

Abstract
INK4A/ARF mutations are acquired in bcr/abl+ lymphoid blast phase chronic myelogenous leukemia (CML) and bcr/abl+ acute lymphoblastic leukemia (ALL). Donor lymphocyte infusion and graft versus leukemia are generally ineffective in such ALL’s, while GVL is highly active against bcr/abl+ CML that does not have a lesion in the INK4A/ARF locus. The mechanisms for the ineffectiveness of GVL are not fully known and it is possible that intrinsic resistance of acute lymphoid leukemias to immune effectors associated with allogeneic GVL may contribute to ineffectiveness. This work tested the hypothesis that INK4A/ARF mutations that are associated with transformation of bcr/abl+ CML to an ALL phenotype and that are associated with increased resistance to apoptosis render ALL cells insensitive to allogeneic immune responses to minor histocompatibility antigens (mHA). Murine acute pre-B ALL’s were induced by transfer of the human p210 bcr/abl gene into bone marrow of INK4A/ARF null mice. These ALL lines were then studied in a murine model of MHC-matched, mHA-mismatched allogeneic BMT. In vivo growth of these ALL’s was inhibited in allogeneic transplants characterized by active allogeneic immune responses compared to their behavior in syngeneic transplants. In vitro ALL’s with INK4A/ARF, p210 bcr/abl, or p190 bcr/abl mutations remained sensitive to anti-mHA cytolytic T cells. In addition, the ALL’s were capable of inducing primary immune responses to mHA’s in vivo. Thus, ALL’s with INK4A/ARF or bcr/abl mutations are not intrinsically resistant to allogeneic T cell responses suggesting that active immunotherapies against mHA have potential to control such acute lymphoblastic leukemias.
INTRODUCTION
Bcr/abl mutations are the defining genetic feature in chronic myelogenous leukemia (CML). The natural history of CML is that the leukemia can persist for a long period as a chronic disease, but that it eventually acquires additional genetic lesions and is transformed into an acute leukemia, either lymphoid or myeloid. Allogeneic hematopoietic stem cell transplantation is a potentially curative treatment of choice for patients with CML. Clinical studies involving transplant have demonstrated that the allogeneic graft versus leukemia (GVL) effect is extremely potent in chronic phase CML; relapses after allogeneic transplant are relatively uncommon and many relapses can be effectively treated with infusion of donor lymphocytes. In contrast, GVL is negligible when CML converts to an acute lymphoid blast phase (1–4). The mechanisms of this insensitivity to immune control are not fully understood, and multiple hypotheses have been advanced (5). A single explanation is unlikely and mechanisms may ultimately prove to be related to particular mutations within classes of leukemias. One hypothesis we have considered is that mutations that are acquired by leukemias that drive it toward an acute lymphoid phenotype may also render the leukemia cell intrinsically resistant to effector cells that mediate GVL.
Remarkably, INK4A/ARF mutations occur in 50% of cases of lymphoid blast crisis of CML (6). In addition, one-quarter of bcr/abl+ acute lymphoblastic leukemia (ALL) cases also have lesions near the INK4A/ARF locus (7;8). The INK4A/ARF locus contains two tumor suppressors that have separate promoters but share exons. Mutations or deletion within this gene locus can impact the negative regulation of Rb-mediated cell cycle progression (INK4A) and p53 degradation (ARF), and are associated with both murine and human carcinogenesis (9;10). In addition, loss of function in the INK4A/ARF locus also renders lymphocytes resistant to induction of apoptosis from a variety of stimuli (11;12). These observations suggest the hypothesis that INK4A/ARF null, bcr-abl+ ALL cells are intrinsically resistant to GVL effectors possibly due to apoptotic resistance.
We have employed a murine model that provides an opportunity to test this hypothesis. Pre-B acute lymphoblastic leukemias have been induced by insertion of human bcr/abl cDNAs into nonmalignant marrow cells of INK4A/ARF null mice. The leukemias have short latency and in vivo behave like human ALL. The leukemias are on a C57BL/6 background that allows us to exploit our well-characterized model of MHC-matched, minor histocompatibility antigen (mHA) mismatched BMT in which mHA antigens have been characterized at a genetic and peptide level.
MATERIALS AND METHODS
Mice
The C57BL/6 INK4A/ARF null mouse carries a deletion of the INK4A/ARF locus eliminating genes for both p16 (INK4A) and p19 (ARF) (National Cancer Institute, Frederick, MD) (13). C3.SW mice (Jackson) were transplant donors and C57BL/6 mice (National Cancer Institute, Frederick, MD) were recipients. The mice are MHC antigen matched (H2b) but minor histocompatibility antigen (mHA) mismatched at many loci (H1, H3, H7, H8, H9, H13). C3.SW are H2b and were derived from an 11 generation back cross of C3H against a non-inbred H2b donor strain (14).
