

1. Introduction
The drug tamoxifen, which opposes the action of estrogen in certain tissues and mimics its action in other tissues, has played a major role in the decline of breast cancer mortality rates over the past twenty years (1). This intriguing tissue-specific behavior was demonstrated in the National Surgical Adjuvant Breast and Bowel Project-sponsored Breast Cancer Prevention Trial (BCPT), which was launched in April of 1992. In the group of woman at high risk for breast cancer who were treated with tamoxifen, there was a remarkable 45% reduction in the incidence of breast cancer, demonstrating for the first time, that breast cancer cannot only be treated but also prevented (1). Such a positive outcome led researchers to release the results 14 months earlier than originally planned (1). The BCPT also demonstrated a reduced occurrence in bone fracture incidence (1). However, there was an increased incidence of endometrial cancer (33 cases versus 14 in the placebo group) (2, 3). This troubling result demonstrates the crucial need of fully understanding the potential carcinogenic mechanisms of tamoxifen, as well as related selective estrogen receptor modulators (SERMs), as they are in wide spread use as prophylactic agents in high risk but otherwise healthy individuals.
The most widely recognized mechanism for the carcinogenicity of tamoxifen is estrogen receptor mediated hormonal activity (4). As tamoxifen has been shown to stimulate uterine growth and induce estrogen-like changes in the genital tract of humans and other animals (5, 6), it is likely that tamoxifen stimulates estrogen-responsive growth regulatory genes (7). The regulation of gene transcription may be largely responsible for the carcinogenic effects of tamoxifen in the endometrium; however, an alternative initiation mechanism could involve bioactivation of tamoxifen to form either redox-active or electrophilic metabolites, including a carbocation (8, 9), an o-quinone (10), and a quinone methide (11) (Scheme 1). These metabolites have the potential to act as chemical carcinogens by modifying critical cellular macromolecules (12, 13, 14). A complete understanding of the relevant mechanisms of toxicity would allow for the design of new drugs which do not generate genotoxic or cytotoxic species while still maintaining their beneficial properties.
Scheme 1. Bioactivation of Tamoxifen.

Tamoxifen is hydroxylated by P450 to form 4-hydroxytamoxifen, which can be converted to a quinone methide via a 2-electron oxidation mechanism (11). Alternatively, 4-hydroxytamoxifen can be further hydroxylated at the 3 position to produce 3,4-dihydroxytamoxifen, which can then be oxidized to the o-quinone (10). The tamoxifen carbocation is formed by initial α-hydroxylation followed by O-sulfonation and subsequent sulfate dissociation (8).
Synthetic ligands such as tamoxifen and the related compound, raloxifene, belong to a class of molecules termed “Selective Estrogen Receptor Modulators” (SERMs). This term signifies their ability to act as estrogen agonists in some tissues (bone, liver, and cardiovascular system), antagonists in other tissues (breast and brain), and mixed agonists/antagonists in the uterus. Tamoxifen has greater stimulatory activity in the uterus compared to that of raloxifene (15, 16, 17) and the “Study of Tamoxifen and Raloxifene” (STAR) trial is currently underway to compare the safety and effectiveness of these two compounds for the prevention of breast cancer in post-menopausal women (18). Preliminary results of the STAR trial indicate that raloxifene works just as well as tamoxifen at reducing breast cancer risk, but does not increase the risk of endometrial cancer (19). A great deal of effort is being expended toward improving tissue selectivity of SERMs so that they are optimized for preventing and treating breast cancer while alleviating menopausal symptoms. Specifically, our current understanding of estrogenic physiology suggests that the ideal SERM would display estrogen agonist activity in bone, cardiovascular tissue, and the central nervous system, with antagonist effects in uterus and breast tissue. As a result, this ideal SERM would enhance bone formation, improve lipid profiles, and provide neuroprotection, without increasing risk of estrogen-dependent cancers.
1.1. Estrogen Receptor
Estrogens put forth their regulatory effects by binding to estrogen receptors (ERs), which induces conformational changes in protein structure that allow for receptor dimerization and interaction with coactivator molecules (Scheme 2) (20, 21). The subsequent transcriptional activation of genes occurs either through the binding of liganded ERs directly to estrogen response elements (EREs) in gene promoters, or indirectly through binding to other transcription factors (22, 23, 24, 25, 26, 27, 28, 29).
Scheme 2. Ligand-Dependent Transcriptional Activation by the ER.

Ligands put forth their regulatory effects by binding to ERs, which induces conformational changes in protein structure that allow for receptor dimerization and interaction with coactivator molecules (20, 21). Coregulators associate in a ligand-dependent manner with the ER. Coactivators enhance ligand-dependent transcriptional activation by the ERα (48, 49).
ERs α and β are members of the nuclear receptor superfamily of ligand-inducible transcription factors (30, 31). ERα is predominantly expressed in the breast, uterus, and vagina, whereas higher levels of ERβ relative to ERα are found in the central nervous system, cardiovascular system, gastrointestinal system, immune system, kidney, lungs, and bone (32). ERα regulates the differentiation and maintenance of neural, skeletal, cardiovascular, and reproductive tissues (33, 34). Compounds that modulate ERα transcriptional activity are used to treat osteoporosis, cardiovascular disease, and breast cancer (35, 36). The existence of distinct tissue expression profiles for ERα and ERβ suggests that tissues could be differentially targeted with receptor specific ligands (37). However, to date, the complete therapeutic utility of ERβ-selective ligands remains unclear (37). Several research groups have employed highly selective ERβ ligands to probe the physiological role of this receptor (37, 38, 39, 40). For example, using the ERβ selective antagonist, ERB-041, Harris et al. demonstrated that this receptor might be a useful target of certain inflammatory diseases (40).
ERα and ERβ are 96% homologous in their DNA binding domains and 53% identical in their ligand binding domains (LBDs) (41). All ERα ligands bind to the C-terminal LBD. The LBD recognizes numerous compounds differing in size, shape, and chemical properties (42). Ligands such as the endogenous 17β-estradiol (E2) and the synthetic estrogen diethylstilbestrol (DES) are pure agonists, while others such as ICI-164,384 function as pure antagonists (42).
SERMs function as antagonists in specific tissue and promoter contexts (43). Two different activation factors (AFs) mediate the transcriptional activation of ERα; AF-1 in the N-terminus and AF-2 in the LBD. AF-1 activity is regulated by growth factors through the MAP kinase pathway (44), while AF-2 activity is regulated through ligand binding (45). Numerous structural studies suggest that ligands regulate AF-2 activity by altering the structure of the LBD. Comparison of the structures of unliganded nuclear receptor (NR) LBDs to those of agonist-bound LBDs suggests that an agonist-induced conformational change involving the repositioning of helix 12 is essential for AF-2 activity (46). The structures of the ERα LBD with E2 and raloxifene bound show that both ligands bind at the same site within the LBD, but each ligand induces a different conformation of helix 12 (47). The alteration of helix 12 by raloxifene partially buries residues necessary for AF-2 activity, suggesting raloxifene and other antagonists block AF-2 function by disrupting the conformation of the AF-2 surface.
Several proteins associate in a ligand-dependent manner with the ERs (Scheme 2). These proteins are termed transcriptional coactivators because they enhance ligand-dependent transcriptional activation by the ERα and other NRs (48, 49). Coactivators SRC-1 and GRIP1 bind to agonist-bound LBDs for the ERα (and other NRs) using the putative AF-2 interaction surface (50). Certain coactivators, such as SRC-1 and GRIP1, recognize agonist-bound NR LBDs through a short signature motif, LXXLL (where L is leucine and X is any amino acid), termed the NR box (51, 52, 53, 54). A crystal structure of agonist DES bound to the ERα LBD and a peptide derived from the NR box II region of the coactivator GRIP1 indicates that the peptide binds in a short α helix to a hydrophobic groove on the surface of the LBD (42). In contrast, the structure of ERα bound to 4-hydroxytamoxifen, the active metabolite of tamoxifen, reveals that helix 12 destroys the coactivator recognition site by imitating interactions of the NR box peptide with the LBD.
It is now understood that the pharmacology of estrogens relies not only on the ligand and the estrogen receptors but also on third parties, such as gene promoter elements and coregulatory proteins (55). The ERs can interact with target genes either by binding directly to DNA response elements or through indirect tethering to other DNA binding transcription factors. Taking all of these factors into account, there appears to be a number of combinatorial possibilities for achieving tissue and gene-specific regulation of SERMs. For example, Shang and Brown revealed that the agonism of tamoxifen could be specifically attributed to the coactivator SRC1, which is present at a higher concentration in uterine cells compared to mammary gland cells (29). Therefore, the agonistic activity of tamoxifen in the uterus is a result of the conformation of the ligand-receptor complex, the promoter context (tethered interaction instead of direct DNA binding), and the level of specific coactivator. This estrogenic activity could be a major contributor to tamoxifen’s carcinogenic effects in the endometrium. However, there is a great deal of interest in additional carcinogenic mechanisms involving the bioactivation of tamoxifen to reactive metabolites.
1.2. Bioactivation of SERMs
There are at least four different classes of electrophilic metabolites that can be formed from SERMs; carbocations, quinone methides, diquinone methides, and o-quinones. The contribution of each electrophile to a particular SERM’s metabolic profile depends on the structure and reactivity of the SERM. Cytochrome P450 can hydroxylate tamoxifen and related compounds (toremifene, droloxifene, and idoxifene) at the α-position. Upon conjugation with sulfate and subsequent loss of the sulfate group, these SERMs form highly reactive carbocations which can react with DNA (Scheme 1) (8, 9); however, the detection of endometrial DNA-adducts in exposed women remains highly controversial (56, 57, 58). The second bioactivation pathway is exemplified by the hydroxylation of tamoxifen by P450 to form a 4-hydroxylated metabolite, which can be further oxidized to a classical quinone methide (11) (Scheme 1). The SERM acolbifene is also converted by P450 to a diquinone methide (59) (Scheme 3). In addition, the SERMs raloxifene (Scheme 4) (60) and desmethylated arzoxifene (DMA) (61) can be converted to diquinone methides. The raloxifene metabolite has recently been implicated in the inhibition of P450 3A4 and the alkylation of many other liver microsomal proteins (62, 63). The fourth bioactivation pathway for SERMs involves o-quinone formation and has been implicated in tamoxifen-mediated protein binding (Scheme 1) (64, 65). Clearly, the structure of a compound determines the formation, reactivity, and selectivity of the reactive intermediates formed. Understanding the influence of SERM structure on these properties would provide the valuable insight necessary for the design of new SERMs.
Scheme 3. Bioactivation of Acolbifene.

Acolbifene is converted to both a classical quinone methide and a diquinone methide by P450 (59).
Scheme 4. Bioactivation of Raloxifene.
