22:6 > 18:0,20:4 ≥ 18:0,18:1 > 18:0,22:5-PS) also showed tissue-specific substrate preference (Kim et al., 2004). While both brain and liver microsomes favored 18:0,22:6-PC, the overall PSS activity in brain (normalized to the protein content) was more than twice that of liver. The most striking difference observed was PS synthesis from 18:0,22:5-PC which was efficiently converted to the corresponding PS species only by brain microsomes as long as serine was present in the medium. In the absence of serine, however, PS synthesis by brain microsomes from 18:0,20:4-, 18:0,18:1- and 18:0,22:5-PC was negligible, and only 18:0,22:6-PC was converted to PS species. These data suggested that the preferred synthesis of 18:0,22:6-PS, rather than unfavored degradation of this PS species by decarboxylation, was primarily responsible for the observed accumulation of 18:0,22:6-PS in neuronal membranes. In order to maintain a high level of PS content in neuronal tissues during n-3 fatty acid deficiency, an increase of DPAn6 in neuronal tissues may be warranted since phospholipid species containing DPAn6 is the next best substrate for PS biosynthesis.
Figure 3.

PE formation as a function of PS substrate concentration by (a) liver or (b) brain cortex mitochondrial protein. Kevala and Kim, Anal. Biochem., 292, 130-138, 2001
Figure 4.

PS formation by brain cortex microsomes as a function of serine concentration and time. Kim et al., Biochemistry, 43, 1030-1036, 2004
C. Regulation of phosphatidylserine accumulation in cultured cells
The PS level in Neuro-2A cells was also increased by supplementing cells with DHA and DPAn6 while oleic acid (OA, 18:1n-9) did not have any effect. Consistent with the brain microsomal PS biosynthetic activity shown above, DHA was more effective in accumulating PS in comparison to DPAn6. Non-neuronal cell lines including CHO-K1, NIH-3T3, HEK-293 and HELA did not increase total PS in response to DHA or DPAn6, suggesting that the phospholipid profile is differently regulated in neuronal cells (Guo et al., 2007). The molecular species analysis by electrospray HPLC/MS indicated that DHA was incorporated into PS mainly as 18:0,22:6 species also in non-neuronal cells. However, the increase of 18:0,22:6-PS was compensated by the decrease of other species such as 18:0, 18:1- or 18:1,18:1-PS similar to the product inhibition of PS synthesis observed in CHO cells (Nishijima et al. 1986; Kuge et al. 1999). In neuronal cells, the product feed back was not as prominent, resulting in the increase of the total PS content after DHA supplementation. Silencing pss1 and pss2 genes for the enzyme responsible for PS biosynthesis affected neither the PSS enzyme level nor DHA-induced PS accumulation in Neuro-2A cells, although mRNA levels were successfully manipulated (Figure 5). Only the cells enriched with DHA in a serine depleted condition showed inhibited PS accumulation in agreement with the serine requirement for the mammalian PS synthesis using the serine-base exchange reaction. Similarly, over-expressing pss1 and pss2 genes cloned from the liver (Stone and Vance 1999, Sturbois-Balcerzak et al., 2001) in non-neuronal cells did not increase the PS accumulation (Guo et al., 2007). These data suggest that PS levels are tightly regulated in living cells and these highly unsaturated very long chain fatty acids are uniquely capable of expanding the PS pool specifically in neuronal cells. This observation is consistent with the fact that neuronal tissues which are abundant in DHA generally contain higher levels of PS in comparison to non-neuronal tissues. It is also in agreement with the PS reduction observed selectively in neuronal tissues with n-3 fatty acid deficiency.
Figure 5.

Effect of silencing pss1 and pss2 on PS accumulation in Neuro 2A cells. Akbar et al., Proc. Natl. Acd. Sci. 102, 10858-10863, 2005
3. Does DHA support neuronal survival?
To test the hypothesis that DHA provided by astroglia is neurotrophic, the effect of DHA on apoptotic neuronal cell death caused by serum starvation was evaluated in comparison to other fatty acids using PC-12 or Neuro 2A cells (Kim et al., 2000a). Direct exposure of these cells to DHA during the 5h serum starvation period did not influence cell death while AA decreased the DNA fragmentation in a dose-dependent manner under the same conditions. The effect of AA was not affected by the cyclooxygenase inhibitor indomethacin or the lipoxygenase inhibitor NDGA, indicating that the observed effect was likely due to free AA (Kim et al., 2001). The protection by DHA, as evidenced by the reduced caspase-3 activity and DNA fragmentation, became evident only after at least 24h of enrichment prior to serum starvation. During this period, DHA was gradually incorporated into membrane phoispholipids, and after 48h over 90% of the exogenously added DHA was found in phospholipids. The enrichment of cells with AA was also able to reduce the DNA fragmentation; however, the protective effect was sensitive to the duration of serum starvation. After a prolonged serum starvation period (more than 24h), the protective effect of AA was lost and only DHA remained protective as shown for the caspase-3 activation time course and DNA ladder formation in Figures 6. The Neuro 2A cells enriched with DHA for 48h showed much less fragmented nuclei after 48h of serum starvation as evidenced by Hoechst staining (Figure 7), consistently indicating the protective effect of DHA. These results established that DHA as a membrane component prevented apoptotic cell death induced by serum starvation.
Figure 6.
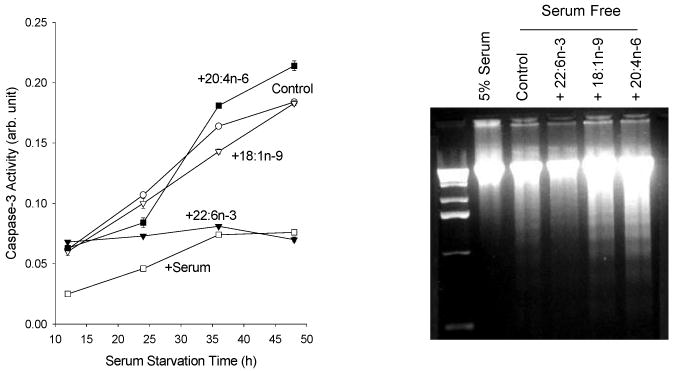
Effect of fatty acid enrichment on caspase-3 activation and DNA ladder formation. Neuro-2A cells were enriched with fatty acids for 48h, and subsequently serum starved. DNA ladder formation was tested after 48h of serum starvation. Kim et al., J. Biol. Chem. 275, 35215-35223, 2000.
Figure 7.