Cell lines
Table 1 summarizes relevant features of the leukemia cell lines used in these experiments. Primary marrow cells from INK4A/ARF null mice were transduced with a retroviral vector encoding the human p210 bcr/abl cDNA (15). The MSCV-BCR/ABL-IRES-GFP vector was kindly provided by Dr. Richard Van Etten. The neo gene was removed from this vector by digestion with Nco I and Cla I, and the YFP gene was inserted by standard cloning procedure to yield the MSCV-Nup98/HoxA9-YFP vector used in the present study. Retroviral vector plasmids were transfected into phoenix-eco cells (ATCC) using lipofectamine 2000 per manufacturer’s instructions (10 micrograms DNA per 100,000 cells in a six-well tissue culture dish). At 36 hours post-transfection, viral supernatants were collected, filtered, and stored at −80 degrees centigrade. Retrovirus treated marrow cells (2×105) were infused iv into irradiated (600 cGy) C57BL/6 recipients and spontaneous acute lymphoblastic leukemias (ALL) emerged within three weeks. NSTY lines were derived by in vitro culture of splenocytes from these leukemia bearing mice; no cytokine supplementation was required (16;17). The male acute myeloid leukemia line (AML-m) was derived from a C57BL/6 male infused with wild type male C57BL/6 marrow transduced in vitro with vectors encoding p210 bcr/abl and NUP98/HOXA9 (15;15;18). C1498 (ATCC) is a spontaneous C57BL/6 acute NKT cell leukemia (19). ASLN cells were derived from a C57BL/6 transgenic mouse created by insertion of a C57BL/6 oocyte of a human p190 bcr/abl cDNA under the control of the immunoglobulin heavy chain enhancer E-mu and the murine Mb-1 promoter. YAC (TIB-160, ATCC) is an A/Sn strain lymphoma line that is sensitive to NK cells and is the conventional target in murine NK cytolysis assays. P815 cells are a DBA/2 strain (H2d) mastocytoma (TIB-64, ATCC).
Table 1.
Leukemia cell lines used in these experiments.
| Name | Phenotype | BCR/ABL | INK/ARF null | Female/Male |
|---|---|---|---|---|
| NSTY-1 | pre-B ALL | p210 | Yes | Female |
| NSTY-6 | pre-B ALL | p210 | Yes | Male |
| NSTY-7 | pre-B ALL | p210 | Yes | Male |
| AML-f | Poorly differentiated AML | p210 | No | Female |
| AML-m | Poorly differentiated AML | p210 | No | Male |
| ASLN | pre-B ALL | p190 | No | Female |
| C1498 | NKT | Normal | Unknown | Female |
Bone marrow transplant
Following 1100 cGy TBI (given in two equal divided doses 14–16 hours apart) and ip 5-fluoruracil (125 mg/kg) C57BL/6 recipients were infused with 4 × 106 marrow cells plus 10 × 106 splenocytes from normal C57BL/6 mice or C3.SW mice immunized against C57BL/6 splenocytes (20–22). They were housed in conventional rooms with food and water ad libitum. From 2 days before BMT until day 14, the water was acidified (pH 2.5) and supplemented with 2 g/L neomycin sulfate (Sigma, St. Louis, MO). We and other investigators have observed that with use of C3.SW donors (with 5–10 × 106 T cells in the graft) and C57BL/6 recipients fatal graft versus host disease is observed in a significant number of recipients over a 6 week period (22). In experiments in which leukemia progression in vivo was measured C57BL/6 mice underwent myeloablation with radiation and 5-FU and then were simultaneously infused with 4×106 bone marrow cells, 1×107 splenocytes, and 1×104 NSTY-1 acute lymphoblastic leukemia cells. The source of splenocytes varied between experimental groups with some recipients receiving marrow and spleen cells from C3.SW mice that had been vaccinated against C57BL/6 splenocytes to enhance alloreactivity, while other control mice received cells from C3.SW that had not been vaccinated or from normal C57BL/6 mice.
Generation of alloreactive T cells
C3.SW mice were vaccinated sc with 107 25 Gy irradiated splenocytes two to three times at two week intervals. Splenocytes from C3.SW mice immunized against C57BL/6 splenocytes were restimulated in vitro four days at a 5:3 ratio at 107 cells/ml with 25 Gy-irradiated C57BL/6 splenocytes that express all the known minor histocompatibility antigens. In other experiments designed to generate CTL with a single specificity for one minor antigen peptide C3.SW responder cells were stimulated with syngeneic C3.SW splenocytes preincubated with exogenous minor histocompatibility antigen peptide. Cells were incubated at 37° C in R10S media (RPMI, 10% FCS, 200 mM glutamine, 104 U/ml penicillin/streptomycin, non-essential amino acids (1 ml per 100 ml medium), 100 mM Na pyruvate, 50 mM beta-mercaptoethanol). No cytokines were added to the culture (23). T cells generated from these cultures were used as effector cells with target leukemia cells and a flow cytometry based leukemia cell inhibition assay was used to measure the capacity of the effector cells to inhibit the leukemia target cells.