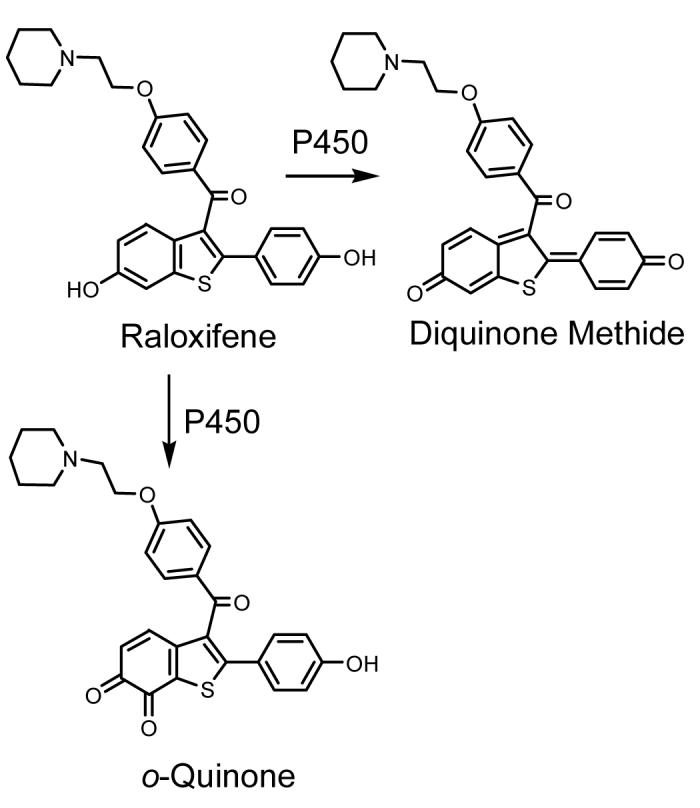
Raloxifene is oxidized by P450 to produce a diquinone methide and an o-quinone (60).
2. Classes of Selective Estrogen Receptor Modulators
SERMs are often categorized as second-, third-, and fourth-generation suggesting a progressive development in a process intended to improve the beneficial effects while reducing the harmful side effects associated with the earlier SERMs (41). This general classification of SERMs serves little purpose because the numbering is highly subjective and does not describe a logical transition from first to fourth. The prototype SERM, tamoxifen, and its derivatives, toremifene, droloxifene, and, idoxifene, belong to the chemical class of triphenylethylene compounds (Figure 1A). The second-generation SERM raloxifene and the third-generation SERM arzoxifene are benzothiophene compounds (Figure 1B), while the fourth-generation SERM alcolbifene is a benzopyran derivative (Figure 1C). As a result, for the purpose of this review, it is preferable to classify SERMs according to chemical family.
Figure 1. Chemical Structures of SERMs.

A) Triphenylethylene SERMs; B) Benzothiophene SERMs; C) Benzopyran SERMs
2.1. Triphenylethylene SERMs
SERMs of the triphenylethylene type have been extensively used for the treatment of hormone-dependent breast cancer. Tamoxifen is an oral drug first used in metastatic breast cancer in the early 1970s (66). Large clinical trials were initiated in the late 1970s and early 1980s to test the drug’s role as adjuvant therapy in early stage breast cancer. Observations of marked decreases in the development of contralateral breast cancer among tamoxifen recipients suggested potential for the drug in chemoprevention of breast cancer, and a large clinical trial to test the efficacy of tamoxifen in prevention of invasive breast cancer among women at increased risk was implemented in the United States in 1992 (66). There are a number of reports that have emphasized an increased incidence of premalignant and neoplastic endometrial changes (67, 68) which questioned the risk-benefit ratio for this medication (69, 70). In addition, several studies in animal models have raised concern over the safety of chronic treatment with this drug (71). For example, tamoxifen induces hepatocarcinomas (71), frequent and specific mutations in the p53 tumor suppresser gene (72), and mammary tumors (73) in rats.
Triphenylethylene derivatives (Figure 1A), with small structural changes compared to tamoxifen, are also in use for the treatment of breast cancer. Droloxifene, which has a 3-hydroxy substituent, has antiestrogenic activity in vitro which is equivalent or slightly greater than that of tamoxifen (74). Interestingly, unlike tamoxifen, droloxifene does not appear to induce liver tumors in rats and no DNA adducts have been detected from this analog (75). Similarly, the 4-iodo derivative of tamoxifen, idoxifene, has comparable antiestrogenic potency to tamoxifen without generating hepatocarcinogenic effects in rodents (76, 77). Toremifene also displays antiestrogenic activity in the mammary gland and its beneficial effects on bone mineral density and lipid profiles are similar to that of tamoxifen (41). However, also like tamoxifen, toremifene has stimulatory effects in the endometrium and is presently only used in postmenopausal women with metastatic breast cancer (41). Toremifene was found to form DNA adducts in rats, although the levels were significantly lower for toremifene compared to that of tamoxifen (78). The annual hazard rate (per 1,000 patients/year) of developing endometrial carcinoma in patients on toremifene therapy is 1.14, which is much less compared to that of tamoxifen (2.0) (79). It should be noted however, that in the absence of extensive epidemiology studies of all of these tamoxifen derivatives, the potential remains for genotoxic/cytotoxic effects to surface in the future.
2.2. Benzothiophene SERMs
Greater optimism has surrounded the profile of benzothiophene SERMs (Figure 1B) (80). Much of the enthusiasm relates to the fact that these drugs appear devoid of any agonist activity in the endometrium, while at the same time appearing to be potent antiestrogens in the breast and agonists in bone (15, 16, 17). A benzothiophene SERM, raloxifene, is in clinical use for the prevention and treatment of postmenopausal osteoporosis and is currently in the STAR trial, the largest breast cancer chemoprevention trial ever conducted (18). Very recent results of the STAR trial indicate that regular use of raloxifene works just as well as tamoxifen at reducing breast cancer risk in postmenopausal women at high risk, but appears to cause less of the dangerous side effects associated with tamoxifen use (19). Raloxifene is not being pursued in chemotherapy of breast cancer due to low efficacy in women with advanced breast cancer (81). The Raloxifene Use for The Heart trial (RUTH) is also currently being conducted to investigate whether raloxifene will decrease the incidence of coronary events and the risk of invasive breast cancer in postmenopausal women with heart disease or at risk of heart attack (82). Preliminary results of this trial indicate that raloxifene does not decrease or increase the risk of coronary events but could potentially reduce the risk of invasive breast cancer (82).
Arzoxifene is another member of the benzothiophene SERM family, and it has been reported to have a more favorable therapeutic and safety profile as compared to raloxifene (83, 84, 85, 86). In addition, unlike tamoxifen, arzoxifene acts as an estrogen antagonist in the uterus (87). In a phase II trial of arzoxifene in advanced endometrial cancer, arzoxifene showed high antitumor activity (88). Until very recently, the use of arzoxifene versus tamoxifen as a first-line therapy in metastatic breast cancer was being studied in a multicenter phase III trial (89). However, the trial was terminated early after initial results suggested that arzoxifene is inferior to tamoxifen with respect to time to progression (a measure of time from when the disease is treated until the disease starts to get worse) (89).
Despite the mixed results obtained from in vivo studies, several cell-based assays have demonstrated arzoxifene’s efficacy (90). Both arzoxifene and its active metabolite desmethylated arzoxifene (DMA) were shown to bind the ER with high affinity and inhibit estrogen dependent growth of MCF-7 breast cancer cells (90). When assayed for the inhibition of proliferation of these cells, Suh et al. reported arzoxifene (IC50, 0.4 nM) to be more potent than tamoxifen (IC50, 480 nM) or its active metabolite, 4-hydroxytamoxifen (IC50, 1.2 nM), with DMA being the most potent agent tested (IC50, 0.05 nM) and raloxifene being equal to that of arzoxifene (90).
2.3. Benzopyran SERMs
The so-called fourth generation SERM, acolbifene (Figure 1C), is an active benzopyran derivative of EM-800. Acolbifene is the most potent antiestrogen in terms of inhibition of both ERα and ERβ (91), and EM-800 is an extremely potent inhibitor of the proliferation of breast and endometrial cancer cells in culture (92, 93). EM-800 has also been shown to prevent the growth of estrogen-stimulated tumor xenografts in athymic mice (94) and prevent bone loss and cardiovascular disease in rats (95).
3. Bioactivation Pathways
Herein, we discuss four different electrophilic metabolites that can be formed from the SERMs shown in Figure 1. The triphenylethylene SERMs can form highly reactive carbocations, which have been implicated in DNA binding (8, 9, 56, 57, 58, 96). The second bioactivation pathway involves oxidation of tamoxifen and acolbifene to classical quinone methides (11). In addition, raloxifene (60), DMA (61), and acolbifene (59) can be converted to diquinone methides. The fourth bioactivation pathway involves o-quinone formation, which is generally a minor pathway for SERMs. In the following sections, we review these bioactivation pathways along with an example of formation of more complex reactive intermediates formed from SERMs.
3.1. Carbocations
Hydroxylation of tamoxifen at the α-carbon is predominately carried out by P450s 3A, 2D6, 2C9, 1A1, 1A2, and 1B1 (97). Following O-sulfonation, α-hydroxytamoxifen reacts preferentially with the exocyclic amino group of guanine in DNA to form two cis and two trans epimers of α-(N2-deoxyguanosyl)tamoxifen (98). Shibutani et al. reported the detection of these adducts in the endometrial tissue of 8 out of 16 women treated with tamoxifen (9), and more recently Kim et al. observed the same adducts in primary cultured human endometrial explants incubated with α-hydroxytamoxifen (99). However, due in part to the high concentrations of α-hydroxytamoxifen that Kim et al. used in their experiments (25 and 100 μM), several research groups have questioned the validity of this data (56, 100). As α-hydroxytamoxifen is a very minor metabolite of tamoxifen, it is most likely present at very low concentrations (ca. 0.2 nM) (101). In a separate study, Sharma et al. reported the use of high performance liquid chromatography (HPLC) with online postcolumn photochemical activation and fluorescence detection to detect the presence of (E)-α-(N2-deoxyguanosyl)tamoxifen in human endometrial explants incubated with tamoxifen (102). These results were also highly controversial due to the ambiguous characterization of the tamoxifen-DNA adducts (58). Specifically, this method could potentially lead to erroneous detection of tamoxifen-DNA adducts as the ability to distinguish between these adducts and noncovalently bound metabolites depends on the resolution of the HPLC system (100). To address this issue, two additional techniques were employed by Beland et al. to assay for tamoxifen-DNA adducts in human endometrial explants (58). Both HPLC coupled with electrospray ionization tandem mass spectrometry and 32P-postlabeling analysis were used and tamoxifen-DNA adducts were not detected with either method. The authors conclude that although these data are in conflict with that reported by Sharma et al., they are consistent with the limited amount of α-hydroxytamoxifen produced in human tissue and, more importantly the small amount of α-hydroxytamoxifen that is sulfonated (58).
Toremifene is significantly less potent than tamoxifen at causing DNA adduct formation in rats (78). Interestingly, human P450 3A4 was found to α-hydroxylate toremifene approximately six times faster than tamoxifen (103). However, α-hydroxytoremifene is not a good substrate for human hydroxysteroid sulfotransferase (hHST) (104). Therefore, O-sulfonation does not occur to a significant extent with this metabolite (104). Furthermore, the resulting toremifene α-sulfate is not as reactive with DNA compared with the tamoxifen α-sulfate (79). It has been proposed that this low reactivity may be brought on by steric hinderance caused by the bulky chlorine atom on the ethyl moiety of toremifene (79). Additionally, the electron-withdrawing chlorine atom may make elimination of the α-sulfate group less effective and in turn, slow the formation of the reactive carbocation that reacts with DNA (105). Although further experimental and clinical studies are required, the use of toremifene may be accompanied with less of the toxic effects associated with tamoxifen use in breast cancer patients and women receiving chemoprevention (79).
