

Abstract
In view of the worldwide spread of multidrug resistance of Mycobacterium tuberculosis, there is an urgent need to discover antituberculosis agent with novel structures. InhA, the enoyl acyl carrier protein reductase (ENR) from Mycobacterium tuberculosis is one of the key enzymes involved in the mycobacterial fatty acid elongation cycle and has been validated as an effective antimicrobial target. We report here discovery through high throughput screening of a series of pyrrolidine carboxamides as a novel class of potent InhA inhibitors. Crystal structures of InhA complexed with three inhibitors have been used to elucidate the inhibitor binding mode. The potency of the lead compound was improved over 160-fold by subsequent optimization through iterative microtiter library synthesis followed by in situ activity screening without purification. Resolution of racemic mixtures of several inhibitors indicate that only one enantiomer is active as an inhibitor of InhA.
Tuberculosis (TB) is the leading cause of morbidity and mortality among the infectious diseases. The World Health Organization (WHO) has estimated that one-third of the world's population, nearly 2 billion people mostly in the developing countries,1 have been infected with Mycobacterium tuberculosis, the causative agent of TB. Among the infected individuals eight million develop active TB and nearly 2 million people die from the disease annually.2 In recent years, the pandemic of AIDS has had a major impact on the worldwide TB problem. On one hand, HIV infection is the most potent risk factor for converting latent TB into the active, transmissible form, thus fueling the spread of TB; on the other hand, TB bacteria can accelerate the progress of AIDS infection. One third of the increase in the incidence of TB in the past 5 years can be attributed to coinfection with HIV.2 This situation has been further exacerbated by the emergence of multidrug-resistant tuberculosis (MDR-TB) strains that are resistant to some or most current anti-TB drugs.3 Over the decade, it is estimated that as many as 50 million people worldwide have been infected with MDR-TB strains. According to WHO, from 2002 to 2020, there will be about one billion more people newly infected with TB and approximately 36 million deaths if the worldwide ravage of tuberculosis is left unchecked.3 Despite the increasing worldwide incidence of TB and its alarming threat towards the public health, no novel antituberculosis drugs have been introduced into clinical practice over the past four decades. The impact of ever increasing drug resistance, the serious side effects of some current anti-TB drugs, and the lack of efficacy of current treatments in immunodepressed patients, combine to make the development of new antimycobacterial agents an urgent priority.
The enzymes involved in the bacterial fatty acid biosynthetic pathway, the fatty acid synthase system, are attractive targets for the design of new antibacterial agents.4-7 Fatty acid biosynthesis in bacteria is catalyzed by a set of distinct, mono-functional enzymes collectively known as the type II FAS (FASII). These enzymes differ significantly from the type I FAS (FASI) in mammalians, in which all of the enzymatic activities are encoded in one or two multifunctional polypeptides. This distinctive difference in the FAS molecular organization between most bacteria and mammals makes possible the design of specific inhibitors of increased selectivity and lower toxicity. Mycobacterium tuberculosis contains unique signature fatty acids, the mycolic acids, that are unusually long chain α-alkyl, β-hydroxy fatty acids of 60−90 carbons.8 The TB-specific drugs isoniazid (isonicotinic acid hydrazide (INH)) and ethionamide (Figure 1) have been shown to target the synthesis of these mycolic acids, which are central constituents of the mycobacterial cell wall. The biosynthesis of mycolic acids is achieved by the FAS in M. tuberculosis. Unlike other bacteria, M. tuberculosis is unique in that it possesses both type I and type II fatty acid biosynthetic pathways. FASI in M. tuberculosis is responsible for generation of the shorter saturated alkyl chain fatty acids, including the 24 carbon α-branch of mycolic acids. Some of the products from the FASI system, such as the C16–C26 fatty acid products, are later transferred to the FASII system, where they are further elongated to up to C56, forming the meromycolate chain that serves as the precursor for the final mycolic acids.
Figure 1.

Chemical structures of InhA inhibitors (R represents various substituents).
Among the enzymes involved in FASII, the NADH-dependent enoyl-ACP reductase encoded by the Mycobacterium gene inhA is a key catalyst in mycolic acid biosynthesis. Studies over the years have established that InhA is the primary molecular target of INH,9 the drug that for the past 40 years has been, and continues to be, the frontline agent for the treatment of TB. As a prodrug, INH must first be activated by KatG, a catalase-peroxidase that oxidizes INH to an acyl-radical that binds covalently to NADH, the co-substrate for InhA.10 The INH-NADH adduct then functions as a potent inhibitor of InhA. The requirement for INH activation opened a backdoor for the development of drug resistance by M. tuberculosis. Indeed, KatG-associated mutations accounts for 50% of the INH-resistant clinical isolates.11 Thus, direct InhA inhibitors that avoid this activation requirement would be promising candidates for the development of novel antitubercular agents. Other than the well-known diazoborines12 and triclosan,13 both of which are non-selective and relatively weak agents, two series of InhA direct inhibitors, pyrazole derivatives 1 (Genz 8575)14 and indole-5-amides 2 (Genz 10850)14 (Figure 1), have been reported to have both in vivo and in vitro activity. In this study, we report the discovery of pyrrolidine carboxamides as a novel series of InhA inhibitors by means of high throughput screening, followed by the application of a microtiter synthetic strategy on the focused library combined with in situ screening without purification for further structural optimization.