Natural killer cells
NK cells were selected from freshly isolated splenocytes using positive selection with paramagnetically labeled DX5 monoclonal antibody (Miltenyi). NK cells were then directly placed as effector cells in the in vitro leukemia cell inhibition assays. In some experiments mice were injected ip twice at 24 hour intervals with 100 micrograms of polyI:C (Sigma) in 0.5 ml sterile PBS. DX5 positive cells were selected one day after the second injection. Isolated NK cells were used as effector cells with target leukemia cells and a flow cytometry based leukemia cell inhibition assay was used to measure the capacity of the effector cells to inhibit the leukemia target cells.
Leukemia cell inhibition assay
The assay measures the relative size of leukemia target cell populations cocultured for 3 days with potential cytolytic effector cells in microcultures in 96 well plates. Five thousand target cells were placed with effector cells at specified effector-to-target cells ratios. Following three days of culture 4000 fluorescent microbeads were added to each well and immediately thereafter the well was harvested and examined flow cytometrically. Regions were defined corresponding to the beads and viable leukemia cells and the number of events in each region was recorded. The number of viable leukemia cells in a well was calculated using the following formula: (leukemia cells in well) = (leukemia cells counted) × (constant number of fluorescent beads added to well/number of fluorescent beads counted). The percent leukemia cells surviving in wells with effector cells was calculated with the following formula: Percent leukemia cells surviving = (number of leukemia cells in well with effectors/number of leukemia cells in wells without effector cells) × 100. Duplicates or triplicates of each condition were measured.
ELISPOT
106 splenocytes were added to wells preincubated with IFN-γ capture antibody and stimulated for 48 h with HY peptides (Uty, Dby, and Smcy) or irrelevant peptide (WPRPQIPP)(5 µg/well) (23;24). IFN- γ release was detected with biotinylated anti-IFN- γ antibody (Caltag) and streptavidin peroxidase (24).
Flow cytometry
Conventional analytic flow cytometry was performed using a Becton Dickinson FACScan with analysis performed by Cellquest software or WinMDI. Directly labeled monoclonal antibodies for the cell surface markers specified in the text were obtained from Pharmingen or Caltag.
RESULTS
Introduction of the p210 bcr-abl gene into INK4A/ARF null bone marrow produces acute pre-B lymphoblastic leukemia
Like Williams et al. (10) we have observed that insertion of a human p210 bcr-abl cDNA into marrow of INK4A/ARF null mice induces acute leukemias in vivo. The leukemias (NSTY) grow well in vitro without cytokine support. They express B lineage markers (B220+, CD19+), but are negative for T cell (CD3−), granulocyte (Gr-1-), monocyte (CD11b−) and erythroid (Ter119−) markers (Figure 1). They express MHC I (H2-D+ and H2-K+), but are negative for MHC II (Ia) and surface immunoglobulin (data not shown). In addition, the detailed surface antigen expression profile surface is characteristic of murine late pro-B/early pre-B cells (25;26): AA4.1 +, IL7R+, c-Kit-, CD43 +/−, CD2 + (data not shown). When 104 NSTY-1 cells are intravenously injected into normal C57BL/6, 100% of mice develop leukemia within 3 weeks with infiltration of marrow, blood, spleen and brain.
Figure 1. Flow cytometric phenotype of the p210 bcr/abl+, INK4A/ARF null acute lymphoblastic leukemia.
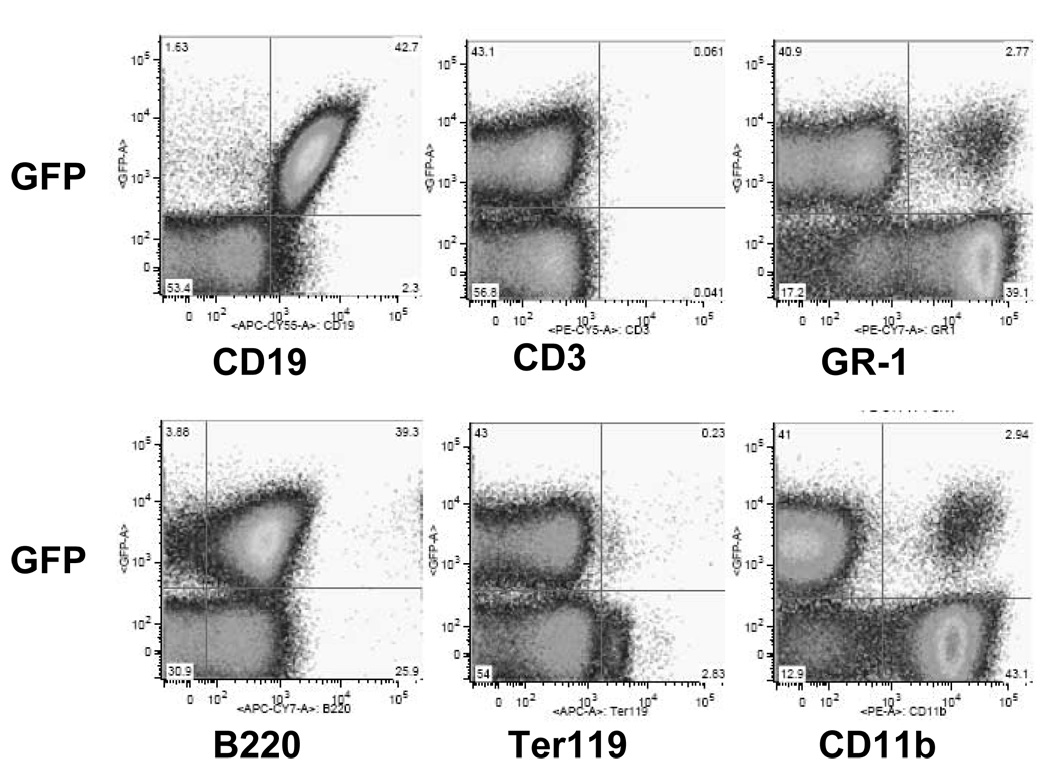
Bone marrow from INK4A/ARF null mice developing acute leukemia following injection of p210 bcr/abl GFP+ retrovirus treated syngeneic bone marrow was assessed by flow cytometry. GFP is on the Y-axis and identifies leukemia cells expressing bcr/abl. (The bcr/abl vector coexpresses GFP through an IRES element and thus GFP can be used as a marker gene for vector expressing cells.) Staining with fluorochrome labeled monoclonal antibodies specific for various hematopoietic cell surface markers is displayed on the X-axis.