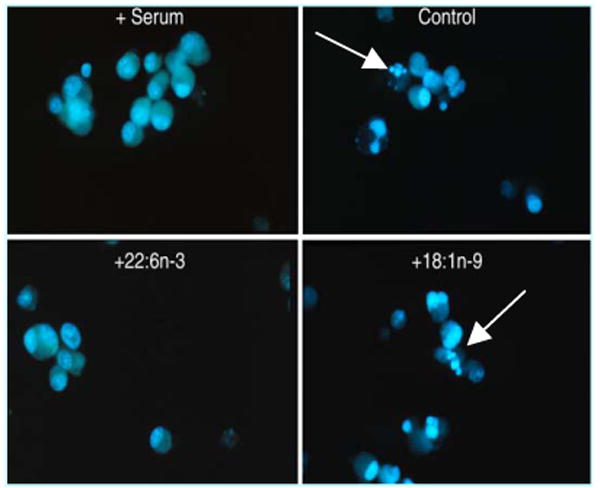
Effect of fatty acid enrichment on DNA fragmentation detected by Hoechst staining. Kim et al., J. Biol. Chem. 275, 35215-35223, 2000.
4. Is PS responsible for the anti-apoptotic effect of DHA?
A. Involvement of PS in the anti-apoptotic effect of DHA
To provide a mechanistic basis supporting the beneficial effects of DHA, the involvement of PS in the anti-apoptotic effect of DHA was first determined by evaluating the cell death in relation to the PS content modulated by varying the enrichment period or by depleting the serine supply needed for the PS biosynthesis. The anti-apoptotic effect of DHA was potentiated as a function of enrichment period. During enrichment, DHA incorporated into PS steadily, resulting in a significant increase in the total PS content. Similar treatment with oleic acid (OA, 18:1n-9) neither altered PS content nor resulted in a protective effect. Hindering PS accumulation by enriching cells in a serine-free medium diminished the protective effect of DHA concomitantly inhibiting PS accumulation, strongly suggesting that PS mediates the anti-apoptotic effect of DHA (Kim et al, 2000a).
B. Differential effect of DHA and DPAn6 on neuronal survival
To gain an insight into the essential role of DHA and the adverse effects of n-3 fatty acid deficiency, we also examined the differential effect of DHA and DPAn6 on survival in vitro and extended the investigation to primary cell culture model in conjunction with in vivo depletion of DHA. Neuro 2A cells contained DHA and DPAn6 as minor components, each less than 1% of the total fatty acid. By enriching the cells with 25 μM DHA for 48h, the DHA level was raised to 15-16%, the approximate level commonly found in neuronal membranes. Since DPAn6 replaces DHA in n-3 fatty acid deficiency, the cells were supplemented with DHA and DPAn6 at various ratios to model the varying extent of n-3 fatty acid deficiency. DHA and DPAn6 differentially affected the PS accumulation and neuronal survival under a serum starvation condition (Figure 8). The cells enriched with DHA or DPAn6 alone showed significantly higher levels of the total PS in comparison to non-enriched control cells. However, the level of PS attained by DPAn6 was less than 80% of that observed with DHA enrichment. As the proportion of DHA with respect to DPAn6 increased, a gradual enhancement in the total PS content was observed, in agreement with our finding that DPAn6-phospholipids are less effective than DHA species as substrates in PS biosynthesis (Kim et al. 2004). The protective effect against apoptotic cell death as evaluated by caspase-3 activity gradually increased with an increasing proportion of DHA, indicating that the PS content and anti-apoptotic effect were well-correlated. The apoptotic cell death evaluated by in situ DNA fragmentation showed consistent results. DHA enrichment significantly decreased the serum starvation-induced TUNEL positive cells in comparison to non-enriched control. Cells enriched with DPAn6 also contained a reduced number of TUNEL positive cells, however, to a lesser extent, again confirming the correlation between the PS content and an anti-apoptotic effect. When PS accumulation was inhibited by depleting serine from the enrichment media, the extent of protection as well as the differential effect of these polyunsaturated fatty acids was diminished, indicating that PS accumulation is primarily responsible for the protective effect of both DHA and DPAn6 (Akbar et al., 2005).
Figure 8.

Differential effect of DHA and DPAn6 on PS accumulation and caspase-3 activation. Akbar et al., Proc. Natl. Acd. Sci. 102, 10858-10863, 2005
C. Neuronal cell death promoted by in vivo DHA depletion
Based on the PS-dependent protective effect observed in transformed cells, the implication of an in vivo reduction in PS, resulting from the depletion of DHA under n-3 fatty acid deficient conditions, in neuronal survival was examined using E-18 hippocampal primary cultures (Figure 9). When pregnant rats were fed with an n-3 fatty acid deficient diet from day 2 of pregnancy, a reciprocal replacement of DHA by DPAn6 was observed in E-18 fetal hippocampi although the DHA accretion was low (3.3%) at this stage of development. Consistently, the PS content in the fetal hippocampi from the n-3 fatty acid deficient group was lower by 15% in comparison to the n-3 fatty acid adequate group. The spontaneous apoptosis under basal conditions was slightly but significantly higher in hippocampal primary culture from n-3 fatty acid deficient animals in comparison to the n-3 fatty acid adequate culture (32% vs. 25%). Under trophic factor removed conditions (-TF), a dramatic increase in cell death (42%) was observed in n-3 fatty acid deficient cultures while n-3 fatty acid adequate cultures did not show a significant increase from the basal level. The increased susceptibility to cell death in DHA-deficient neurons, where PS was reduced, is consistent with the PS-dependent protection by DHA against neuronal apoptosis observed in Neuro 2A cells. Similarly, it has been demonstrated that DHA supplementation prevented hippocampal cell death induced by ischemia-reperfusion in an animal model (Cao et al., 2004). However, it still remains to be tested whether this in vivo protection against hippocampal cell death by DHA is consistent with the PS-dependent protective effect of DHA on neuronal survival demonstrated in our study using cells in culture.
Figure 9.

Effect of in vivo depletion of n-3 fatty acids on apoptotic cell death and the fatty acid and PS profile in E18 hippocampal cultures. Akbar et al., Proc. Natl. Acd. Sci. 102, 10858-10863, 2005
5. What are the signaling mechanisms responsible for the anti-apoptotic effect of DHA?
A. PI(3) kinase-dependent signaling mechanism for protective effects of DHA : membrane translocation of Akt
In addition to the apoptotic cell death induced by serum starvation, neuronal apoptosis induced by staurosporine (ST) treatment was also inhibited by DHA in a PS-dependent manner (Akbar and Kim, 2002). These findings suggested a unique role of DHA in the membrane-related signaling events involved in cell survival through modulating PS levels in neuronal membranes. To understand the signaling mechanisms underlying the observed PS-dependent anti-apoptotic effect of DHA, effects of specific kinase inhibitors were examined. The protective effect of DHA enrichment was abolished when the apoptosis was induced in the presence of PI(3)K inhibitors such as wortmanin (WM) and LY-294002 (LY), indicating that PI(3)K pathway is required for the protection by DHA. Concurrently, phosphorylation of Akt at Thr-308 and Ser-473, the consequence of PI(3)K activation, was significantly reduced by serum starvation and enrichment of cells with DHA prior to serum starvation partially prevented the reduction of Akt phosphorylation at Thr-308 and Ser-473. In vitro kinase assay using immuno-precipitated Akt consistently revealed the reduction of Akt activity under serum starvation, which was partially rescued by DHA-enrichment. These data indicate that the anti-apoptotic effect of DHA was mediated at least partly via the PI(3)K/ Akt pathway.