Results and Discussion
Identification of Novel InhA Inhibitors
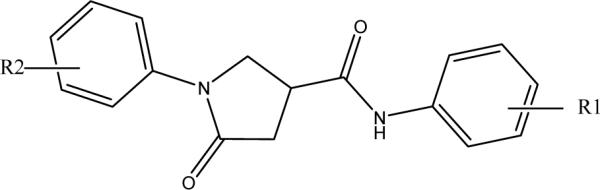
To identify lead compounds targeting the M. tuberculosis InhA, we performed a high throughput screen of 30,000 compounds, from which 30 compounds were identified as potential InhA inhibitors exhibiting at least 50% InhA inhibitory activity at 30 μM. In order to eliminate potential false positive hits that might result from aggregate-forming inhibitors (AFI),15 it is necessary to submit all the hits identified to a subsequent verification test. Consequently, the potencies of each compound in the presence or absence of 0.01% X-Triton were compared. As reported earlier, this is a simple approach for the identification of promiscuous inhibitors.16 None of the 30 hits displayed AFI behavior. In addition, none of the hits was found to be active against the enoyl ACP reductase from E. coli (ecENR) or the malaria parasite Plasmodium falciparum (pfENR), which further rules out the possibility that they are promiscuous inhibitors since AFI should inhibit these enzymes indiscriminately (data not shown). These compounds can be classified into 13 structurally diverse groups. The presence of substantial scaffold differences among the inhibitor groups suggests a high tolerance of the InhA binding pocket, a finding consistent with the broad substrate specificity required to accommodate C26 to C56 acyl CoA derivatives as substrates. Among them, a pyrrolidine carboxamide d6 (Figure 2) the first representative of a new class of InhA inhibitors showing an IC50 of 10.05 μM (Table 1). Given the potency, the commercial availability of this series, and the relatively accessible chemical synthesis of amide compounds, the pyrrolidine carboxamide series was chosen for optimization.
Figure 2.

Structure of lead compound d6 (left) and general structure of pyrrolidine carboxamides.
Table 1.
InhA inhibition activities of pyrrolidine carboxamide compounds (cyclohexyl)
 | |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) |
| s1 | H | H | 10.66 ± 0.51 |
| s2 | H | 2-COOMe | 34.88 ± 2.06 |
| s3 | H | 2-Br | >100 |
| s4 | H | 3-Br | 0.89 ± 0.05 |
| s5 | H | 4-Br | 28.02 ± 4.29 |
| s6 | H | 3-Cl | 1.35 ± 0.05 |
| s7 | H | 4-Cl | >100 |
| s8 | H | 3-I | 26%c |
| s9 | H | 4-I | 14.50 ± 0.79 |
| s10 | H | 3-Me | 16.79 ± 0.54 |
| s11a | H | 3- CF3 | 3.51 ± 0.09 |
| s12 | H | 3-NO2 | 10.59 ± 0.48 |
| s13 | H | 3-NHAC | >75 |
| s14 | H | 3-CH2(CH3)2 | 56%c |
| s15b | H | 3-CH2(CH3)2 | 5.55 ± 0.21 |
| s16 | H | 4-NHAC | >100 |
| s17 | H | 4-Ac | 73.58 ± 9.97 |
| s18 | H | 4-OEt | >100 |
| s19 | H | 4-OPh | >100 |
| d1 | 2-Cl | 4-Cl | 56.02± 10.23 |
| d2 | 2-Cl | 5-Cl | 56.50 ±11.09 |
| d3 | 2-Me | 5-Cl | 0.97 ± 0.03 |
| d4 | 3-Me | 4-Br | 37.41 ± 1.76 |
| d5 | 2-Me | 6-Me | >100 |
| d6 | 2-Me | 5-Me | 10.05 ± 0.33 |
| d7b | 3-Me | 5-Me | 3.14 ± 0.12 |
| d8 | 2-Me | 3-Cl | 23.12 ± 1.00 |
| d9 | 2-Me | 4-NO2 | 31.37 ± 1.45 |
| d10a | 3-F | 5-F | 1.49 ± 0.05 |
| d11 | 3-Cl | 5-Cl | 0.39 ± 0.01 |
| d12a | 3-Br | 5-CF3 | 0.85 ± 0.05 |
| d13a | 3-OMe | 5-CF3 | 1.30 ± 0.04 |
| d14a | 3-CF3 | 5-CF3 | 3.67 ± 0.17 |
| d15 | 2-OMe | 5-Cl | 1.60 ± 0.06 |
| d16 | 3-Cl | 4-F | 14.83 ± 0.98 |
| d17 | 3-Br | 4-CH3 | 51%c |
| d18 | 3,4 | —OCH2CH2O- | >100 |
Synthesized by the oxalyl chloride method
Synthesized by the HBTU/DIEA method
Percentage of InhA inhibition at 15 μM
Initial in silico docking studies of this compound into the InhA active site suggested the formation of a hydrogen-bonding network between d6, enzyme active site residues, and the NAD+ cofactor that probably serves as the key feature that governs the orientation of the compound within the active site. The dual hydrogen bonding network involved the oxygen atom on the pyrrolidine carbonyl group, InhA catalytic residue Tyr158, and the NAD+. This hydrogen bonding network seems to be a conserved feature among all the InhA-inhibitor complexes identified so far.14 The same hydrogen bonding network was observed in the crystal complexes of triclosan and 2 with InhA.14 Since the molecular modeling study suggested a central role of the pyrrolidine structure in formation of the hydrogen bonding network, we initially focused on exploration of the effect of substituents on the phenyl ring (ring A) and the cyclohexyl ring (ring C) while keeping the core feature of a 5-oxo-pyrrolidine (ring B) (Figure 2).