Growth of p210 bcr/abl+ INK4A/ARF null acute pre-B ALL cells (NSTY) is inhibited in recipients undergoing bone marrow transplantation with alloreactive grafts
A simple but direct test of the hypothesis that INK4A/ARF null leukemias are resistant to immune effectors is to compare their progression from a state of minimal residual disease at the time of bone marrow transplant in transplant recipients experiencing alloreactivity with that in groups that experience less alloreactivity. If the leukemias were intrinsically resistant to allogeneic effects, the rate of leukemia progression and time to mortality from leukemia should be similar among the transplant groups. We tested this in a well characterized model of MHC-matched, mHA mismatched transplant. C3.SW donors primed against C57BL/6 splenocytes generate anti-C57BL/6 T cell responses including peptide-epitope defined CD8 T cell responses directed against mHAs H7, H3 and H13 (23). Such vaccination substantially increases the alloreactivity of the donor graft (22). Two control groups that have much less alloreactivity were also studied: unprimed C3.SW donors or normal C57BL/6 donors. In the transplants normal C57BL/6 females were transplanted with cells from either syngeneic normal C57BL/6, unprimed C3.SW, or C3.SW previously primed against C57BL/6. To establish a uniform burden of minimal residual disease 1×104 NSTY-1 acute leukemia cells were mixed with each graft and infused on the day of transplant. Recipients in the control groups died of progressive leukemia within 14 days. In contrast, mice receiving primed allogeneic grafts exhibited prolonged survival (Figure 2). Leukemia burden was assessed by flow cytometry on peripheral blood and/or marrow in randomly selected mice from either cohort at 14 days. In syngeneic transplants (sampled mice n=3) marrow leukemia content was 65 ± 4% (average ± sem), significantly greater than in the primed allogeneic transplant group (sampled mice n=6) where it was 16 ± 6 % (p < 0.001 by two tailed unpaired t test). Surviving animals were euthanized at 4 weeks, two weeks after the last animal in the syngeneic group had died. Leukemia cell content in marrow was measured in two of these mice, with 0.3 and 5% leukemia cells detected (data not shown). Histological assessment of animals for GVHD was not performed since the purpose of the experiment was to measure the rate of leukemia progression in the two environments and because other published work from our lab has shown that fatal GVHD is consistently observed at 5–10 weeks using this transplant protocol (22).
Figure 2. Mice receiving allogeneic transplant survive longer with the acute lymphoblastic leukemia compared to controls receiving syngeneic transplant.

Following myeloablation C57BL/6 mice were simultaneously infused with 4 × 106 bone marrow cells, 1 × 107 splenocytes, and 1 × 104 NSTY-1 acute lymphoblastic leukemia cells (to establish a uniform burden of minimal residual disease) on the day of transplant. “Primed Allogeneic” mice (n=19) received marrow and spleen cells from C3.SW mice that had been vaccinated against C57BL/6 splenocytes (not NSTY-1) to enhance alloreactivity. “Unprimed Allogeneic” mice (n=14) received cells from C3.SW that had not been vaccinated. “Syngeneic” mice (n=17) received cells from normal C57BL/6 mice. Mice were followed for survival for up to one month and then were sacrificed. Flow cytometry performed on blood and marrow from samples from each group demonstrated NSTY-1 cells in animals that appeared ill. Data are pooled from three experiments. Survival proportions were compared using the logrank test.