To determine the target of the PS- and PI(3)K/Akt pathway-dependent protection by DHA, we examined the translocation of Akt, the prerequisite step for the phosphorylation and activation of Akt. Akt translocation, resulting from the interaction between the PH domain of Akt and the plasma membrane PIP3 generated by PI(3)K activation, was evaluated using the PH domain tagged with the enhanced green fluorescence protein (GFP-AktPH). Upon stimulation with IGF, Neuro 2A cells enriched with DHA and DPAn6 showed considerably faster translocation in comparison to the non-supplemented control (Figure 10). While translocation of GFP-AktPH to the plasma membrane reached a plateau at around 5.5 min in control cells, cells enriched with DHA or DPAn6 GFP-AktPH showed such translocation within 2.5 or 3.5 min, respectively. The faster translocation observed in DHA enriched cells was due to the PS concentration, since the inhibition of PS accumulation by depleting serine from the enrichment media prevented the facilitated translocation observed in these cells. Therefore, the differential ability to raise the PS concentration by DPAn6 with respect to DHA is likely responsible for the slower translocation observed in DPAn6-enriched cells in comparison to DHA-enriched cells. We also found that PS is highly localized in the cytosolic face of the plasma membrane (Calderon and Kim, in press-a), further supporting the involvement of PS in the modulation of the translocation of Akt to the plasma membrane.
Figure 10.
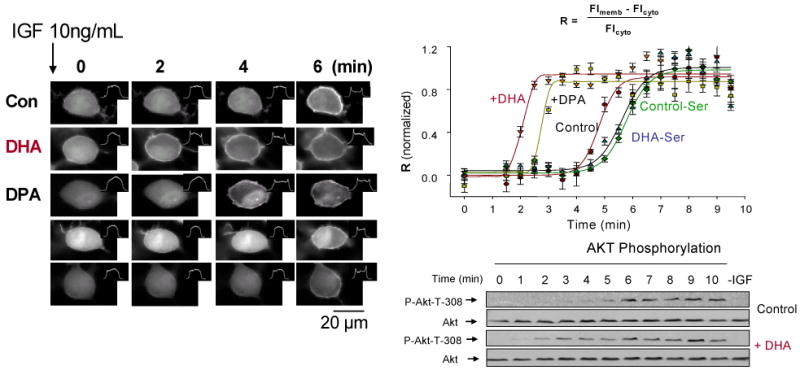
Effect of DHA enrichment on IGF-stimulated Akt translocation and phosphorylation. Akbar et al., Proc. Natl. Acd. Sci. 102, 10858-10863, 2005
The high resolution crystal structure has indicated that besides the binding pocket for PIP3, the PH domain of Akt contains basic residues, Arg15, Lys20, Arg67 and Arg69, which form a flat surface for a possible interaction with acidic membrane phospholipids (Thomas et al., 2002). We observed that the mutation of any one of these residues resulted in the abolition of or incomplete GFP-AktPH translocation in response to IGF at concentrations up to 1μg/mL. DHA supplementation did not produce any significant effect in these mutants, supporting the suggested interaction of these basic residues with acidic membrane components, in this case PS, and its importance for the observed acceleration of Akt translocation by DHA.
In agreement with the observed faster translocation of GFP-AktPH, Akt phosphorylation, the consequence of Akt translocation from cytosol to plasma membrane, was considerably faster in DHA enriched cells in comparison to non-enriched control cells. The faster translocation and phosphorylation of Akt observed in DHA enriched cells was not due to the DHA effect on the signaling events which are upstream of Akt translocation, as in vitro PI(3)K activity as well as endogenous PIP3 formation monitored by metabolic 32P labeling followed virtually the same time course for both control and DHA-enriched cells. In summary, we found that membrane modification by the DHA status affects Akt signaling, impacting neuronal survival. DHA promoted neuronal survival by facilitating Akt translocation/activation through its capacity to increase PS in neuronal membranes. DPAn6 was not as effective as DHA in accumulating PS and translocating Akt, and thus, less effective in preventing apoptosis. Consistently, in vivo reduction of DHA by n-3 fatty acid deficiency increased neuronal susceptibility to apoptotic cell death tested in culture.
B. Raf-1 kinase dependent mechanism for DHA effects
In addition to PI(3)K/Akt, Raf-1 kinase has been suggested to be involved in cell survival (Neshat et al., 2000). It has been shown that membrane translocation is required for the activation of Raf-1 (Stokoe et al., 1994) and the regulatory domain interacting with PS plays an important role in this process (Improta-Brears et al., 1999). We found that Raf-1 membrane translocation in response to BDNF was significantly enhanced by DHA enrichment in Neuro 2A cells (Kim et al., 2000a). Both anti-apoptotic effect of DHA and Raf-1 translocation was sensitive to the DHA-induced PS accumulation. Moreover, in vitro biomolecular interaction analysis by surface plasmon resonance indicated that the Raf-1 interaction with PS/PE/PC liposomes was PS-dependent as shown in Figure 11. These data suggested that Raf-1 activation facilitated by the increased PS after DHA enrichment may also contribute to the down-regulation of caspase-3 and preventing neuronal apoptosis. As the Raf-1 activation has been implicated in cell survival, proliferation, differentiation and migration depending on the type of cells (Galabova-Kovacs et al, 2006), modulation of Raf-1 activation by DHA may affect neuronal cells with similar implications.
Figure 11.

Interaction of Raf-1 with PS evaluated in vitro using biomolecular interaction analysis. Kim et al., J. Biol. Chem. 275, 35215-35223, 2000.
5. Does ethanol exposure in vivo affect DHA-dependent PS accumulation and neuronal survival?
Long-term ethanol exposure has been shown to interfere with PS accumulation induced by DHA in cultured cells (Hamilton and Kim, 2000), and this finding has been extended to an animal model (Wen and Kim, 2004). It has been suggested that hippocampus-related cognitive processes are especially sensitive to ethanol (Matthews and Morrow, 2000; Silvers et al., 2003). To provide an insight for the biochemical basis supporting the hippocampus-related functional deficits associated with prenatal ethanol exposure, we investigated the effects of chronic ethanol exposure on the phospholipid profile in developing rat hippocampi. Using reversed phase HPLC-electrospray ionization mass spectrometry (ESI-MS), we found that ethanol exposure during prenatal and developmental period lowered the total PS level by 15∼20% at postnatal day 0 (P0) and 21 (P21), primarily due to the reduction of 18:0, 22:6-PS species (Figure 12).