3.2. Classical Quinone Methides
The second bioactivation pathway involves oxidation of SERMs to quinone methides. Quinone methides are reactive metabolites of 4-alkylphenols and are most likely responsible for their cytotoxic effects (106, 107, 108). Like all reactive intermediates, the selectivity of a quinone methide depends on its rate of formation as well as its reactivity. Quinone methides differ structurally from quinones in that one of the carbonyl oxygens is replaced by a methylene or substituted methylene group. This renders the quinone methide a much more reactive electrophile with a reduced capacity for redox cycling (107, 108). Therefore, quinone methides generally react in biological systems via non-enzymatic Michael additions. With simple quinone methides, these reactions produce benzylic adducts of peptides, proteins, and nucleic acids (107, 108, 109). Theoretically, quinone methides can be characterized as resonance-stabilized carbocations (110, 111), due to the important contribution of the charged aromatic resonance form. This electron deficiency at the methylene carbon, along with inter- or intra-molecular factors which stabilize this resonance structure, lead to increased reactivity with cellular nucleophiles.
A relatively stable quinone methide is produced by initial P450-catalyzed aromatic hydroxylation of tamoxifen to yield 4-hydroxytamoxifen, followed by a P450-catalyzed direct 2-electron oxidation (Scheme 1) (112). This quinone methide is extremely long lived at physiological pH and temperature (t1/2 = 3 h) (Table 1) (11), most likely due to stabilization imposed by the two aryl substituents and the π system of the additional vinyl group. The vinyl substituent alone can decrease the reactivity of o-methoxyquinone methides by a factor of 100 (113) and an aryl substituent leads to a 230-fold increase in stability relative to quinone methides with an unsubstituted exocyclic methylene group (109). This π-stabilization, in addition to steric factors, completely changes the chemistry of the 4-hydroxytamoxifen quinone methide. Most quinone methides react instantaneously with GSH, whereas the 4-hydroxytamoxifen quinone methide has a half-life in the presence of GSH of approximately 4 min (11). This reaction with GSH is reversible as the GSH conjugates slowly decompose to regenerate the quinone methide (11). Tamoxifen-GSH conjugates were detected in liver microsomal incubations with 4-hydroxytamoxifen; however, none were observed in incubations with breast cancer cells (MCF-7) (11).
Table 1. Relative Reactivities of SERM Intermediates.
| SERM metabolite | Half-lifea | Reference |
|---|---|---|
| Tamoxifen Quinone Methideb | 3 h | (11) |
| Tamoxifen 3,4-o-Quinonec | 80 min | (10) |
| Toremifene Quinone Methideb | 60 min | (11) |
| Raloxifene 6,7-o-Quinoneb | 69 min | (60) |
| Acolbifene Quinone Methideb | 32 s | (59) |
| DMA Diquinone Methideb | 15 s | (61) |
| Raloxifene Diquinone Methideb | < 1 s | (60) |
| Tamoxifen Carbocationd | 22 μse | (143) |
Approximate half-life.
The disappearance of the quinoid species was followed by monitoring the decrease in UV absorbance at the appropriate wavelength in phosphate buffer (pH 7.4, 37 °C) (11, 59, 60, 61).
The rate of disappearance of the o-quinone was determined by monitoring the rate of apparent disappearance of the corresponding GSH conjugates by HPLC (10).
The carbocation was generated by laser flash photolysis in 40% acetonitrile (143).
Refers to the decay in acid where the amine is protonated (143).
Toremifene also undergoes oxidative metabolism to form a quinone methide. The 4-hydroxytoremifene quinone methide has a half-life at physiological pH and temperature of 1 h, while its half-life in the presence of GSH is approximately 6 min (Table 1) (11). The 4-hydroxytoremifene quinone methide reacts with two molecules of GSH and loses chlorine to yield the corresponding di-GSH conjugate (11). This reaction mechanism likely involves an electrophilic episulfonium ion intermediate, which could contribute to the potential cytotoxicity of toremifene (11). In microsomal incubations, 4-hydroxytoremifene was metabolized to di-GSH conjugates at 3 times the rate of 4-hydroxytamoxifen, although no GSH conjugates were observed in the incubations with MCF-7 cells (11). These data imply that the quinone methide may not play a significant role in the cytotoxicity or genotoxicity of tamoxifen and toremifene.
Interestingly, it has been shown that the polymorphic P450 isozyme 2D6 primarily catalyzes 4-hydroxylation of tamoxifen in the human liver (114). This result has been confirmed using human liver microsomes and expressed P450s (115), as well as by molecular modeling studies (116). There also appears to be a contribution from P450 2C9 and P450 3A4 as tamoxifen 4-hydroxylases in some individuals. As 5-10% of the population have defects in P450 2D6 (117) and there are several allelic variants of 2C9 (118), it is possible that these individuals will not experience the effects, if any, of the tamoxifen quinone methide. Alternatively, breast cancer patients with defective 2D6 or 2C9 who are receiving tamoxifen chemotherapy, are less likely to respond to the drug since 4-hydroxytamoxifen is 100 times more potent as an estrogen receptor antagonist (119, 120).
Acolbifene is also metabolized to form a classical quinone methide (Scheme 3) (59). The classical quinone methide is formed by oxidation at the C-17 methyl group and has a half life of 32 s at physiological pH and temperature (59). This is considerably shorter than that of the 4-hydroxytamoxifen quinone methide (3 h) (11) and indicates that the acolbifene quinone methide is an electrophile of intermediate stability (Table 1). The acolbifene quinone methide reacts with GSH to form five mono-GSH conjugates and five di-GSH conjugates (59). In addition, incubations of acolbifene with either tyrosinase or rat or human liver microsomes also yield the classical quinone methide-GSH conjugates (59).
In addition to reaction with GSH, the acolbifene quinone methide was determined to react with deoxynucleosides. In contrast to what was observed with raloxifene and DMA, the acolbifene quinone methide formed adducts with each of the four deoxynucleosides, with one of the major adducts resulting from reaction with the exocylic amino group of adenine (59). LC-MS-MS analysis of incubations of the DMA diquinone methide with deoxynucleosides yielded a negligible amount of one deoxyguanosine adduct (61), while no adducts were observed in incubations of deoxynucleosides with the raloxifene diquinone methide (unpublished results). Experiments were also performed to assess the toxicity of acolbifene, DMA, and raloxifene in the S30 cell line. DNA damage detected by the comet assay showed that both acolbifene and DMA induced more DNA damage than raloxifene (59). Both the stability and reactivity of the quinone methides might account for this difference. However, as these experiments were performed at very high SERM concentrations (μM), the results are not indicative of cellular induction of DNA damage in vivo.
3.3. Diquinone Methides
The third mechanism of bioactivation is the formation of diquinone methides, which occurs with the benzothiophene SERMs raloxifene (60) and arzoxifene (61), and the benzopyran SERM acolbifene (59). Raloxifene undergoes rapid absorption, extensive first-pass glucuronidation, and enterohepatic cycling after oral administration, resulting in an absolute bioavailability of just 2% (121, 122). Raloxifene can be bioactivated by rat and human liver microsomes to an electrophilic diquinone methide and in a very minor pathway to o-quinones (Scheme 4) (60, 63). The raloxifene diquinone methide is relatively short-lived, with a half-life of less than 1 s at physiological pH and temperature (Table 1) (60). This highly reactive metabolite has the potential to contribute to cytotoxicity likely through alkylation of proteins in vivo.
Raloxifene is associated with decreases in cytochrome P450 aromatase activity in human colon carcinoma cells (123) and is an irreversible inhibitor of P450 3A4 (63). As reported by Chen et al., the P450 inhibition in human liver microsomes was NADPH- and preincubation time-dependent, indicating that metabolism was required for inactivation. The KI and kinact values were about 9.9 μM and 6 min-1, respectively (63). The authors found that the loss of P450 activity was partially attenuated by the addition of GSH to the incubation mixture. Furthermore, raloxifene-GSH conjugates were detected in the incubation mixtures, derived from either a raloxifene arene oxide and/or a raloxifene diquinone methide. As the GSH adducts were almost completely abolished when the liver microsomes were pretreated with ketoconozole or an anti-P450 3A4 IgG, the bioactivation of raloxifene is most likely catalyzed by the 3A4 isoform (63). This suggests that bioactivation of raloxifene by P450 3A4 results in electrophilic intermediates that covalently modify the enzyme.
To identify microsomal proteins covalently modified by raloxifene metabolites, we synthesized a novel raloxifene covert oxidatively activated tag (COATag) in which the SERM was linked to biotin (Figure 2A) (62). In this study, the raloxifene COATag allowed identification of covalently modified proteins by immunoblotting and LC-MS-MS analysis (Figure 2B). The COATag was unreactive toward the selected model protein, purified human glutathione-S-transferase (hGST PI-1); however, upon co-incubation with tyrosinase, a covalently COATagged hGST protein adduct was isolated and identified by mass spectrometric analysis as modified at cys-47 (62). Similarly, the formation of COATagged proteins on incubation with rat liver microsomes was dependent upon the concentration of NADPH. These COATagged proteins were quantified and identified by SDS-PAGE and western blot analysis. Four major bands were observed in the blots from microsomal incubations, for which peptide mass spectrometry maps were obtained using in-gel digestion, followed by MALDI-TOF or ESI mass spectral analysis of the resulting peptide mixtures. Cytosolic glucose regulated protein (78 kDa, GRP78/BiP), protein disulfide isomerase isozyme A4 precursor (72 kDa, ERp72), protein disulfide isomerase isozyme A1 (57 kDa, PDIA1), protein disulfide isomerase isozyme A3 (58 kDa, ER-60) and microsomal glutathione S-transferase, (17 kDa, mGST1) were identified as targets (62). These data show that raloxifene produces a highly reactive intermediate that modifies tissue microsomal proteins with a low degree of selectivity, which might be an expected feature of a reactive intermediate with a relatively short lifetime.
Figure 2. Analysis of Raloxifene COATag Modified Proteins.

A) Structure of the raloxifene covert oxidatively activated tag (COATag) in which raloxifene is linked to biotin (62). B) Identification of covalently modified proteins by the raloxifene COATag was achieved by immunoblotting and LC-MS-MS analysis (62).
DMA is a structural analogue of raloxifene in which the carbonyl group of raloxifene has been replaced by an ether linkage (Figure 1B). Arzoxifene is oxidatively desmethylated in vivo to yield DMA, an active metabolite that is a more potent ligand for both ERα and ERβ than both arzoxifene and raloxifene (61). However, it is unclear as to how much DMA is actually formed from arzoxifene, since in women, reported DMA plasma concentrations are highly variable (124). The methoxy group of arzoxifene is designed both to reduce glucuronidation and increase the ClogP value and is proposed to limit brain penetration. The intended effect of these modifications is to increase the bioavailability of the active drug relative to raloxifene (125). DMA can be oxidized to a diquinone methide, in the presence of rat or human liver microsomes (61). The half-life of the DMA diquinone methide was found to be 15 s at physiological pH and temperature (61), which indicates that it is an electrophile of intermediate stability, along with the acolbifene quinone methide discussed above (Table 1).