Susceptibility to mHA antigen specific allogeneic T cells
The slower progression of the pre-B leukemias suggested that INK4A/ARF null leukemia cells were sensitive to some component of the allogeneic response. Within this transplant model CD8 mHA-specific T cells play a significant role in the allogeneic response. In our transplant model the donor C3.SW and recipient C57BL/6 mHA differences at H3, H7 and H13 are defined at a peptide level, and peptide-specific CD8 CTL’s can be readily generated. We tested the hypothesis that INK4A/ARF null leukemia cells remain sensitive to such mHA-specific CD8 T cells. Primed C3.SW donor or naïve C57BL/6 splenocytes were stimulated in vitro with mHA peptides and used as effectors in leukemia inhibition assays in vitro. Donor strain allogeneic T cells significantly inhibited the growth of INK4A/ARF null leukemia cells while syngeneic T cells did not (Figure 3A). The sensitivity of NSTY-1 was similar to that of C1498, another C57BL/6 NKT leukemia line known to be sensitive to mHA-specific T cells (panel 3B).
Figure 3. Sensitivity of acute lymphoblastic leukemia cells to allogeneic alloreactive T cells.

Leukemia cell inhibition assays were performed using alloreactive T cells from C3.SW mice immunized against C57BL/6 splenic allogeneic minor histocompatibility antigens as effectors. In panel (A) female NSTY-1 were the leukemia targets while in panel (B) C1498 cells were the targets. Leukemia cells were cocultured with alloreactive T cells for 48 hours and leukemia cell number was compared to the number of leukemia cells in wells that did not have T cells. “Allogeneic” effectors were derived from allogeneic C3.SW mice previously sensitized to C57BL/6 splenocytes while “syngeneic” effectors were from normal syngeneic C57BL/6 splenocytes. Effector cells had been restimulated in vitro with a single mHA peptide (H3, H7 or H13) prior to their use in the leukemia cell inhibition assays. Each condition was performed in duplicate in this experiment performed once. Error bars represent standard error of the mean. Inhibition with allogeneic cells was compared to inhibition with syngeneic cells. In all comparisons p < 0.05 by two tailed unpaired t test with the exception of C1498 target 12.5:1 where p = 0.056.
We extended these experiments examining intrinsic resistance of p210+ bcr/abl INK4A/ARF null ALL leukemia cells by comparing their sensitivity to CTL’s to that of p210+ bcr/abl acute myeloid leukemia cells induced in normal C57BL/6 mice. Figure 4 demonstrates that the sensitivity profiles of the ALL and AML lines were very similar.
Figure 4. p210 bcr/abl+ INK4A/ARF null pre-B ALL cells and p210 bcr/abl+ AML cells exhibit similar sensitivity to allogeneic alloreactive T cells.

Leukemia cell inhibition assays were performed using NSTY-1 or AML-f leukemia cells as targets. Effector cells were generated in vitro using intact C57BL/6 splenocytes since these cells express all of the known minor histocompatibility antigens. Either primed allogeneic C3.SW or normal syngeneic C57BL/6 splenocytes were restimulated in vitro for 4 days and then used as effector cells. The experiment was performed once. Each condition was performed in triplicate. Error bars represent standard error of the mean. Inhibition with allogeneic cells was compared to inhibition with syngeneic cells. In all comparisons p < 0.01 by two tailed unpaired t test with the exception of AML-f target at 25:1 where p = 0.78.
These experiments had suggested that neither the p210 bcr/abl nor the INK/ARF mutations conferred substantial resistance to CTL’s. While the p210 bcr/abl mutation is the most common, a substantial number of bcr/abl+ ALL’s harbor an alternative mutation, the p190 bcr/abl. We assessed the sensitivity of a p190 bcr/abl ALL line (ASLN) and observed that its sensitivity to anti-mHA CTL’s was very similar to that of the control C1498 leukemia line (Figure 5). In additional experiments we positively selected CD8 and CD4 T cells from alloreactive donors and used them as effectors in leukemia cell inhibition assays. The CD8 fractions were significantly more potent than CD4 fractions in inhibition of both ASLN and NSTY-1 ALL lines; however, the CD4 fraction still mediated some inhibition of the leukemia cells. Neither T cell population inhibited the unrelated DBA/2 strain (H2d) P815 mastocytoma (Figure 6).
Figure 5. p190 bcr/abl+ pre-B ALL cells exhibit sensitivity to allogeneic alloreactive T cells.

Leukemia cell inhibition assays were performed using pre-B ASLN or C1498 leukemia cells as targets. Effector T cells were from short term cultures in which irradiated C57BL/6 splenocytes were used to restimulate spleen cells from either allogeneic C3.SW mice immunized against normal C57BL/6 splenocytes or from normal C57BL/6 mice. Similar results for ASLN have been seen in three leukemia inhibition assays. Error bars represent standard error of the mean. Values for allogeneic were compared to syngeneic for each effector to target ratio. In all comparisons p < 0.002 by two tailed unpaired t test.