Figure 12.

Effect of ethanol on total PS and PS molecular species in rat hippocampus at P0 and P21. Wen and Kim, J. Neurchem. 89, 1368-1377, 2004
An in vitro bioassay employing reversed phase HPLC-ESI-MS and brain microsomes incubated with deuterium labeled exogenous substrates revealed that 18:0, 22:6 species is the best substrate for brain microsomal PS synthesis from both PC and PE, most likely representing PSS1 and PSS2 activities. The PS biosynthetic activity in the brain, especially for 18:0, 22:6-PS production from both PC and PE, was significantly hampered by maternal exposure to ethanol as shown in Figure 13 (Wen and Kim, 2007). Under our experimental conditions, the expression of PSS1 and PSS2 was not reduced by the ethanol exposure based on the mRNA level. Similarly, the PSS1 enzyme level did not change after ethanol exposure although PSS2 could not be probed since the proper antibody is not currently available. Degradation of PS by mitochondrial PS decarboxylation was not enhanced but also inhibited. These data indicated that attenuated PS biosynthetic activity was largely responsible for the PS reduction observed in developing rat brains after maternal exposure to ethanol.
Figure 13.

Effect of ethanol on brain microsomal PS biosynthetic activities at P0 and P21. Wen and Kim, J. Neurosci. Res. 85, 1568-1578, 2007
Based on our findings that ethanol inhibited the accumulation of PS, especially DHA-containing PS species, and DHA prevented neuronal apoptosis through promoting PS accumulation, we examined the effect of ethanol on DHA-dependent neuronal survival in Neuro 2A cells. Also, the effect of in vivo exposure to ethanol on the hippocampal neuronal survival in culture was examined in relation to the DHA and PS status. We found that Neuro 2A cells exposed to ethanol accumulated considerably less PS in response to DHA enrichment, and were less effective in phosphorylating Akt and suppressing caspase-3 activity under serum starved or staurosporine-treated conditions (Akbar et al., 2006). The in vivo paradigm correlated well with the in vitro findings. As was the case for P0 and P21 hippocampi, the total PS content in the E-18 fetal hippocampus was slightly (14%) but significantly (p<0.02) lowered by the prenatal ethanol exposure, primarily due to the reduction in the 18:0, 22:6-PS species. Fetal hippocampal neuronal cultures obtained at E-18 from ethanol-treated pregnant rats contained significantly higher apoptotic cells after 7 days in vitro under basal conditions. These ethanol-exposed neuronal cultures were particularly susceptible to apoptotic cell death induced by overnight trophic factor removal in comparison to the pair-fed control group (Figure 14). From these data it may be suggested that the loss of PS and the resulting neuronal cell death inappropriately escalated during development can contribute to the defects in brain function often observed in fetal alcohol syndrome.
Figure 14.

Effect of prenatal ethanol exposure on hippocampal neuronal survival under basal and trophic factor removed conditions (-TF). Cell death is indicated by TUNEL staining. Akbar et al., J. Neurosci. Res. 83, 432-440, 2006.
6. Does DHA have other neurotrophic functions?
Although the precise molecular mechanisms of DHA actions in the brain are still unknown, several studies have shown that DHA deficiency during development is associated with impairment of hippocampus-dependent neuronal function such as learning and memory (Yoshida et al., 1997; Moriguchi et al., 2000), suggesting another important trophic control by DHA in hippocampal development. Therefore, we examined the effect of DHA on neuronal differentiation, more specifically neurite development in hippocampal neurons.
A natural deficiency of highly polyunsaturated fatty acids has been shown to occur to brain-derived cells when they are transferred from the tissue to cell culture in chemically defined medium (Bourre et al., 1983), serving as a convenient in vitro model of n-3 fatty acid deficiency. This paradigm together with in vitro supplementation of DHA, allowed us to modulate the DHA content in hippocampal neurons in a controlled manner without affecting the content of other polyunsaturated fatty acids. DHA supplementation for 6 days in vitro enhanced the neurite growth in hippocampal neuronal cells (Figure 15). Control and DHA treated hippocampal neurons were stained with MAP2 and traced using camera Lucida and the individual neurite lengths were measured with the NIH Image Software. The cultures treated with DHA showed a significant increase in the population of neurons with longer total neurite lengths and a remarkable decrease in the number of neurons with shorter total neurite lengths in comparison to the control culture. The observed increase in total neurite length was found to be due to both longer individual neurites and more dendritic branches. DHA supplemented neurons showed a tendency to contain more neurites with longer length. More dramatically, DHA supplementation increased the number of branches per neuron as the population of neurons with more branches increased with DHA. With DHA supplementation, the highest frequency was observed for the group of neurons with 13-15 branches/neuron. In contrast, without DHA, the population of neurons with less number of branches was more frequent as the highest frequency was observed for the group of neurons with 4-6 branches.
Figure 15.
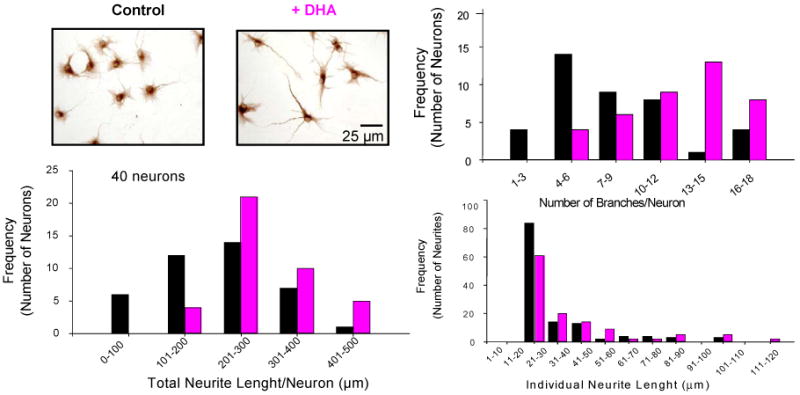
Hippocamplal neurite growth promoted by DHA supplementation. Calderon and Kim, J. Neurochem. 90, 979-988, 2004
Supplementation of hippocampal neurons in culture with arachidonic (AA), oleic (OA), or docosapentaenoic acid (DPAn6) did not have any effect, indicating specificity of the DHA action on neurite growth. It is particularly interesting to note that DPAn6, which replaces DHA during n-3 fatty acid deficiency, was not effective in promoting neurite outgrowth. The in vivo reduction of DHA in fetal hippocampi induced by n-3 fatty acid deficiency during prenatal period also produced similar results. Although accretion of DHA at E18 was not significant (3.3±0.3%), an n-3 fatty acid deficient diet from day 2 of pregnancy caused reciprocal replacement of DHA with DPAn6 (4.1±0.5%) in fetal hippocampi. The culture obtained from the deficient hippocampi contained a greater number of neurons with considerably shorter total neurite length per neuron, which was contributed by both shorter individual neurite length and fewer branches per neuron. Supplementation of the deficient culture with DHA at 1.5 μM reversed the effect of n-3 fatty acid deficiency, increasing the number of neurons with longer neurites comparable to the control culture from the animals fed the n-3 fatty acid adequate diet. These data suggested that DHA has a unique role in promoting hippocampal development. According to our results, depletion of DHA in fetal hippocampus by n-3 fatty acid deficiency has an adverse effect on neurite growth in culture and this effect can be reversed by DHA supplementation. It is possible that n-3 fatty acid deficiency may similarly decrease the dendritic length and number of branches in vivo, which in turn would affect the number and quality of synaptic connections during development and in adulthood. As in the case with in vitro situation, DHA supplementation may promote differentiation in hippocampal neurons in vivo.