Given the need to develop new SERMs with attenuated toxicity and increased bioavailability while maintaining their beneficial effects, we synthesized a fluorinated DMA derivative (4′F-DMA) which is incapable of forming a diquinone methide (Figure 1B). 4′F-DMA showed similar ER binding affinity compared to that of DMA, whereas the antiestrogenic activity was 10-fold lower for than that of DMA; however, comparable to that of raloxifene (61). No GSH conjugates were detected in microsomal incubations with 4′F-DMA in the presence of GSH. Furthermore, DMA significantly decreased the GSH levels within 30 min, whereas 4′F-DMA had no effect on GSH levels (61). 4′F-DMA also induced much less DNA damage compared to DMA in cryopreserved rat hepatocytes. Phase I and phase II metabolism and toxicological behavior of DMA and 4′F-DMA were also compared using cryopreserved rat hepatocytes. Two DMA GSH conjugates, two DMA glucuronide conjugates, and one DMA sulfate conjugate were detected in incubations of DMA with rat hepatocytes. In contrast, 4′F-DMA did not yield GSH conjugates in incubations with rat hepatocytes, demonstrating that 4′-fluoro substitution of DMA blocked quinoid formation at the cellular level (61). Moreover, 4′F-DMA was observed to form two-fold less glucuronide conjugates relative to DMA in hepatocytes, which might be expected by the absence of the 4′OH group, since in raloxifene this is the major site of glucuronidation.
Intestinal glucuronidation of raloxifene is the major contributor to the presystemic clearance of raloxifene in vivo (126). Therefore, microsomes prepared from human small intestine were used to further evaluate the glucuronidation of DMA and 4′F-DMA. 4′F-DMA formed four-fold less glucuronide conjugates as compared to DMA in the incubation with human small intestinal microsomes in the presence of UDP-glucuronic acid (61). In human intestinal Caco-2 cells, a 24 h incubation of 4′F-DMA afforded thirty-fold less glucuronide conjugates and five-fold less sulfate conjugates compared with that of DMA (61). Therefore, 4′F-DMA should demonstrate improved bioavailability relative to DMA and raloxifene.
The ER-dependent activity of 4′F-DMA was examined by measuring ERE-luciferase activity in transiently transfected MCF-7 cells and Ishikawa cells (61). None of the SERMs studied showed estrogenic activity in either cell line; however, estradiol-induced luciferase activity was significantly antagonized by raloxifene, DMA, 4′F-DMA, and the pure antiestrogen, ICI 182780, in both cell lines (61). 4′F-DMA showed less antiestrogenic activity as compared to DMA, but comparable activity compared to raloxifene and ICI 182780 in both cell lines. Furthermore, 4′F-DMA antagonized the actions of estradiol in the MCF-7 cell proliferation assay without itself showing estrogenic activity and again, the antiestrogenicity of 4′F-DMA was comparable to raloxifene and ICI 182780. Taken together, these data suggest that 4′F-DMA represents a promising SERM with comparable antiestrogenic activity to raloxifene, but improved metabolic stability and an attenuated potential for toxicity compared to the current benzothiophene SERMs.
Acolbifene is also metabolized through a minor pathway leading to a diquinone methide (Scheme 3) (59). The acolbifene diquinone methide results from the oxidation of two phenol substituents (59). Like the classical quinone methide, the diquinone methide reacts with GSH to form five mono-GSH conjugates and five di-GSH conjugates, although the majority of the GSH adducts are formed by reaction of the classical quinone methide with GSH (59). Furthermore, incubations of acolbifene with either tyrosinase or rat or human liver microsomes also yield the classical quinone methide-GSH conjugates (59). Therefore, the formation of the acolbifene classical quinone methide might represent the major toxic pathway relative to that of the diquinone methide.
3.4.o-Quinones
In addition to the formation of the tamoxifen carbocation and quinone methide, tamoxifen can be oxidized to the corresponding o-quinone (127). Both tamoxifen and 4-hydroxytamoxifen can be metabolized to 3,4-dihydroxytamoxifen (Scheme 1). As catechols are readily oxidized to o-quinones by a variety of oxidative enzymes, metal ions, and in some cases molecular oxygen, it is possible that alkylation/oxidation of cellular macromolecules by tamoxifen o-quinone could contribute to the toxic effects of tamoxifen. However, as this is a minor pathway for tamoxifen, its contributions to tamoxifen’s overall toxicity are most likely minimal.
The o-quinone formed from 3,4-dihydroxytamoxifen has a half-life of approximately 80 min under physiological conditions (Table 1) (10). Incubation of rat liver microsomes with 3,4-dihydroxytamoxifen in the presence of GSH produced several o-quinone GSH conjugates, which were also observed in the incubations with breast cancer cells (MCF-7). Incubation of 3,4-dihydroxytamoxifen o-quinone with deoxynucleosides produced four thymidine and four deoxyguanosine adducts, which may have derived from the addition at different sites on the o-quinone ring and/or E, Z isomers of tamoxifen skeleton (10). In addition to acting as electrophilic Michael addition acceptors to form conjugates with GSH and deoxynucleosides, the o-quinones can also undergo redox cycling with the semiquinone radicals to generate superoxide anion radicals, which is mediated through cytochrome P450 and P450 reductase (109). The reaction of superoxide anion radicals with hydrogen peroxide (formed enzymatically or via spontaneous dismutation of superoxide anion radical in the presence of trace amounts of iron or other transition metals) yields hydroxyl radicals. Hydroxyl radicals are powerful oxidizing agents that may be responsible for the damage to essential cellular macromolecules. The detection of DNA single strand breaks in incubations of 3,4-dihydroxytamoxifen with various breast cancer cell lines suggests that the 3,4-dihydroxytamoxifen o-quinone could cause oxidative DNA damage through redox cycling with semiquinone radicals (128).
Like tamoxifen, the 4-hydroxylation also represents a major phase I metabolic pathway of toremifene (129, 130). In rat liver microsomal incubations, both 4-hydroxytoremifene and droloxifene were metabolized by P450s to the corresponding catechol metabolites 3,4-dihydroxytoremifene and 3,4-dihydroxydroloxifene to yield o-quinone derived GSH conjugates. In the 4-hydroxytoremifene incubation, the amount of quinone methide derived GSH conjugates was higher than that of o-quinone derived GSH conjugates (131), possibly because the β-Cl substitution lowered the energy barrier for α- hydrogen atom abstraction, easing the 2-electron oxidation pathway leading to the 4-hydroxytoremifene quinone methide. Compared with 3,4-dihydroxytamoxifen o-quinone, the 3,4-dihydroxytoremifene o-quinone showed higher reactivity toward deoxynucleosides. The toremifene-o-quinone reacted with all four deoxynucleosides to form the corresponding adducts at physiological pH and temperature. Three of the adducts were detected in incubations with thymidine, five in incubations with deoxyguanosine, two in incubations with deoxyadenosine, and eight in incubations with deoxycytosine (131). When incubated with breast cancer cell lines, 3,4-dihydroxytoremifene also caused DNA single strand breaks (128). Furthermore, different amounts of DNA damage were observed in ER positive cell lines and ER negative cell lines, suggesting the ERs might play a role in this process (128).
Raloxifene is metabolized by rat or human liver microsomes to an electrophilic diquinone methide and an o-quinone (Scheme 4) which can be trapped by GSH to form mono-and/or di-GSH conjugates (60). Hydroxylated catechols such as 7-hydroxyraloxifene and 3′-hydroxyraloxifene were also detected in the incubation mixture. The half-life of raloxifene di-quinone methide is less than 1 s; however, the half-life of the raloxifene 6,7-o-quinone is 69 min (Table 1). The stability offered by 6,7-o-quinone implies that it may be more toxic than the raloxifene diquinone methide. However, as this pathway is a minor one for raloxifene, physiological concentrations of the raloxifene o-quinone are probably insufficient to induce toxicity.
Very recently, Eli Lilly reported the discovery of two new SERMs for the potential treatment of uterine leiomyoma (LY2066948) and hot flushes (LSN2021310) (Figure 3). LY2066948 has a methyl sulfone group at 4′ position, which renders this molecule less susceptible to metabolic conjugation and limits brain penetration. In several in vivo studies in rat models, LSN2021310 increased bone mineral density, lowered serum cholesterol, exhibited minimal uterine agonist activity, and displayed dose-dependent activity of hot flush efficacy. However, both LY2066948 and LSN2021310 share the same naphthol structure with equilenin (Figure 3), the equine estrogen present in hormone replacement therapy preparations. The presence of an unsaturated B-ring generally leads to exclusive 4-hydroxylation, which, in turn leads to the formation of a highly reactive o-quinone (132). It is known that both P450s 1A1 and 1B1 catalyze the 4-hydroxylation of equilenin (Figure 3) (133) and the catechol formed will autoxidize to the 3,4-o-quinone (134). Catalyzed by NAD(P)H, P450 reductase, or quinone reductase, the o-quinone readily enters into a redox couple with the semiquinone radical, resulting in the generation of reactive oxygen species (ROS) (Scheme 5) (135). In in vitro studies, the 3,4-equilenin o-quinone, semiquinone, and ROS displayed high reactivity to nucleosides and DNA, leading to the formation of unusual cyclic nucleoside adducts, oxidative damage to DNA nucleobases, DNA single strand breaks, and apurinic sites(135, 136, 137). In a rat model, 4-hydroxyequilenin was shown to cause DNA damage in vivo (138). In addition, 4-hydroxyequilenin can inhibit detoxification enzymes of the cell, such as glutathione S-transferase (GST) (139) and catechol O-methyltransferase (COMT) (140, 141). Since the naphthol structure is responsible for the carcinogenic bioactivation of equilenin, it is likely that LY2066948 and LSN2021310 are also metabolized to 3,4-o-quinones and will display similar carcinogenic potential.
Figure 3. Chemical Structures of Naphthol SERMs and Equine Estrogens.

Naphthol SERMs LY2066948 and LSN2120310 are similar in structure to the equine estrogen equilenin. Equilenin is hydroxylated by P450 on the B-ring to form 4-hydroxyequilenin (132, 133).
Scheme 5. Bioactivation of Equilenin.

P450s catalyze the exclusive 4-hydroxylation of equilenin and the catechol formed can auto-oxidize to the 3,4-o-quinone via a semiquinone intermediate (134). Catalyzed by NAD(P)H, P450 reductase, or quinone reductase, the o-quinone readily enters into a redox couple with the semiquinone radical, resulting in the generation of reactive oxygen species (ROS) (135).
3.5. Miscellaneous Bioactivation Pathways
The final bioactivation pathways we will discuss includes the formation of quinone metabolites, an iminium ion, and an aldehyde from two recently discovered SERMs (Dihydrobenzoxathiin compounds I and II in Scheme 6 and 7). Dihydrobenzoxathiin I was synthesized as a selective ERα antagonist and its bioactivation in both human liver microsomes and recombinant P450 3A4 has been studied extensively by Zhang et al. (142). This compound is metabolized to a hydroquinone and an o-quinone (Scheme 6). The hydroquinone is obtained from oxidative cleavage of the dihydrobenzoxathiin moiety and has the potential to undergo oxidation to the corresponding quinone. Both the o-quinone and the hydroquinone could contribute to the observed protein binding. In addition, since a dinitrile compound (bis-cyano adduct) was detected when the incubation was performed in the presence of sodium cyanide, an iminium ion intermediate derived from the piperidine side chain of (I) is most likely formed in the incubation. Therefore, compound (I) is subject to at least three bioactivation pathways, two of them leading to quinone metabolites, and the third producing an iminium ion.
Scheme 6. Reactive Metabolite Formation from Dihydrobenzoxathiin I.

Three bioactivation pathways were observed when dihydrobenzoxathiin I was incubated with human liver microsomes and recombinant P450 3A4. Oxidative cleavage of the dihydrobenzoxathiin moiety led to the formation of a hydroquinone. Phenol hydroxylation at either ortho position followed by further oxidation formed an o-quinone. Additionally, an iminium ion intermediate was formed on the piperidine side chain (142).