Figure 6. Inhibition of NSTY-1 and ASLN by alloreactive CD8 and CD4 T cells.

Spleen cells from a donor strain C3.SW mouse previously vaccinated against recipient strain C57BL/6 spleen cells were restimulated in vitro with irradiated C57BL/6 spleen cells for 4 days. CD4 and CD8 T cells were positively selected from the cultures using paramagnetically labeled monoclonal antibodies (Miltenyi) and used as effector cells at a 10:1 effector:target ratio in a leukemia inhibition assay. Percent viable leukemia cells is plotted versus control leukemia cells not incubated with T cells. One of three similar experiments is presented. Mean and standard error of the mean are shown. Both CD8 and CD4 fractions inhibited ASLN and NSTY (p < 0.001 by two tailed unpaired t test), while P815 was not inhibited by either. CD8 inhibition was significantly greater than CD4 inhibition for both ASLN and NSTY (p < 0.01 by two tailed unpaired t test). DX5+ NK cells represented less than 0.6% of the pre-purification samples and less than 0.4% of the post-purification samples. The post-purification CD4 fraction contained only 1.3% CD8 cells. The post-purification CD8 fraction contained only 3.1% CD4 cells.
Natural killer cells emerge early after allogeneic transplant and could play a significant role in controlling leukemia in the early post-transplant period. We examined the sensitivity of the leukemia cell lines to NK cells. Neither the NSTY-1 ALL (p210 bcr/abl+, INK4A/ARF null) nor the ASLN ALL (p190 bcr/abl+) were inhibited in vitro by NK cells, unlike the NK sensitive YAC control cells (Figure 7). The AML-f cells (p210 bcr/abl+, INK4/ARF normal) exhibited modest sensitivity to NK cells.
Figure 7. p210 bcr/abl+, INK4A/ARF pre-B ALL (NSTY-1) and p190 bcr/abl+ pre-B ALL (ASLN) do not exhibit sensitivity to NK cells in vitro.

Acute leukemia lines were assessed for sensitivity to NK cells in vitro in leukemia cell inhibition assays. Yac cells, a known NK sensitive cell line, were used as positive control targets. NK cells were directly isolated from female C57BL/6 mice and used at 100:1 effector to target cell ratios. Mice were not pretreated with poly I:C. One of three representative experiments is presented. Error bars represent standard error of the mean. Two tailed unpaired t-test was used to determine if the mean for each cell line was less than 100% (i.e., significantly reduced by NK cells). Both Yac (p < 0.005) and AML-f ( p< 0.022) were inhibited by NK cells. NSTY-1, ASLN and C1498 were not inhibited by NK cells.
Pre-B ALL cells with the p210 bcr/abl and INK4A/ARF mutations are capable of inducing primary T cell responses in vivo
While the studies clearly demonstrated that the ALL cells were not intrinsically or even relatively resistant to allogeneic cytolytic T cells, it remained possible that the combinations of mutations had rendered the cells incapable of effective priming of an immune response. This would be an alternate hypothesis for explaining the poor immune control of ALL. Leukemia specific antigens have not been functionally or molecularly defined to date in our ALL models, and thus direct measurement of T cell responses to such putative antigens independent of responses to widely distributed minor histocompatibility antigens is not yet feasible. To circumvent this technical obstacle we chose to employ a physiologically relevant leukemia cell surrogate antigen. T cell responses to male histocompatibility antigens (HY) can be induced and readily measured in female mice, and provide a straightforward experimental approach for testing this alternate hypothesis. Live male leukemia cells were injected intravenously and two weeks later interferon-gamma release in response to HY peptides was directly assessed in freshly isolated splenocytes. Male bcr/abl+ AML cells that are known to elicit a male mHA HY-antigen specific T cell response in vivo were used as a positive control. We compared two male NSTY lines (NSTY-6 and NSYT-7) to this control (AML-m). Normal female C57BL/6 mice were injected with 104 male leukemia cells, and naïve female C57BL/6 served as a negative control. Two weeks later splenocytes were assayed directly in IFN- γ ELISPOT using HY peptide antigens. Figure 8 demonstrates a clear primary HY specific response in normal mice injected with male NSTY-6 or NSTY-7 cells.
Figure 8. Acute lymphoblastic leukemia cells are capable of inducing primary T cell responses in vivo.