The trophic action of DHA on neuronal differentiation may be derived from the facilitated membrane interaction and activation of Raf-1 or Akt due to PS increase, as observed for neuronal survival. Alternatively, the role of DHA as a free fatty acid may also be important as DHA has been indicated to be a ligand for the retinoid X receptor (RXR), a nuclear receptor that acts as a ligand-activated transcription factor (Mata de Urquiza et al., 2000). In fact, four different isoforms of RXR were found to be present in hippocampal neurons with distinctive intracellular localization (Calderon and Kim, in press). Our data using the luciferase reporter assay indicated that DHA can activate the RXRα but at 10-30 μM concentrations while a similar activation can be achieved with 9-cis-retinoic acid at low nM concentrations (Calderon and Kim, in press-b). The increase of neurite outgrowth in hippocampal culture was observed at 1.5 μM, suggesting that RXRα activation by DHA may not be directly responsible for the observed effect of DHA on neurite growth. However, the involvement of other RXR isoforms has not been tested and may need to be considered. It is also possible that a derivative (or derivatives) of DHA rather than DHA itself may mediate the observed effect of DHA on promoting neurite growth, as novel docosatrienes produced from DHA have been shown to exert variety of biological actions at nM concentrations (Hong et al., 2003; Mukherjee et al. 2004). The capacity to produce 10,17-dihydroxy docosatriene has been demonstrated in glial cells (Hong et al., 2003).
7. How does membrane interaction affect the conformation of signaling proteins?
We have established that the protective action of DHA in neuronal survival is targeted at the membrane translocation of Akt (Akbar et al., 2005) and Raf-1 (Kim et al., 2000a). The immediate consequence of translocation is conformational changes of the signaling protein for subsequent activation steps. This conformational change, which is pivotal for proper function of many translocating kinases, can be influenced by the nature of the membranes which these proteins interact with. Interaction of Akt with plasma membrane will also result in conformational changes of Akt required for phosphorylation at T308 and S473 in the kinase and regulatory domains, respectively.
Although partial crystal structures of Akt have been reported, neither the 3D structure of the entire molecule nor its conformational changes accompanying each activation step have been demonstrated. We used the cross-linking strategy (Huang et al., 2004, Huang et al., 2005) to probe the conformational changes of Akt in various activation states. The inactive Akt molecule was cross-linked with lysine-specific cross-linkers, digested with trypsin and analyzed by mass spectrometry. Through-space cross-linked pairs were confirmed using O18 labeling, by identifying the incorporation of four O18 atoms from two H2O18 molecules to the two carboxy termini during tryptic hydrolysis. Characterization of cross-liked peptides by tandem mass spectrometry revealed the presence of two inter-domain cross-linked pairs, K30 of the PH domain linked to K389 of the kinase domain and K284 of the kinase domain to K426 of the regulatory domain.
Monitoring these inter-domain cross-linked pairs, we were able to monitor dramatic inter-domain conformational changes of Akt at different activation stages (Figure 16). When inactive Akt was interacted with liposomes containing phospholipids (PE/PC/PS/PIP3 50/18/30/2), typically found in the inner leaflet of plasma membrane, and cross-linked with a lysine specific cross-linker, K30-K389 and K284-K426 were no longer observed within the 24 Å´ distance constraint, suggesting that the PH domain and regulatory domain moved farther away from the kinase domain. Individual lysine residues were modified by cross-linkers, indicating that these residues were still accessible. For active Akt which is phosphorylated at both T308 and S473, K30-K389 cross-linking was observed but K284-K426 cross-linking was still missing. When active Akt was bound to substrate, GSK-3β peptide and an ATP analogue, K284-K426 cross-linking reappeared in the active form.
Figure 16.

Nano-ESI-QqTOF mass spectra from cross-linked Akt at different stages of Akt activation sequence. Huang and Kim, Mol. Cell. Proteom. 5, 1045-1053, 2006.
Our results strongly suggested that Akt undergoes distinctive conformational changes at each successive activation step. Based on our data, the following activation scheme was proposed (Figure 17). Inactive Akt exists as a folded structure in cytoplasm with the PH and regulatory domains covering the kinase domain, at least in part. Membrane translocation causes conformational changes of Akt so that both PH and regulatory domains become open to expose T308 and S473 for phosphorylation. Phosphorylation of Akt presumably triggers the separation of the PH domain from the membrane, releasing Akt to the cytoplasm. The open conformation of the regulatory domain in the activated form of cytosolic Akt allows substrate entry, which is an important determinant for the activated state of Akt. Our results provided the first demonstration for the dramatic inter-domain conformational change in the full-length Akt structure in solution during activation and substrate binding. More importantly, our results set up a stage for the next investigation on Akt-membrane and/or Akt-protein interaction which is the key to understand molecular mechanisms involved in survival signaling modulated by DHA and ethanol.
Figure 17.

Proposed conformational changes during Akt activation and substrate binding. Huang and Kim, submitted to Mol. Cell. Proteom. 5, 1045-1053, 2006.