Scheme 7. Reactive Metabolite Formation from Dihydrobenzoxathiin II.

Dihydrobenzoxathiin II was incubated with human liver microsomes and recombinant P450 3A4. The major metabolite biphenyl hydroquinone resulted from the rearrangement of the parent compound. Oxidative cleavage of the dihydrobenzoxathiin moiety led to the formation of hydroquinone. A reactive aldehyde metabolite was also detected (142).
Extensive studies of these benzoxathiin compounds led to the discovery of (II), which has a 3′-OH instead of a 4′-OH, and the substitution of the piperidine side chain with a pyrolidine moiety. Compared with (I), the new compound exhibited optimal in vitro and in vivo potency and selectivity toward ERα. Scheme 7 shows the reactive metabolites obtained in the incubation with human liver microsomes and recombinant P450 3A4. The major metabolite was a biphenyl hydroquinone derivative, which came from the rearrangement of the parent compound. In addition, the dihydrobenzoxathiin oxidative cleavage metabolite was also a hydroquinone and both of these metabolites could potentially covalently modify proteins via reactive quinone intermediates. An aldehyde was also detected and has to potential to form Schiff base adducts with proteins. Co-incubation with N-acetylcysteine (NAc) led to four NAc adducts, providing evidence that dihydrobenzoxathiin (II) also underwent bioactivation to form reactive quinone species. Since, the degree of protein covalent binding by (II) was much lower than (I), (II) was chooses as the lead compound.
4. Concluding Remarks
The foreseen long-term clinical use of SERMs and risk of carcinogenesis require study of potential mechanisms of toxicity including covalent modification of cellular macromolecules. Adducts with glutathione, proteins, nucleosides, and DNA have been reported for many SERMs in vitro and in some cases in vivo. Several different bioactivation mechanisms are involved in converting SERMs to reactive intermediates, all of which can be initiated via oxidation by cytochrome P450. These bioactivation pathways include carbocation formation, the classical quinone methide and diquinone methide pathways, and o-quinone formation. The triphenylethylene SERMs, such as the prototype tamoxifen, are hydroxylated by P450 at the α-position. Upon conjugation with sulfate and subsequent loss of the sulfate group, these molecules form highly reactive carbocations which have the potential to react with DNA (8, 9). However, tamoxifen-DNA adducts in endometrial tissue have only been detected with some ambiguity (56, 99). Therefore, the physiological relevance of this mechanism have been questioned by several research groups (56, 57, 58).
The second bioactivation pathway is exemplified by the hydroxylation of tamoxifen by P450 to form 4-hydroxylated metabolites, which can be further oxidized to classical quinone methides (11). The amount of the tamoxifen quinone methide produced as well as its stability suggests it is unlikely to result in cytotoxicity or genotoxicity (11). However, acolbifene is also converted to a classical quinone methide, which may be its major pathway of bioactivation (59). In addition, raloxifene (60), DMA (61), and acolbifene (59) can all be converted to diquinone methides. The raloxifene diquinone methide has recently been implicated in the inhibition of P450 3A4 (63), and the alkylation of many other liver microsomal proteins (62).
The acolbifene quinone methide has a half-life that is considerably shorter (32 s) (59) than that of the 4-hydroxytamoxifen quinone methide (3 h) (11), but longer than that of the raloxifene diquinone methide (1 s) (Table 1) (60). According to Thompson et al., quinone methides with half-lives in the range of 10 s to 10 min have time to diffuse away from the site of formation to react with cellular nucleophiles (107). These quinone methides are more likely to cause toxicity compared to more reactive ones that will react immediately with another quinone methide, solvent, oxygen, or the enzyme responsible for their formation (59). According to this hypothesis, the acolbifene quinone methide can be considered an electrophile of intermediate stability and therefore might be a contributor to the potential genotoxicity or cytotoxicity of acolbifene through depletion of GSH and modification of cellular macromolecules (59).
The Thompson hypothesis is persuasive in its simplicity and would have the benefit of being a predictive tool of use in drug discovery. However, the observation that the short-lived raloxifene diquinone methide is able to escape its oxidative enzyme to modify a variety of proteins, although is does so with a low degree of selectivity, clearly indicates that the original hypothesis must be modified. This evolution can only be effected through further studies on the reaction mechanisms and targets of reactive intermediates formed from oxidative metabolism of SERMs and related compounds.
The fourth bioactivation pathway for SERMs involves o-quinone formation and has been implicated in tamoxifen-mediated protein binding (64, 65). This is generally a minor pathway for SERMs but might be a major pathway for β-napthol compounds. Finally, quinone metabolites, an iminium ion, and an aldehyde formed from two recently discovered SERMs (Dihydrobenzoxathiin compounds I and II in Scheme 5 and 6) have all been implicated in protein binding (142).
Understanding how the structure of SERMs influences the formation and reactivity of reactive intermediates is an important tool for the discovery of potential new SERMs that are not associated with genotoxic or cytotoxic effects. A complete understanding of these toxic metabolites is necessary as SERMs are already in wide spread use as prophylactic agents in high risk but otherwise healthy individuals.
Acknowledgements
This work is supported by NIH Grants CA102590, CA79870 and CA73638.
Glossary
- AF
activation factor
- BCPT
National Surgical Adjuvant Breast and Bowel Project-sponsored Breast Cancer Prevention Trial
- COATag
covert oxidatively activated tag
- COMT
catechol-O-methyltransferase
- DES
diethylstilbestrol
- DMA
desmethylated arzoxifene
- E2
17β-estradiol
- ER
estrogen receptor
- ER-60
protein disulfide isomerase isozyme A3
- ERE
estrogen response element
- ERp72
protein disulfide isomerase isozyme A4 precursor
- ESI
electrospray ionization
- 4′F-DMA
4-fluorodesmethylated arzoxifene
- GRIP1
glutamate receptor interacting protein 1
- GRP78/BiP
cytosolic glucose regulated protein
- GST
glutathione-S-transferase
- hGST PI-1
human glutathione-S-transferase
- hHST
human hydroxysteroid sulfotransferase
- LBD
ligand binding domain
- MALDI-TOF
matrix-assisted laser desorption ionization-time of flight
- MAP
mitogen activated protein
- mGST1
microsomal glutathione S-transferase
- NR
nuclear receptor
- PDIA1
protein disulfide isomerase isozyme A1
- ROS
reactive oxygen species
- RUTH trial
Raloxifene Use for The Heart trial
- SERM
Selective Estrogen Receptor Modulator
- SRC-1
steroid receptor coactivator-1
- STAR trial
Study of Tamoxifen and Raloxifene trial.
References
- (1).Smigel K. Breast Cancer Prevention Trial shows major benefit, some risk. J. Natl. Cancer Inst. 1998;90:647–648. doi: 10.1093/jnci/90.9.647. [DOI] [PubMed] [Google Scholar]
- (2).Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- (3).Jordan VC, Morrow M. Tamoxifen, raloxifene, and the prevention of breast cancer. Endocr. Rev. 1999;20:253–278. doi: 10.1210/edrv.20.3.0368. [DOI] [PubMed] [Google Scholar]
- (4).Henderson BE, Ross R, Bernstein L. Estrogens as a cause of human cancer: the Richard and Hinda Rosenthal Foundation award lecture. Cancer Res. 1988;48:246–253. [PubMed] [Google Scholar]
- (5).Friedl A, Jordan VC. What do we know and what don’t we know about tamoxifen in the human uterus. Breast Cancer Res. Treat. 1994;31:27–39. doi: 10.1007/BF00689674. [DOI] [PubMed] [Google Scholar]
- (6).Cohen I, Rosen DJ, Shapira J, Cordoba M, Gilboa S, Altaras MM, Yigael D, Beyth Y. Endometrial changes with tamoxifen: comparison between tamoxifen-treated and nontreated asymptomatic, postmenopausal breast cancer patients. Gynecol. Oncol. 1994;52:185–190. doi: 10.1006/gyno.1994.1029. [DOI] [PubMed] [Google Scholar]
- (7).Roy RN, Gerulath AH, Cecutti A, Bhavnani BR. Effect of tamoxifen treatment on the endometrial expression of human insulin-like growth factors and their receptor mRNAs. Mol. Cell. Endocrinol. 2000;165:173–178. doi: 10.1016/s0303-7207(00)00248-3. [DOI] [PubMed] [Google Scholar]
- (8).Shibutani S, Suzuki N, Terashima I, Sugarman SM, Grollman AP, Pearl ML. Tamoxifen-DNA adducts detected in the endometrium of women treated with tamoxifen. Chem. Res. Toxicol. 1999;12:646–653. doi: 10.1021/tx990033w. [DOI] [PubMed] [Google Scholar]
- (9).Shibutani S, Ravindernath A, Suzuki N, Terashima I, Sugarman SM, Grollman AP, Pearl ML. Identification of tamoxifen-DNA adducts in the endometrium of women treated with tamoxifen. Carcinogenesis. 2000;21:1461–1467. [PubMed] [Google Scholar]
- (10).Zhang F, Fan PW, Liu X, Shen L, van Breeman RB, Bolton JL. Synthesis and reactivity of a potential carcinogenic metabolite of tamoxifen: 3,4-dihydroxytamoxifen-o-quinone. Chem. Res. Toxicol. 2000;13:53–62. doi: 10.1021/tx990145n. [DOI] [PubMed] [Google Scholar]
- (11).Fan PW, Zhang F, Bolton JL. 4-Hydroxylated metabolites of the antiestrogens tamoxifen and toremifene are metabolized to unusually stable quinone methides. Chem. Res. Toxicol. 2000;13:45–52. doi: 10.1021/tx990144v. [DOI] [PubMed] [Google Scholar]
- (12).Bolton JL. Quinoids, quinoid radicals, and phenoxyl radicals formed from estrogens and antiestrogens. Toxicology. 2002;177:55–65. doi: 10.1016/s0300-483x(02)00195-6. [DOI] [PubMed] [Google Scholar]
- (13).Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem. Res. Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- (14).Kim SY, Suzuki N, Laxmi YR, Shibutani S. Genotoxic mechanism of tamoxifen in developing endometrial cancer. Drug Metab. Rev. 2004;36:199–218. doi: 10.1081/dmr-120033997. [DOI] [PubMed] [Google Scholar]
- (15).Katzenellenbogen BS, Montano MM, Ediger TR, Sun J, Ekena K, Lazennec G, Martini PG, McInerney EM, Delage-Mourroux R, Weis K, Katzenellenbogen JA. Estrogen receptors: selective ligands, partners, and distinctive pharmacology. Recent Prog. Horm. Res. 2000;55:163–193. discussion 194-165. [PubMed] [Google Scholar]
- (16).Katzenellenbogen BS, Sun J, Harrington WR, Kraichely DM, Ganessunker D, Katzenellenbogen JA. Structure-function relationships in estrogen receptors and the characterization of novel selective estrogen receptor modulators with unique pharmacological profiles. Ann. N. Y. Acad. Sci. 2001;949:6–15. doi: 10.1111/j.1749-6632.2001.tb03998.x. [DOI] [PubMed] [Google Scholar]
- (17).McKenna NJ, O’Malley BW. An issue of tissues: divining the split personalities of selective estrogen receptor modulators. Nat. Med. 2000;6:960–962. doi: 10.1038/79637. [DOI] [PubMed] [Google Scholar]
- (18).Rhodes DJ, Hartmann LC, Perez EA. Breast cancer prevention trials. Curr. Oncol. Rep. 2000;2:558–565. doi: 10.1007/s11912-000-0110-0. [DOI] [PubMed] [Google Scholar]
- (19).Phillips C. NCI Cancer Bulletin. National Cancer Institute; 2006. STAR Results: Raloxifene as Effective as Tamoxifen, Better Safety Profile; pp. 1–2. [Google Scholar]
- (20).McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- (21).McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- (22).Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J. Steroid. Biochem. Mol. Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- (23).Kushner PJ, Agard D, Feng WJ, Lopez G, Schiau A, Uht R, Webb P, Greene G. Oestrogen receptor function at classical and alternative response elements. Novartis. Found. Symp. 2000;230:20–26. doi: 10.1002/0470870818.ch3. discussion 27-40. [DOI] [PubMed] [Google Scholar]