ELISPOT assays were performed measuring interferon gamma secretion from mice that had been injected with 104 live male NSTY cells or live male AML-m cells. Ten days after leukemia cell injection spleen cells were removed and 106 spleen cells were placed in each well. HY peptides or irrelevant control peptides were also added to each well. HY specific spots = (spots in HY pulsed wells) − (spots in irrelevant peptide pulsed wells). One of two similar experiments is presented. The assay was performed in triplicate for each animal. “C57” indicates the animal was a normal female C57BL/6 mouse. “NSTY-6” and “NSTY-7” are male bcr/abl+, INK4A/ARF null acute lymphoid leukemia lines that were independently derived from male INK4A/ARF null mice. “AML-m” is a male bcr/abl+ AML line from a mouse that does not have the INK4A/ARF deletion. “n” is the number of mice in each group. The mean is presented and error bars represent standard error of the mean. T-test was used to compare mean spots in leukemia injected groups versus no leukemia group; p < 0.05 for each of the groups injected with leukemia.
DISCUSSION
These studies examined potential mechanisms for the relative ineffectiveness of allogeneic donor lymphocyte infusion and allogeneic graft versus leukemia effects in pre-B acute lymphoblasatic leukemias that harbor bcr/abl and INK4A/ARF mutations, common high-risk genetic lesions in human ALL. Specifically, experiments using murine leukemias induced by human mutations tested the hypothesis that such mutations render leukemias resistant to allogeneic effectors. Both in vivo and in vitro p210 bcr/abl+, INK4A/ARF null ALL lines were inhibited by allogeneic immune effectors. The leukemias remained exquisitely sensitive to allogeneic T cells directed against minor histocompatibility antigens. ALL cells harboring the p190 bcr/abl mutation also remained sensitive to anti-mHA T cells. The sensitivity of the p210 bcr/abl+ ALL’s was similar to p210 bcr/abl+ AML’s, contrary to results in clinical transplantation in which more potent GVL effects are observed in AML compared to ALL. In addition, we demonstrated that in vivo the ALL cells could effectively induce T cell responses to antigens in the ALL cells.
There are several limitations to these studies. First, the experiments examined the sensitivity of the leukemias to widely distributed minor histocompatibilty antigens, rather than hematopoietic specific or leukemia specific T cell responses. Thus, it remains conceivable that while the ALL’s remain sensitive to T cells directed against these ubiquitous mHA, they may be resistant to T cells directed against tissue specific or leukemia specific antigens. However, such resistance would not be related to lack of MHC class I expression, resistance to CTL effector mechanisms, blockade to induction of apoptosis, or inability to present antigens in general. The experimental model effectively rules out these mechanisms. Untested is the possibility that the ALL’s fail to synthesize and present potential hematopoietic antigens. We cannot test this hypothesis since we do not know the nature of such antigens and the nature of T cell responses to such antigens in this model. Second, the experiments assessing the rates of leukemia progression in the allogeneic transplants (Figure 1) did not characterize the GVHD in vivo. This was a conscious decision of experimental design based on years of experience with this model in which GVHD is reliably induced and GVHD related mortality occurs in 6–8 weeks. Our goal was not to determine if there would be long term survivors of the transplants or if leukemia-selective GVL could be induced. Rather, the goal of the experiments was to determine if the leukemias with the specific human genetic mutations were intrinsically resistant to allogeneic effects.
Thus, contrary to common belief (1–4) acute lymphoid leukemias remain susceptible to alloreactive donor T cells and can stimulate mHA specific T cell responses in vivo, suggesting that vaccines or adoptive cellular immunotherapies directed against mHA might be useful under some circumstances. However, still undefined are those circumstances in which this might be feasible. One hypothesis that is emerging from our studies is that relative kinetics of leukemia and T cell growth may be critical in determining the window of opportunity for immune intervention. In humans ineffective unmanipulated GVL may not be due to intrinsic resistance to immune control, but may be due to leukemia cell proliferation outpacing in vivo generated anti-mHA T cell responses. The INK4A/ARF mutation prevents cell cycle exit, producing shorter doubling times. Thus, effective immune control of such acute leukemias may require not only T cells that recognize leukemia cells in vivo but also agents that reduce leukemia growth rates until T cell responses peak kinetically. We are evaluating a variety of agents including conventional antimetabolites and leukemia specific tyrosine kinase inhibitors for their impact on the balance between generation of anti-mHA immune responses and leukemia population growth.