IV. Conclusions
Our studies established specific biochemical and biological functions of DHA, i.e., promoting PS accumulation, neuronal differentiation and survival. Preferred microsomal PS synthesis of DHA-containing species appears to be a mechanism for accumulating PS. Depletion of DHA by n-3 fatty acid deficiency depletes PS selectively from neuronal membranes due to the fact that DPAn6, the substitute for DHA under this condition, can not fully support the original level of PS biosynthesis. Long-term exposure to ethanol also interferes with PS accumulation in neuronal tissues, primarily due to impaired PS biosynthetic activities rather than depletion of preferred substrates. The biochemical function of DHA to promote PS accumulation in the central nervous system is an important underpinning of the maintenance of neuronal survival. The protection by DHA is PS- and PI(3) kinase-dependent. Specifically, facilitated PS-dependent Akt translocation is a target event where the protective effect of DHA is derived. The PS-dependent acceleration of Akt translocation is particularly vital under suboptimal conditions where normal survival signal is compromised. In addition, Raf-1 translocation which is also facilitated by DHA may also contribute to the neuronal survival and differentiation. In this regard, the loss of PS either by n-3 fatty acid deficiency or by ethanol exposure and thus increased susceptibility to cell death may have significant implications in neuronal dysfunction. Our current understanding of DHA action is depicted in Figure 18.
Figure 18.

Positive effect of DHA on neuronal survival.
Although the effects of DHA on neuronal survival and differentiation have been repeatedly demonstrated in cultured cells, the current understanding has yet to be verified in vivo. In addition, the molecular mechanism involved in the membrane interaction of the signaling proteins and subsequent modulation by DHA and ethanol has not been fully investigated. As the PS accumulation is an important aspect for the observed effects of DHA, continued characterization of the PS regulation in the neuronal system is warranted. The observation that DHA increases PS specifically in neuronal cells may suggest the existence of a unique PSS isoform in neuronal cells. Alternatively, specific post translational modification of PSS enzymes may confer the characteristics of PS synthetic activity in neuronal cells. Although the protective effect of DHA on neuronal survival observed in our study was PS-dependent, the involvement of DHA metabolites such as 10,17S-docosatriene (neuroprotectin D) may also be considered. The mechanism for the trophic action of DHA on neuronal differentiation still remains an important topic for future research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbar M, Kim HY. Protective effects of docosahexaenoic acid (DHA) in staurosporine-induced apoptosis: Involvement of wortmanin-sensitive pathway. J Neurochem. 2002;82:655–665. doi: 10.1046/j.1471-4159.2002.01015.x. [DOI] [PubMed] [Google Scholar]
- Akbar M, Calderon F, Wen Z, Kim HY. Docosahexaenoic Acid: A Positive Modulator of Akt Signaling in Neuronal Survival. Proc Natl Acad Sci USA. 2005;102:10858–10863. doi: 10.1073/pnas.0502903102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar M, Baick J, Calderon F, Wen Z, Kim HY. Ethanol Promotes Neuronal Apoptosis by Inhibiting Phosphatidylserine Accumulation. J Neurosci Res. 2006;83:432–440. doi: 10.1002/jnr.20744. [DOI] [PubMed] [Google Scholar]
- Alling C, Liljequist S, Engel J. The effect of chronic ethanol administration on lipids and fatty acids in subcellular fractions of rat brain. Med Biol. 1982;60:149–154. [PubMed] [Google Scholar]
- Ansell GB, Spanner S. Phosphatidylserine, phosphatidylethanolamine and phosphatidylcholine. In: Hawthorne JN, Ansell GB, editors. Phospholipids. Elsevier Biomedical Press; New York, N.Y.: 1982. pp. 1–49. [Google Scholar]
- Banker G, Cowan N. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1997;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Belayev L, Marcheselli VL, Khoutorova L, Rodriguez de Turco EB, Busto R, Ginsberg MD, Bazan NG. Docosahexaenoic acid complexed to albumin elicits high-grade ischemic neuroprotection. Stroke. 2005;36:118–123. doi: 10.1161/01.STR.0000149620.74770.2e. [DOI] [PubMed] [Google Scholar]
- Bourre JM, Faivre A, Dumont O, Nouvelot A, Loudes C, Puymirat J, Tixier-Vidal A. Effect of polyunsaturated fatty acids on fetal mouse brain cells in culture in a chemically defined medium. J Neurochem. 1983;41:1234–1242. doi: 10.1111/j.1471-4159.1983.tb00817.x. [DOI] [PubMed] [Google Scholar]
- Calderon F, Kim HY. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J Neurochem. 2004;90:979–988. doi: 10.1111/j.1471-4159.2004.02520.x. [DOI] [PubMed] [Google Scholar]
- Calderon F, Kim HY. Role of RXR in Neurite Outgrowth Induced by Docosahexaenoic Acid. Prostaglandins Leukot Essent Fatty Acids. doi: 10.1016/j.plefa.2007.10.026. In press a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon F, Kim HY. Detection of Intracellular Phosphatidylserine in Living Cells. J Neurochem. doi: 10.1111/j.1471-4159.2007.05079.x. In press b. [DOI] [PubMed] [Google Scholar]
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Obeda O, Rostaing P, Triller A, Salem N, Jr, Ashe KH, Frautschy SA, Cole GM. Docosahexaenoic acid protects from dendritic pathology in an alzheimer's disease mouse model. Neuron. 2004;43:633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DH, Xu JF, Xue RH, Zheng WF, Liu ZL. Protective effect of chronic ethyl docosahexaenoate administration on brain injury in ischemic gerbils. Pharmacol Biochem Behav. 2004;79:651–659. doi: 10.1016/j.pbb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Chu F, Shan SO, Moustakas DT, Alber F, Egea PF, Stroud RM, Walter P, Burlingame AL. Unraveling the interface of signal recognition particle and its receptor by using chemical cross-linking and tandem mass spectrometry. Proc Natl Acad Sci USA. 2004;101:16454–16459. doi: 10.1073/pnas.0407456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW. Isolation of synaptosomal and synaptic plasma membrane fractions. In: Fleischer S, Packer L, editors. Methods of enzymology. Vol. 31. Academic Press; New York: 1974. pp. 445–452. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galabova-Kovacs G, Kolbus A, Matzen D, Meissl K, Piazzolla D, Rubiolo C, Steinitz K, Baccarini M. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle. 2006;5:1514–1518. doi: 10.4161/cc.5.14.2981. [DOI] [PubMed] [Google Scholar]