- (24).Watanabe T, Inoue S, Hiroi H, Orimo A, Kawashima H, Muramatsu M. Isolation of estrogen-responsive genes with a CpG island library. Mol. Cell. Biol. 1998;18:442–449. doi: 10.1128/mcb.18.1.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dubik D, Shiu RP. Mechanism of estrogen activation of c-myc oncogene expression. Oncogene. 1992;7:1587–1594. [PubMed] [Google Scholar]
- (26).Umayahara Y, Kawamori R, Watada H, Imano E, Iwama N, Morishima T, Yamasaki Y, Kajimoto Y, Kamada T. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J. Biol. Chem. 1994;269:16433–16442. [PubMed] [Google Scholar]
- (27).Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- (28).Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- (29).Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- (30).Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- (31).Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- (32).Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- (33).Korach KS. Insights from the study of animals lacking functional estrogen receptor. Science. 1994;266:1524–1527. doi: 10.1126/science.7985022. [DOI] [PubMed] [Google Scholar]
- (34).Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N. Engl. J. Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- (35).Gradishar WJ, Jordan VC. Clinical potential of new antiestrogens. J. Clin. Oncol. 1997;15:840–852. doi: 10.1200/JCO.1997.15.2.840. [DOI] [PubMed] [Google Scholar]
- (36).Jordan VC. Antiestrogenic action of raloxifene and tamoxifen: today and tomorrow. J. Natl. Cancer Inst. 1998;90:967–971. doi: 10.1093/jnci/90.13.967. [DOI] [PubMed] [Google Scholar]
- (37).Manas ES, Unwalla RJ, Xu ZB, Malamas MS, Miller CP, Harris HA, Hsiao C, Akopian T, Hum WT, Malakian K, Wolfrom S, Bapat A, Bhat RA, Stahl ML, Somers WS, Alvarez JC. Structure-based design of estrogen receptor-beta selective ligands. J. Am. Chem. Soc. 2004;126:15106–15119. doi: 10.1021/ja047633o. [DOI] [PubMed] [Google Scholar]
- (38).Hillisch A, Peters O, Kosemund D, Muller G, Walter A, Schneider B, Reddersen G, Elger W, Fritzemeier KH. Dissecting physiological roles of estrogen receptor alpha and beta with potent selective ligands from structure-based design. Mol. Endocrinol. 2004;18:1599–1609. doi: 10.1210/me.2004-0050. [DOI] [PubMed] [Google Scholar]
- (39).Gungor T, Chen Y, Golla R, Ma Z, Corte JR, Northrop JP, Bin B, Dickson JK, Stouch T, Zhou R, Johnson SE, Seethala R, Feyen JH. Synthesis and characterization of 3-arylquinazolinone and 3-arylquinazolinethione derivatives as selective estrogen receptor beta modulators. J. Med. Chem. 2006;49:2440–2455. doi: 10.1021/jm0509389. [DOI] [PubMed] [Google Scholar]
- (40).Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC., Jr. Evaluation of an estrogen receptor-beta agonist in animal models of human disease. Endocrinology. 2003;144:4241–4249. doi: 10.1210/en.2003-0550. [DOI] [PubMed] [Google Scholar]
- (41).Shang Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nat. Rev. Cancer. 2006;6:360–368. doi: 10.1038/nrc1879. [DOI] [PubMed] [Google Scholar]
- (42).Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- (43).Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, Matsumoto K, Palkowitz AD, Sato M, Termine JD, Winter MA, Yang NN, Dodge JA. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc. Natl. Acad. Sci. U. S. A. 1997;94:14105–14110. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- (45).Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–951. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- (46).Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr. Opin. Cell. Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- (47).Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- (48).Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Nuclear receptor coactivators and corepressors. Mol. Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- (49).Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr. Opin. Cell Biol. 1997;9:222–232. doi: 10.1016/s0955-0674(97)80066-x. [DOI] [PubMed] [Google Scholar]
- (50).Feng W, Ribeiro RC, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- (51).Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- (52).Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- (53).Ding XF, Anderson CM, Ma H, Hong H, Uht RM, Kushner PJ, Stallcup MR. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificities. Mol. Endocrinol. 1998;12:302–313. doi: 10.1210/mend.12.2.0065. [DOI] [PubMed] [Google Scholar]
- (54).Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, Chambon P. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. Embo. J. 1996;15:6701–6715. [PMC free article] [PubMed] [Google Scholar]
- (55).Katzenellenbogen BS, Katzenellenbogen JA. Biomedicine. Defining the “S” in SERMs. Science. 2002;295:2380–2381. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- (56).Beland FA, Marques MM, Gamboa da Costa G, Phillips DH. Tamoxifen-DNA adduct formation in human endometrium. Chem. Res. Toxicol. 2005;18:1507–1509. doi: 10.1021/tx050255w. author reply 1509-1511. [DOI] [PubMed] [Google Scholar]
- (57).Carmichael PL, Sardar S, Crooks N, Neven P, Van Hoof I, Ugwumadu A, Bourne T, Tomas E, Hellberg P, Hewer AJ, Phillips DH. Lack of evidence from HPLC 32P-post-labelling for tamoxifen-DNA adducts in the human endometrium. Carcinogenesis. 1999;20:339–342. doi: 10.1093/carcin/20.2.339. [DOI] [PubMed] [Google Scholar]
- (58).Beland FA, Churchwell MI, Hewer A, Phillips DH, daCosta GG, Marques MM. Analysis of tamoxifen-DNA adducts in endometrial explants by MS and 32P-postlabeling. Biochem. Biophys. Res. Commun. 2004;320:297–302. doi: 10.1016/j.bbrc.2004.05.168. [DOI] [PubMed] [Google Scholar]
- (59).Liu J, Liu H, van Breemen RB, Thatcher GR, Bolton JL. Bioactivation of the selective estrogen receptor modulator acolbifene to quinone methides. Chem. Res. Toxicol. 2005;18:174–182. doi: 10.1021/tx0497752. [DOI] [PubMed] [Google Scholar]
- (60).Yu L, Liu H, Li W, Zhang F, Luckie C, van Breemen RB, Thatcher GR, Bolton JL. Oxidation of raloxifene to quinoids: potential toxic pathways via a diquinone methide and o-quinones. Chem. Res. Toxicol. 2004;17:879–888. doi: 10.1021/tx0342722. [DOI] [PubMed] [Google Scholar]
- (61).Liu H, Liu J, van Breemen RB, Thatcher GR, Bolton JL. Bioactivation of the selective estrogen receptor modulator desmethylated arzoxifene to quinoids: 4′-fluoro substitution prevents quinoid formation. Chem. Res. Toxicol. 2005;18:162–173. doi: 10.1021/tx049776u. [DOI] [PubMed] [Google Scholar]
- (62).Liu J, Li Q, Yang X, van Breemen RB, Bolton JL, Thatcher GR. Analysis of Protein Covalent Modification by Xenobiotics Using a Covert Oxidatively Activated Tag: Raloxifene Proof-of-Principle Study. Chem. Res. Toxicol. 2005;18:1485–1496. doi: 10.1021/tx0501738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Chen Q, Ngui JS, Doss GA, Wang RW, Cai X, DiNinno FP, Blizzard TA, Hammond ML, Stearns RA, Evans DC, Baillie TA, Tang W. Cytochrome P450 3A4-mediated bioactivation of raloxifene: irreversible enzyme inhibition and thiol adduct formation. Chem. Res. Toxicol. 2002;15:907–914. doi: 10.1021/tx0200109. [DOI] [PubMed] [Google Scholar]
- (64).Dehal SS, Kupfer D. Cytochrome P-450 3A and 2D6 catalyze ortho hydroxylation of 4-hydroxytamoxifen and 3-hydroxytamoxifen (droloxifene) yielding tamoxifen catechol: involvement of catechols in covalent binding to hepatic proteins. Drug. Metab. Dispos. 1999;27:681–688. [PubMed] [Google Scholar]
- (65).Dehal SS, Kupfer D. Evidence that the catechol 3,4-Dihydroxytamoxifen is a proximate intermediate to the reactive species binding covalently to proteins. Cancer Res. 1996;56:1283–1290. [PubMed] [Google Scholar]
- (66).Fisher B, Redmond C. New perspective on cancer of the contralateral breast: a marker for assessing tamoxifen as a preventive agent. J. Natl. Cancer Inst. 1991;83:1278–1280. doi: 10.1093/jnci/83.18.1278. [DOI] [PubMed] [Google Scholar]
- (67).Killackey MA, Hakes TB, Pierce VK. Endometrial adenocarcinoma in breast cancer patients receiving antiestrogens. Cancer Treat. Rep. 1985;69:237–238. [PubMed] [Google Scholar]
- (68).Fornander T, Rutqvist LE, Cedermark B, Glas U, Mattsson A, Silfversward C, Skoog L, Somell A, Theve T, Wilking N, et al. Adjuvant tamoxifen in early breast cancer: occurrence of new primary cancers. Lancet. 1989;1:117–120. doi: 10.1016/s0140-6736(89)91141-0. [DOI] [PubMed] [Google Scholar]
- (69).Cohen CJ, Rahaman J. Endometrial cancer. Management of high risk and recurrence including the tamoxifen controversy. Cancer. 1995;76:2044–2052. doi: 10.1002/1097-0142(19951115)76:10+<2044::aid-cncr2820761323>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- (70).Nease RF, Jr., Ross JM. The decision to enter a randomized trial of tamoxifen for the prevention of breast cancer in healthy women: an analysis of the tradeoffs. Am. J. Med. 1995;99:180–189. doi: 10.1016/s0002-9343(99)80138-7. [DOI] [PubMed] [Google Scholar]
- (71).King CM. Tamoxifen and the induction of cancer. Carcinogenesis. 1995;16:1449–1454. doi: 10.1093/carcin/16.7.1449. [DOI] [PubMed] [Google Scholar]
- (72).Vancutsem PM, Lazarus P, Williams GM. Frequent and specific mutations of the rat p53 gene in hepatocarcinomas induced by tamoxifen. Cancer Res. 1994;54:3864–3867. [PubMed] [Google Scholar]
- (73).Fendel KCZ. Role of tamoxifen in the induction of hormone-independent rat mammary tumors. Cancer Res. 1992:235–237. S.J. [PubMed] [Google Scholar]
- (74).Roos W, Oeze L, Loser R, Eppenberger U. Antiestrogenic action of 3-hydroxytamoxifen in the human breast cancer cell line MCF-7. J. Natl. Cancer Inst. 1983;71:55–59. [PubMed] [Google Scholar]
- (75).White IN, de Matteis F, Davies A, Smith LL, Crofton-Sleigh C, Venitt S, Hewer A, Phillips DH. Genotoxic potential of tamoxifen and analogues in female Fischer F344/n rats, DBA/2 and C57BL/6 mice and in human MCL-5 cells. Carcinogenesis. 1992;13:2197–2203. doi: 10.1093/carcin/13.12.2197. [DOI] [PubMed] [Google Scholar]
- (76).Pace P, Jarman M, Phillips D, Hewer A, Bliss J, Coombes RC. Idoxifene is equipotent to tamoxifen in inhibiting mammary carcinogenesis but forms lower levels of hepatic DNA adducts. Br. J. Cancer. 1997;76:700–704. doi: 10.1038/bjc.1997.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Johnston SR, Riddler S, Haynes BP, A’Hern R, Smith IE, Jarman M, Dowsett M. The novel anti-oestrogen idoxifene inhibits the growth of human MCF-7 breast cancer xenografts and reduces the frequency of acquired anti-oestrogen resistance. Br. J. Cancer. 1997;75:804–809. doi: 10.1038/bjc.1997.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).White IN, Martin EA, Mauthe RJ, Vogel JS, Turteltaub KW, Smith LL. Comparisons of the binding of [14C]radiolabelled tamoxifen or toremifene to rat DNA using accelerator mass spectrometry. Chem. Biol. Interact. 1997;106:149–160. doi: 10.1016/s0009-2797(97)00063-x. [DOI] [PubMed] [Google Scholar]
- (79).Kim SY, Suzuki N, Laxmi YR, Shibutani S. Genotoxic mechanism of tamoxifen in developing endometrial cancer. Drug. Metab. Rev. 2004;36:199–218. doi: 10.1081/dmr-120033997. [DOI] [PubMed] [Google Scholar]
- (80).Johnston SR. Endocrine manipulation in advanced breast cancer: recent advances with SERM therapies. Clin. Cancer Res. 2001;7:4376s–4387s. discussion 4411s-4412s. [PubMed] [Google Scholar]
- (81).Gradishar W, Glusman J, Lu Y, Vogel C, Cohen FJ, Sledge GW., Jr. Effects of high dose raloxifene in selected patients with advanced breast carcinoma. Cancer. 2000;88:2047–2053. doi: 10.1002/(sici)1097-0142(20000501)88:9<2047::aid-cncr10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- (82).Eli Lilly and Company Lilly announces preliminary coronary and breast cancer. Results from Raloxifene Use for The Heart (RUTH) study. 2006 April 12; Issued. Available online at http://newsroom.lilly.com/ReleaseDetail.cfm?ReleaseID=192692.