Acknowledgments
This work was supported in part by grant support from the National Institutes of Health (1R01CA10628)(C.A.M.), U.S. Department of Defense (W81XWH-05-1-0608)(C.T.J.), and the Brockport High School Leukemia Dance Marathon (C.A.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Collins RHJ, Shpilberg O, Drobyski WR, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J Clin Oncol. 1997;15:433–444. doi: 10.1200/JCO.1997.15.2.433. [DOI] [PubMed] [Google Scholar]
- 2.Collins RH, Jr, Goldstein S, Giralt S, et al. Donor leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplantation. 2000;26:511–516. doi: 10.1038/sj.bmt.1702555. [DOI] [PubMed] [Google Scholar]
- 3.Kolb HJ, Schattenberg A, Goldman JM, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. European Group for Blood and Marrow Transplantation Working Party Chronic Leukemia. Blood. 1995;86:2041–2050. [PubMed] [Google Scholar]
- 4.Peggs KS, Mackinnon S. Cellular therapy: donor lymphocyte infusion. Current Opinion in Hematology. 2001;8:349–354. doi: 10.1097/00062752-200111000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Kolb H-J, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. 2004;103:767–776. doi: 10.1182/blood-2003-02-0342. [DOI] [PubMed] [Google Scholar]
- 6.Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–4022. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- 7.Heerema NA, Harbott J, Galimberti S, et al. Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia. 2004;18:693–702. doi: 10.1038/sj.leu.2403324. [DOI] [PubMed] [Google Scholar]
- 8.Primo D, Tabernero MD, Perez JJ, et al. Genetic heterogeneity of BCR/ABL+ adult B-cell precursor acute lymphoblastic leukemia: impact on the clinical, biological and immunophenotypical disease characteristics. Leukemia. 2005;19:713–720. doi: 10.1038/sj.leu.2403714. [DOI] [PubMed] [Google Scholar]
- 9.Sharpless NE, Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutation Research. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 10.Williams RT, Roussel MF, Sherr CJ, Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shank-Calvo JA, Draheim K, Bhasin M, et al. p16Ink4a or p19Arf loss contributes to Tal1-induced leukemogenesis in mice. Oncogene. 2006;25:3023–3031. doi: 10.1038/sj.onc.1209326. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi T, Rufer N, MacDonald HR, et al. The tumor suppressor p16Ink4a regulates T lymphocyte survival. Oncogene. 2006;25:4110–4115. doi: 10.1038/sj.onc.1209437. [DOI] [PubMed] [Google Scholar]
- 13.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 14.Green MC, Witham BA. Handbook on genetically standardized Jax mice. 4 ed. Bar Harbor, Maine: Jackson Laboratory; 1991. [Google Scholar]
- 15.Neering SJ, Bushnell T, Sozer S, Ashton J, Rossi RM, Wang PY, et al. Leukemia stem cells in a genetically defined murine model of blast phase CML. Blood. 2007;110(7):2578–2585. doi: 10.1182/blood-2007-02-073031. Ref Type: Journal (Full) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Case SS, Price MA, Jordan CT, et al. Stable transduction of quiescent CD34(+)CD38(−) human hematopoietic cells by HIV-1-based lentiviral vectors. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2988–2993. doi: 10.1073/pnas.96.6.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jordan CT. Unique molecular and cellular features of acute myelogenous leukemia stem cells. Leukemia. 2002;16:559–562. doi: 10.1038/sj.leu.2402446. [DOI] [PubMed] [Google Scholar]
- 18.Dash AB, Williams IR, Kutok JL, et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7622–7627. doi: 10.1073/pnas.102583199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LaBelle JL, Truitt RL, LaBelle JL, Truitt RL. Characterization of a murine NKT cell tumor previously described as an acute myelogenous leukemia. Leukemia & Lymphoma. 2002;43:1637–1644. doi: 10.1080/1042819021000002974. [DOI] [PubMed] [Google Scholar]
- 20.Anderson LD, Savary CA, Mullen CA. Immunization of allogeneic bone marrow transplant recipients with tumor cell vaccines enhances graft-versus-tumor activity without exacerbating graft-versus-host disease. Blood. 2000;95:2426–2433. [PubMed] [Google Scholar]
- 21.Anderson LD, Mori S, Mann S, Savary CA, Mullen CA. Pretransplant tumor antigen-specific immunization of allogeneic bone marrow transplant donors enhances graft-versus-tumor activity without exacerbation of graft-versus-host disease. Cancer Res. 2000;60:5797–5802. [PubMed] [Google Scholar]
- 22.Anderson LJ, Petropoulos D, Everse LA, Mullen CA. Enhancement of graft-versus-tumor activity and graft-versus-host disease by pretransplant immunization of allogeneic bone marrow donors with a recipient-derived tumor cell vaccine. Cancer Res. 1999;59:1525–1530. [PubMed] [Google Scholar]
- 23.Mori S, El-Baki H, Mullen CA. An analysis of immunodominance among minor histocompatibility antigens in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2003;31:865–875. doi: 10.1038/sj.bmt.1704021. [DOI] [PubMed] [Google Scholar]
- 24.Natzke AM, Shaw JL, McKeller MR, et al. Hematopoietic stem cell recipients do not develop post-transplantation immune tolerance to antigens present on minimal residual disease. Biology of Blood & Marrow Transplantation. 2007;13:34–45. doi: 10.1016/j.bbmt.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Hardy RR, Kincade PW, Dorshkind K, Hardy RR, Kincade PW, Dorshkind K. The protean nature of cells in the B lymphocyte lineage. Immunity. 2007;26:703–714. doi: 10.1016/j.immuni.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Young F, Ardman B, Shinkai Y, et al. Influence of immunoglobulin heavy- and light-chain expression on B-cell differentiation. Genes & Development. 1994;8:1043–1057. doi: 10.1101/gad.8.9.1043. [DOI] [PubMed] [Google Scholar]