- Garcia M, Kim KY, Kim HY. Effect of chronic ethanol on the mobilization of arachidonate and docosahexaenoate by the 5-HT2 Receptor agonist DOI in C-6 glioma cells. Alcoholism:Clin Exp Res. 1997;21:1465–1470. [PubMed] [Google Scholar]
- Garcia M, Kim HY. Activation of 5-HT2A receptor is also coupled to the mobilization of arachidonic and docosahexaenoic acids in C6 glioma cells. Brain Res. 1997;768:43–48. doi: 10.1016/s0006-8993(97)00583-0. [DOI] [PubMed] [Google Scholar]
- Garcia M, Ward G, Ma YC, Salem N, Jr, Kim HY. Effect of docosahexaenoic acid on the synthesis of phosphatidylserine in rat brain microsomes and C6 glioma cells. J Neurochem. 1998;70:24–30. doi: 10.1046/j.1471-4159.1998.70010024.x. [DOI] [PubMed] [Google Scholar]
- Guo M, Stockert L, Akbar M, Kim HY. Neuronal Specific Increase of Phosphatidylserine by Docosahexaenoic Acid. J Mol Neurosci. 2007;33:67–73. doi: 10.1007/s12031-007-0046-z. [DOI] [PubMed] [Google Scholar]
- Hamilton J, Greiner R, Salem N, Jr, Kim HY. N-3 fatty acid deficiency decreases phosphatidylserine accumulation selectively in neuronal tissues. Lipids. 2000;35:863–869. doi: 10.1007/s11745-000-0595-x. [DOI] [PubMed] [Google Scholar]
- Harris RA, Baxter DM, Mitchell MA, Hitzemann RJ. Physical properties and lipid composition of brain membranes from ethanol tolerant-dependent mice. Mol Pharmacol. 1984;25:401–409. [PubMed] [Google Scholar]
- Hitzemann R. Developmental changes in the fatty acids of synaptic membrane phospholipids: effect of protein malnutrition. Neurochem Res. 1981;6:935–947. doi: 10.1007/BF00965025. [DOI] [PubMed] [Google Scholar]
- Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. 2003;17:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- Huang XB, Daas C, Kim HY. Probing three dimensional structure of bovine serum albumin by mass spectrometry. J Am Soc Mass Spectrom. 2004;15:1237–1247. doi: 10.1016/j.jasms.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Huang XB, Daas C, Kim HY. Probing Conformational Changes of Human Serum Albumin due to Unsaturated Fatty Acid Binding by Chemical Cross-linking and Mass Spectrometry. Biochem J. 2005;387:695–702. doi: 10.1042/BJ20041624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XB, Kim HY. Interdomain Conformational Changes in Akt Activation Revealed by Chemical Cross-linking and Tandem Mass Spectrometry. Mol Cell Proteom. 2006;5:1045–1053. doi: 10.1074/mcp.M600026-MCP200. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Negishi H, Yamori Y. Antioxidant nutrients and hypoxia/ischemia brain injury in rodents. Toxicology. 2003;189:55–61. doi: 10.1016/s0300-483x(03)00152-5. [DOI] [PubMed] [Google Scholar]
- Improta-Brears T, Ghosh S, Bell RM. Mutational analysis of Raf-1 cysteine rich domain: requirement for a cluster of basic aminoacids for interaction with phosphatidylserine. Mol Cell Biochem. 1999;198:171–178. doi: 10.1023/a:1006981411691. [DOI] [PubMed] [Google Scholar]
- Kevala HJ, Kim HY. Determination of substrate preference in phosphatidylserine decarboxylation by liquid chromatography-electrospray mass spectrometry. Anal Biochem. 2001;292:130–138. doi: 10.1006/abio.2001.5076. [DOI] [PubMed] [Google Scholar]
- Kim HY. Novel Metabolism of Docosahexaenoic Acid in Neural Cells. J Biol Chem. 2007;282:18661–18665. doi: 10.1074/jbc.R700015200. [DOI] [PubMed] [Google Scholar]
- Kim HY, Sawazaki S, Salem N., Jr Lipoxygenation in Rat Brain? Biochem Biophys Res Commun. 1991;174:729–734. doi: 10.1016/0006-291x(91)91478-u. [DOI] [PubMed] [Google Scholar]
- Kim HY, Wang TC, Ma YC. Liquid chromatography/mass spectrometry (LC/MS) of phospholipids using electrospray ionization. Anal Chem. 1994;66:3977–3982. doi: 10.1021/ac00094a020. [DOI] [PubMed] [Google Scholar]
- Kim HY, Edsall L, Ma YC. Specificity of polyunsaturated fatty acid release from rat brain synaptosomes. Lipids. 1996;31:S229–233. doi: 10.1007/BF02637081. [DOI] [PubMed] [Google Scholar]
- Kim HY, Edsall L. The role of polyunsaturates in neuronal signaling. Lipids. 1999;34:S249–250. doi: 10.1007/BF02562308. [DOI] [PubMed] [Google Scholar]
- Kim HY, Edsall L, Garcia M, Zhang H. The release of polyunsaturated fatty acids and their lipoxygenation in brain. Adv Exp Med Biol. 1999;447:75–85. doi: 10.1007/978-1-4615-4861-4_7. [DOI] [PubMed] [Google Scholar]
- Kim HY, Akbar M, Lau A, Edsall L. Inhibition of neuronal apoptosis by docosahexaenoic acid (22:6n-3): Role of phosphatidylserine in anti-apoptotic effect. J Biol Chem. 2000a;275:35215–35223. doi: 10.1074/jbc.M004446200. [DOI] [PubMed] [Google Scholar]
- Kim HY, Hamilton J. Accumulation of docosahexaenoic acid (22:6n-3) in phosphatidylserine is selectively inhibited by chronic ethanol exposure in C-6 glioma cells. Lipids. 2000b;35:187–195. doi: 10.1007/BF02664769. [DOI] [PubMed] [Google Scholar]
- Kim HY, Akbar M, Kim K. Inhibition of neuronal apoptosis by polyunsaturated fatty acids. J Mol Neurosci. 2001;16:139–143. doi: 10.1385/JMN:16:2-3:223. [DOI] [PubMed] [Google Scholar]
- Kim HY, Akbar M, Lau A. Effects of docosapentaenoic acid on neuronal apoptosis. Lipids. 2003;38:453–457. doi: 10.1007/s11745-003-1083-z. [DOI] [PubMed] [Google Scholar]
- Kim HY, Bigelow J, Hamilton J. Determination of substrate preference in phosphatidylserine synthesis by liquid chromatography-electrospray mass spectrometry. Biochemistry. 2004;43:1030–1036. [Google Scholar]
- Kuge O, Saito K, Nishijima M. Cloning of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylserine synthase (PSS) II, overexpression of which suppresses the phosphatidylserine biosynthetic defect of a PSS I-lacking mutant of CHO-K1 cells. J Biol Chem. 1997;272:19133–19139. doi: 10.1074/jbc.272.31.19133. [DOI] [PubMed] [Google Scholar]
- Martinez M, Vazquez E, Garcia-Silva MT, Manzanares J, Bertran JM, Castello F, Mougan I. Therapeutic effects of docosahexaenoic acid ethyl ester in patients with generalized peroxisomal disorders. Am J Clin Nutr. 2000;71:376S–385S. doi: 10.1093/ajcn/71.1.376s. [DOI] [PubMed] [Google Scholar]