- (83).Dardes RC, Bentrem D, O’Regan RM, Schafer JM, Jordan VC. Effects of the new selective estrogen receptor modulator LY353381.HCl (Arzoxifene) on human endometrial cancer growth in athymic mice. Clin. Cancer. Res. 2001;7:4149–4155. [PubMed] [Google Scholar]
- (84).McMeekin DS, Gordon A, Fowler J, Melemed A, Buller R, Burke T, Bloss J, Sabbatini P. A phase II trial of arzoxifene, a selective estrogen response modulator, in patients with recurrent or advanced endometrial cancer. Gynecol. Oncol. 2003;90:64–69. doi: 10.1016/s0090-8258(03)00203-8. [DOI] [PubMed] [Google Scholar]
- (85).Baselga J, Llombart-Cussac A, Bellet M, Guillem-Porta V, Enas N, Krejcy K, Carrasco E, Kayitalire L, Kuta M, Lluch A, Vodvarka P, Kerbrat P, Namer M, Petruzelka L. Randomized, double-blind, multicenter trial comparing two doses of arzoxifene ( LY353381) in hormone-sensitive advanced or metastatic breast cancer patients. Ann. Oncol. 2003;14:1383–1390. doi: 10.1093/annonc/mdg368. [DOI] [PubMed] [Google Scholar]
- (86).Chan S. Arzoxifene in breast cancer. Eur. J. Cancer. 2002;38(Suppl 6):S55–56. doi: 10.1016/s0959-8049(02)00286-1. [DOI] [PubMed] [Google Scholar]
- (87).Vernon MW. Experimental endometriosis in laboratory animals as a research model. Prog. Clin. Biol. Res. 1990;323:49–60. [PubMed] [Google Scholar]
- (88).Thomas W, Burke MD, Cheryl L. Walker. Arzoxifene as therapy for endometrial cancer. Gynecol. Oncol. 2003;90:S40–S46. doi: 10.1016/s0090-8258(03)00343-3. [DOI] [PubMed] [Google Scholar]
- (89).Munster PN. Arzoxifene: the development and clinical outcome of an ideal SERM. Expert Opin. Investig. Drugs. 2006;15:317–326. doi: 10.1517/13543784.15.3.317. [DOI] [PubMed] [Google Scholar]
- (90).Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, Sporn MB. Arzoxifene, a new selective estrogen receptor modulator for chemoprevention of experimental breast cancer. Cancer Res. 2001;61:8412–8415. [PubMed] [Google Scholar]
- (91).Gauthier S, Caron B, Cloutier J, Dory YL, Favre A, Larouche D, Mailhot J, Ouellet C, Schwerdtfeger A, Leblanc G, Martel C, Simard J, Merand Y, Belanger A, Labrie C, Labrie F. (S)-(+)-4-[7-(2,2-dimethyl-1-oxopropoxy)-4-methyl-2-[4-[2-(1-piperidinyl)-ethoxy]phenyl]-2H-1-benzopyran-3-yl]-phenyl 2,2-dimethylpropanoate (EM-800): a highly potent, specific, and orally active nonsteroidal antiestrogen. J. Med. Chem. 1997;40:2117–2122. doi: 10.1021/jm970095o. [DOI] [PubMed] [Google Scholar]
- (92).Simard J, Labrie C, Belanger A, Gauthier S, Singh SM, Merand Y, Labrie F. Characterization of the effects of the novel non-steroidal antiestrogen EM-800 on basal and estrogen-induced proliferation of T-47D, ZR-75-1 and MCF-7 human breast cancer cells in vitro. Int. J. Cancer. 1997;73:104–112. doi: 10.1002/(sici)1097-0215(19970926)73:1<104::aid-ijc16>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- (93).Simard J, Sanchez R, Poirier D, Gauthier S, Singh SM, Merand Y, Belanger A, Labrie C, Labrie F. Blockade of the stimulatory effect of estrogens, OH-tamoxifen, OH-toremifene, droloxifene, and raloxifene on alkaline phosphatase activity by the antiestrogen EM-800 in human endometrial adenocarcinoma Ishikawa cells. Cancer Res. 1997;57:3494–3497. [PubMed] [Google Scholar]
- (94).Couillard S, Gutman M, Labrie C, Belanger A, Candas B, Labrie F. Comparison of the effects of the antiestrogens EM-800 and tamoxifen on the growth of human breast ZR-75-1 cancer xenografts in nude mice. Cancer Res. 1998;58:60–64. [PubMed] [Google Scholar]
- (95).Luo S, Labrie C, Belanger A, Labrie F. Effect of dehydroepiandrosterone on bone mass, serum lipids, and dimethylbenz(a)anthracene-induced mammary carcinoma in the rat. Endocrinology. 1997;138:3387–3394. doi: 10.1210/endo.138.8.5345. [DOI] [PubMed] [Google Scholar]
- (96).Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 2004;5:207–213. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- (97).Sharma M, Shubert DE, Lewis J, McGarrigle BP, Bofinger DP, Olson JR. Biotransformation of tamoxifen in a human endometrial explant culture model. Chem. Biol. Interact. 2003;146:237–249. doi: 10.1016/j.cbi.2003.06.002. [DOI] [PubMed] [Google Scholar]
- (98).Kim SY, Laxmi YR, Suzuki N, Ogura K, Watabe T, Duffel MW, Shibutani S. Formation of tamoxifen-DNA adducts via O-sulfonation, not O-acetylation, of alpha-hydroxytamoxifen in rat and human livers. Drug. Metab. Dispos. 2005;33:1673–1678. doi: 10.1124/dmd.105.005330. [DOI] [PubMed] [Google Scholar]
- (99).Kim SY, Suzuki N, Laxmi YR, McGarrigle BP, Olson JR, Sharma M, Shibutani S. Formation of tamoxifen-DNA adducts in human endometrial explants exposed to alpha-hydroxytamoxifen. Chem. Res. Toxicol. 2005;18:889–895. doi: 10.1021/tx050019l. [DOI] [PubMed] [Google Scholar]
- (100).Brown K, Carmichael PL. “Antioxidant inhibits tamoxifen-DNA adducts in endometrial explant culture”. Biochem. Biophys. Res. Commun. 2003;310:1039. doi: 10.1016/j.bbrc.2003.09.101. Correspondence regarding M. Sharma et al. [DOI] [PubMed] [Google Scholar]
- (101).Poon GK, Walter B, Lonning PE, Horton MN, McCague R. Identification of tamoxifen metabolites in human Hep G2 cell line, human liver homogenate, and patients on long-term therapy for breast cancer. Drug. Metab. Dispos. 1995;23:377–382. [PubMed] [Google Scholar]
- (102).Sharma M, Shubert DE, Rodabaugh KJ, McGarrigle BP, Vezina CM, Bofinger DP, Olson JR. Antioxidant inhibits tamoxifen-DNA adducts in endometrial explant culture. Biochem. Biophys. Res. Commun. 2003;307:157–164. doi: 10.1016/s0006-291x(03)01134-3. [DOI] [PubMed] [Google Scholar]
- (103).Kim SY, Suzuki N, Santosh Laxmi YR, Rieger R, Shibutani S. Alpha-hydroxylation of tamoxifen and toremifene by human and rat cytochrome P450 3A subfamily enzymes. Chem. Res. Toxicol. 2003;16:1138–1144. doi: 10.1021/tx0300131. [DOI] [PubMed] [Google Scholar]
- (104).Shibutani S, Ravindernath A, Terashima I, Suzuki N, Laxmi YR, Kanno Y, Suzuki M, Apak TI, Sheng JJ, Duffel MW. Mechanism of lower genotoxicity of toremifene compared with tamoxifen. Cancer Res. 2001;61:3925–3931. [PubMed] [Google Scholar]
- (105).Kuramochi H. Conformational studies and electronic structures of tamoxifen and toremifene and their allylic carbocations proposed as reactive intermediates leading to DNA adduct formation. J. Med. Chem. 1996;39:2877–2886. doi: 10.1021/jm960255g. [DOI] [PubMed] [Google Scholar]
- (106).Thompson DC, Thompson JA, Sugumaran M, Moldeus P. Biological and toxicological consequences of quinone methide formation. Chem. Biol. Interact. 1993;86:129–162. doi: 10.1016/0009-2797(93)90117-h. [DOI] [PubMed] [Google Scholar]
- (107).Thompson DC, Perera K, Krol ES, Bolton JL. o-Methoxy-4-alkylphenols that form quinone methides of intermediate reactivity are the most toxic in rat liver slices. Chem. Res. Toxicol. 1995;8:323–327. doi: 10.1021/tx00045a001. [DOI] [PubMed] [Google Scholar]
- (108).Peter MG. Chemical Modifications of Bio-Polymers by Quinones and Quinone Methides. Angew. Chem. Int. Edit. 1989;28:555–570. [Google Scholar]
- (109).Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG. Quinone chemistry and toxicity. Toxicol. Appl. Pharmacol. 1992;112:2–16. doi: 10.1016/0041-008x(92)90273-u. [DOI] [PubMed] [Google Scholar]
- (110).Richard JP, Amyes TL, Bei L, Stubblefield V. The Effect of Beta-Fluorine Substituents on the Rate and Equilibrium-Constants for the Reactions of Alpha-Substituted 4-Methoxybenzyl Carbocations and on the Reactivity of a Simple Quinone Methide. J. Am. Chem. Soc. 1990;112:9513–9519. [Google Scholar]
- (111).Hulbert PB, Grover PL. Chemical rearrangement of phenol-epoxide metabolites of polycyclic aromatic hydrocarbons to quinone-methides. Biochem. Biophys. Res. Commun. 1983;117:129–134. doi: 10.1016/0006-291x(83)91550-4. [DOI] [PubMed] [Google Scholar]
- (112).Potter GA, McCague R, Jarman M. A mechanistic hypothesis for DNA adduct formation by tamoxifen following hepatic oxidative metabolism. Carcinogenesis. 1994;15:439–442. doi: 10.1093/carcin/15.3.439. [DOI] [PubMed] [Google Scholar]
- (113).Bolton JL, Comeau E, Vukomanovic V. The influence of 4-alkyl substituents on the formation and reactivity of 2-methoxy-quinone methides: evidence that extended pi-conjugation dramatically stabilizes the quinone methide formed from eugenol. Chem. Biol. Interact. 1995;95:279–290. doi: 10.1016/0009-2797(94)03566-q. [DOI] [PubMed] [Google Scholar]
- (114).Dehal SS, Kupfer D. CYP2D6 catalyzes tamoxifen 4-hydroxylation in human liver. Cancer Res. 1997;57:3402–3406. [PubMed] [Google Scholar]
- (115).Crewe HK, Ellis SW, Lennard MS, Tucker GT. Variable contribution of cytochromes P450 2D6, 2C9 and 3A4 to the 4-hydroxylation of tamoxifen by human liver microsomes. Biochem. Pharmacol. 1997;53:171–178. doi: 10.1016/s0006-2952(96)00650-8. [DOI] [PubMed] [Google Scholar]
- (116).Wiseman H, Lewis DF. The metabolism of tamoxifen by human cytochromes P450 is rationalized by molecular modelling of the enzyme-substrate interactions: potential importance to its proposed anti-carcinogenic/carcinogenic actions. Carcinogenesis. 1996;17:1357–1360. doi: 10.1093/carcin/17.6.1357. [DOI] [PubMed] [Google Scholar]
- (117).Kroemer HK, Eichelbaum M. “It’s the genes, stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56:2285–2298. doi: 10.1016/0024-3205(95)00223-s. [DOI] [PubMed] [Google Scholar]
- (118).Wilkinson GR, Guengerich FP, Branch RA. Genetic polymorphism of S-mephenytoin hydroxylation. Pharmacol. Ther. 1989;43:53–76. doi: 10.1016/0163-7258(89)90047-8. [DOI] [PubMed] [Google Scholar]
- (119).Coezy E, Borgna JL, Rochefort H. Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res. 1982;42:317–323. [PubMed] [Google Scholar]
- (120).Borgna JL, Rochefort H. Hydroxylated metabolites of tamoxifen are formed in vivo and bound to estrogen receptor in target tissues. J. Biol. Chem. 1981;256:859–868. [PubMed] [Google Scholar]
- (121).Hochner-Celnikier D. Pharmacokinetics of raloxifene and its clinical application. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999;85:23–29. doi: 10.1016/s0301-2115(98)00278-4. [DOI] [PubMed] [Google Scholar]
- (122).Snyder KR, Sparano N, Malinowski JM. Raloxifene hydrochloride. Am. J. Health Syst. Pharm. 2000;57:1669–1675. quiz 1676-1668. [PubMed] [Google Scholar]
- (123).Fiorelli G, Picariello L, Martineti V, Tonelli F, Brandi ML. Estrogen synthesis in human colon cancer epithelial cells. J. Steroid Biochem. Mol. Biol. 1999;71:223–230. doi: 10.1016/s0960-0760(99)00144-2. [DOI] [PubMed] [Google Scholar]
- (124).Buzdar A, O’Shaughnessy JA, Booser DJ, Pippen JE, Jr., Jones SE, Munster PN, Peterson P, Melemed AS, Winer E, Hudis C. Phase II, randomized, double-blind study of two dose levels of arzoxifene in patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2003;21:1007–1014. doi: 10.1200/JCO.2003.06.108. [DOI] [PubMed] [Google Scholar]
- (125).Sato M, Turner CH, Wang T, Adrian MD, Rowley E, Bryant HU. LY353381.HCl: a novel raloxifene analog with improved SERM potency and efficacy in vivo. J. Pharmacol. Exp. Ther. 1998;287:1–7. [PubMed] [Google Scholar]
- (126).Kemp DC, Fan PW, Stevens JC. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug. Metab. Dispos. 2002;30:694–700. doi: 10.1124/dmd.30.6.694. [DOI] [PubMed] [Google Scholar]
- (127).Jordan VC. Metabolites of tamoxifen in animals and man: identification, pharmacology, and significance. Breast Cancer Res. Treat. 1982;2:123–138. doi: 10.1007/BF01806449. [DOI] [PubMed] [Google Scholar]
- (128).Liu X, Pisha E, Tonetti DA, Yao D, Li Y, Yao J, Burdette JE, Bolton JL. Antiestrogenic and DNA damaging effects induced by tamoxifen and toremifene metabolites. Chem. Res. Toxicol. 2003;16:832–837. doi: 10.1021/tx030004s. [DOI] [PubMed] [Google Scholar]
- (129).Mani C, Hodgson E, Kupfer D. Metabolism of the antimammary cancer antiestrogenic agent tamoxifen. II. Flavin-containing monooxygenase-mediated N-oxidation. Drug. Metab. Dispos. 1993;21:657–661. [PubMed] [Google Scholar]
- (130).Berthou F, Dreano Y. High-performance liquid chromatographic analysis of tamoxifen, toremifene and their major human metabolites. J. Chromatogr. 1993;616:117–127. doi: 10.1016/0378-4347(93)80478-m. [DOI] [PubMed] [Google Scholar]
- (131).Yao D, Zhang F, Yu L, Yang Y, van Breemen RB, Bolton JL. Synthesis and reactivity of potential toxic metabolites of tamoxifen analogues: droloxifene and toremifene o-quinones. Chem. Res. Toxicol. 2001;14:1643–1653. doi: 10.1021/tx010137i. [DOI] [PubMed] [Google Scholar]
- (132).Sarabia SF, Zhu BT, Kurosawa T, Tohma M, Liehr JG. Mechanism of cytochrome P450-catalyzed aromatic hydroxylation of estrogens. Chem. Res. Toxicol. 1997;10:767–771. doi: 10.1021/tx970021f. [DOI] [PubMed] [Google Scholar]
- (133).Spink DC, Zhang F, Hussain MM, Katz BH, Liu X, Hilker DR, Bolton JL. Metabolism of equilenin in MCF-7 and MDA-MB-231 human breast cancer cells. Chem. Res. Toxicol. 2001;14:572–581. doi: 10.1021/tx000219r. [DOI] [PubMed] [Google Scholar]
- (134).Zhang F, Chen Y, Pisha E, Shen L, Xiong Y, van Breemen RB, Bolton JL. The major metabolite of equilin, 4-hydroxyequilin, autoxidizes to an o-quinone which isomerizes to the potent cytotoxin 4-hydroxyequilenin-o-quinone. Chem. Res. Toxicol. 1999;12:204–213. doi: 10.1021/tx980217v. [DOI] [PubMed] [Google Scholar]
- (135).Shen L, Pisha E, Huang Z, Pezzuto JM, Krol E, Alam Z, van Breemen RB, Bolton JL. Bioreductive activation of catechol estrogen-ortho-quinones: aromatization of the B ring in 4-hydroxyequilenin markedly alters quinoid formation and reactivity. Carcinogenesis. 1997;18:1093–1101. doi: 10.1093/carcin/18.5.1093. [DOI] [PubMed] [Google Scholar]
- (136).Chen Y, Shen L, Zhang F, Lau SS, van Breemen RB, Nikolic D, Bolton JL. The equine estrogen metabolite 4-hydroxyequilenin causes DNA single-strand breaks and oxidation of DNA bases in vitro. Chem. Res. Toxicol. 1998;11:1105–1111. doi: 10.1021/tx980083l. [DOI] [PubMed] [Google Scholar]
- (137).Shen L, Qiu S, Chen Y, Zhang F, van Breemen RB, Nikolic D, Bolton JL. Alkylation of 2′-deoxynucleosides and DNA by the Premarin metabolite 4-hydroxyequilenin semiquinone radical. Chem. Res. Toxicol. 1998;11:94–101. doi: 10.1021/tx970181r. [DOI] [PubMed] [Google Scholar]
- (138).Zhang F, Swanson SM, van Breemen RB, Liu X, Yang Y, Gu C, Bolton JL. Equine estrogen metabolite 4-hydroxyequilenin induces DNA damage in the rat mammary tissues: formation of single-strand breaks, apurinic sites, stable adducts, and oxidized bases. Chem. Res. Toxicol. 2001;14:1654–1659. doi: 10.1021/tx010158c. [DOI] [PubMed] [Google Scholar]
- (139).Yao J, Chang M, Li Y, Pisha E, Liu X, Yao D, Elguindi EC, Blond SY, Bolton JL. Inhibition of cellular enzymes by equine catechol estrogens in human breast cancer cells: specificity for glutathione S-transferase P1-1. Chem. Res. Toxicol. 2002;15:935–942. doi: 10.1021/tx020018i. [DOI] [PubMed] [Google Scholar]
- (140).Li Y, Yao J, Chang M, Nikolic D, Yu L, Yager JD, Mesecar AD, van Breemen RB, Bolton JL. Equine catechol estrogen 4-hydroxyequilenin is a more potent inhibitor of the variant form of catechol-O-methyltransferase. Chem. Res. Toxicol. 2004;17:512–520. doi: 10.1021/tx0342464. [DOI] [PubMed] [Google Scholar]
- (141).Yao J, Li Y, Chang M, Wu H, Yang X, Goodman JE, Liu X, Liu H, Mesecar AD, Van Breemen RB, Yager JD, Bolton JL. Catechol estrogen 4-hydroxyequilenin is a substrate and an inhibitor of catechol-O-methyltransferase. Chem. Res. Toxicol. 2003;16:668–675. doi: 10.1021/tx0340549. [DOI] [PubMed] [Google Scholar]
- (142).Zhang Z, Chen Q, Li Y, Doss GA, Dean BJ, Ngui JS, Silva Elipe M, Kim S, Wu JY, Dininno F, Hammond ML, Stearns RA, Evans DC, Baillie TA, Tang W. In vitro bioactivation of dihydrobenzoxathiin selective estrogen receptor modulators by cytochrome P450 3A4 in human liver microsomes: formation of reactive iminium and quinone type metabolites. Chem. Res. Toxicol. 2005;18:675–685. doi: 10.1021/tx0496789. [DOI] [PubMed] [Google Scholar]
- (143).Sanchez C, Shibutani S, Dasaradhi L, Bolton JL, Fan PW, McClelland RA. Lifetime and reactivity of an ultimate tamoxifen carcinogen: The tamoxifen carbocation. J. Am. Chem. Soc. 1998;120:13513–13514. [Google Scholar]