- Mata de Urquiza A, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, Perlmann T. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science. 2000;290:2140–2144. doi: 10.1126/science.290.5499.2140. [DOI] [PubMed] [Google Scholar]
- Martinez M. Severe deficiency of docosahexaenoic acid in peroxisomal disorders: a defect of delta 4 desaturation? Neurology. 1990;40:1292–1298. doi: 10.1212/wnl.40.8.1292. [DOI] [PubMed] [Google Scholar]
- Matthews DB, Morrow AL. Effects of acute and chronic ethanol exposure on spatial cognitive processing and hippocampal function in the rat. Hippocampus. 2000;10:122–130. doi: 10.1002/(SICI)1098-1063(2000)10:1<122::AID-HIPO13>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Greiner RS, Salem N., Jr Behavioral deficits associated with dietary induction of decreased brain docosahexaenoic acid concentration. J Neurochem. 2000;75:2563–2573. doi: 10.1046/j.1471-4159.2000.0752563.x. [DOI] [PubMed] [Google Scholar]
- Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy M, Hamilton J, Greiner R, Moriguchi T, Salem N, Jr, Kim HY. Differential effects of n-3 fatty acid deficiency on phospholipid molecular species composition in rat hippocampus. J Lipid Res. 2002;43:611–617. [PubMed] [Google Scholar]
- Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20:1179–1186. doi: 10.1128/mcb.20.4.1179-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima M, Kuge O, Akamatsu Y. Phosphatidylserine biosynthesis in cultured Chinese hamster ovary cells. I. Inhibition of de novo phosphatidylserine biosynthesis by exogenous phosphatidylserine and its efficient incorporation. J Biol Chem. 1986;261:5784–5789. [PubMed] [Google Scholar]
- Niu S, Mitchell D, Lim SY, Wen Z, Salem N, Jr, Kim HY, Litman B. N-3 deficiency in rats reduces G-protein coupled signal transduction in retinal rod outer segment membranes. J Biol Chem. 2004;279:31098–31104. doi: 10.1074/jbc.M404376200. [DOI] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Okada M, Amamoto T, Tomonaga M, Kawachi A, Yazawa K, Mine K, Fujiwara M. The chronic administration of docosahexaenoic acid reduces the spatial cognitive deficit following transient forebrain ischemia in rats. Neuroscience. 1996;71:17–25. doi: 10.1016/0306-4522(95)00427-0. [DOI] [PubMed] [Google Scholar]
- Pawlosky R, Salem N., Jr Ethanol exposure causes a decrease in docosahexaenoic acid and an increase in docosapentaenoic acid in feline brain and retinas. Am J Clin Nutr. 1995;61:1284–1289. doi: 10.1093/ajcn/61.6.1284. [DOI] [PubMed] [Google Scholar]
- Rouser G, Nelson GJ, Fleischer S, Simon G. Biological membranes: Physical fact and function. In: Bourne GH, editor. The structure and function of nervous tissue. Academic Press; New York: 1969. p. 52. [Google Scholar]
- Salem N, Jr, Litman B, Kim HY, Gawrisch K. Mechanism of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36:945–959. doi: 10.1007/s11745-001-0805-6. [DOI] [PubMed] [Google Scholar]
- Sawazaki S, Salem N, Jr, Kim HY. Lipoxygenation of docosahexaenoic acid by rat pineal body. J Neurochem. 1994;62:2437–2447. doi: 10.1046/j.1471-4159.1994.62062437.x. [DOI] [PubMed] [Google Scholar]
- Silvers JM, Tokunaga S, Berry RB, White AM, Matthews DB. Impairments in spatial learning and memory: ethanol, allopregnanolone, and the hippocampus. Brain Res Brain Res Rev. 2003;43:275–284. doi: 10.1016/j.brainresrev.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Soderberg M, Edlund C, Kristensson K, Dallner G. Fatty acid composition of brain phospholipids in aging and in Alzheimer's disease. Lipids. 1991;26:421–425. doi: 10.1007/BF02536067. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Vance JE. Cloning and expression of murine liver phosphatidylserine synthase (PSS)-2: differential regulation of phospholipid metabolism by PSS1 and PSS2. Biochem J. 1999;342:57–64. [PMC free article] [PubMed] [Google Scholar]
- Sturbois-Balcerzak B, Stone SJ, Sreenivas A, Vance JE. Structure and expression of the murine phosphatidylserine synthase-1 gene. J Biol Chem. 2001;276:8205–8212. doi: 10.1074/jbc.M009776200. [DOI] [PubMed] [Google Scholar]
- Thomas CC, Deak M, Alessi DR, van Aalten DM. High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol. 2002;12:1256–1262. doi: 10.1016/s0960-9822(02)00972-7. [DOI] [PubMed] [Google Scholar]
- Traynor-Kaplan AE, Thompson BL, Harris AL, Taylor P, Omann GM, Sklar LA. Transient increase in phosphatidylinositol 3,4-bisphosphate and phosphatidylinositol trisphosphate during activation of human neutrophils. J Biol Chem. 1989;264:15668–15673. [PubMed] [Google Scholar]
- Vance JE, Vance DE. Phospholipid biosynthesis in mammalian cells. Biochem Cell Biol. 2004;82:113–128. doi: 10.1139/o03-073. [DOI] [PubMed] [Google Scholar]
- Varticovski L, Harrison-Findik D, Keeler ML, Susa M. Role of PI3-kinase in mitogenesis. Biochim Biophys Acta. 1994;1226:1–11. doi: 10.1016/0925-4439(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Wen Z, Kim HY. Alterations in hippocampal phospholipid profile by prenatal exposure to ethanol. J Neurochem. 2004;89:1368–1377. doi: 10.1111/j.1471-4159.2004.02433.x. [DOI] [PubMed] [Google Scholar]
- Wen Z, Kim HY. Inhibition of Phosphatidylserine Biosynthesis by Ethanol in Developing Rat Brain. J Neurosci Res. 2007;85:1568–1578. doi: 10.1002/jnr.21263. [DOI] [PubMed] [Google Scholar]
- Willatts P, Forsyth JS, Di Modugno MK, Varma S, Colvin M. Effect of long-chain polyunsaturated fatty acids in infant formula on problem solving at 10 months of age. Lancet. 1998;352:688–691. doi: 10.1016/s0140-6736(97)11374-5. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Yasuda A, Kawazato H, Sakai K, Shimada T, Takeshita M, Yuasa S, Kobayashi T, Watanabe S, Okuyama H. Synaptic vesicle ultrastructural changes in the rat hippocampus induced by a combination of alpha-linolenate deficiency and a learning task. J Neurochem. 1997;68:1261–1268. doi: 10.1046/j.1471-4159.1997.68031261.x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Hamilton JH, Salem N, Jr, Kim HY. N-3 Fatty acid deficiency in the rat pineal gland: Effects on phospholipid molecular species composition and endogenous 12-HETE and melatonin levels. J Lipid Res. 1998;39:1397–1403. [PubMed] [Google Scholar]