Derivatization of Ring A
The modifications in this series of compounds focused on the phenyl ring (ring A) of the lead compound while the rest of the structure, including the cyclohexyl ring (ring C), was kept intact. The pyrrolidine carboxamide derivatives with ring A substitutions were obtained in three ways: 1) by purchase from commercial sources; 2) from a focused compound library generated using a microtiter synthesis approach; and 3) through traditional synthetic methods. The initial round of pyrrolidine compounds, selected on the basis of traditional medicinal chemistry principles, diversity calculations, and molecular modeling, was purchased. The subsequent synthesis and evaluation of compounds using the microtiter plate approach were based on the preliminary structure and activity relationship (SAR) study derived from the first round of compounds.
Microtiter Synthesis and In Situ Screening of InhA Inhibitors
Recently, Wu et al reported a rapid diversity-oriented synthesis in microtiter plates for in situ screening of enzyme inhibitors.17-19 Small-scale parallel syntheses were carried out in 96-well plates and the final products were used directly, without product isolation, for biological screening. The advantages of this methodology are that the reaction is fast and in parallel, diversity can be achieved by careful design and choice of reactants, and the small size of the reaction makes the experiment economically viable. Given the benign conditions and high efficiency (more than 80% yield) of amide synthesis, the application of this strategy is ideal for optimization of the pyrrolidine carboxamide series of InhA inhibitors (Figure 3).
Figure 3.

Microtiter amide library synthesis of amines with the core template pyrrolindine carboxylic acid.
To minimize the background noise for the in situ activity screening, it is first necessary to determine the tolerance limits of InhA for the solvent in the reaction mixture (i.e., N,N-dimethylformamide, DMF), the peptide coupling reagent 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and the base N,N-diisopropylethylamine (DIEA). With 0.5% DMF in the assay buffer (v/v), the activity of InhA was inhibited about 10%. With up to 100 μM HBTU or DIEA, the rate of the reaction was the same as that observed in the absence of these agents. For the screening assay, the final concentration of the reaction mixture of these two agents was 15 μM and the concentration of DMF did not exceed 0.5%.
As compounds with substituents at the meta-position exhibited the best inhibitory activity among the initially tested commercially available compounds, we prepared a library focusing on diversification of the meta-substituents on the phenyl ring. The microtiter library was synthesized using the amide-forming reaction catalyzed by peptide coupling reagents (see Supporting Information Figure S1). The cyclohexyl pyrrolidine carboxylic acid, the core structure for the focused library, was reacted with a diversity of amines in the presence of HBTU (1 equiv) and DIEA (2 equiv) in DMF.19 Sixteen aniline derivatives with various meta-substituents (see Supporting Information Figure S1) were generated and evaluated in situ. The reaction was monitored by TLC and the identity of the desired product in each well was confirmed by LC-MS. After aqueous dilution of the reaction mixture, each product was tested without purification in the 96-well plate format at a concentration of 15 μM. The best compounds identified in the in situ enzyme assay were then synthesized individually, purified by preparative TLC, characterized, and the IC50 of the purified product was determined.
Amide Synthesis with Traditional Chemical Synthesis
During the library preparation and reaction monitoring, it was noted that no desirable product was detected by LC-MS with certain type of amines when the mild amide-forming agent HBTU was used as the coupling reagent. Most of these unreactive compounds contained strong electron-withdrawing groups at the meta-position. In contrast, the reaction proceeded smoothly with amines substituted with neutral groups, such as 3,5-dimethylaniline. Furthermore, it was observed that the “inert” anilines were consistently unable to form the desired amide product when other pyrrolidine carboxylic acid analogues were used.
To investigate the potential InhA inhibitory property of compounds derived from the unreactive precursors, other amide synthetic strategies (not applicable for microtiter synthesis due to the highly reactive reactants) were employed, including the oxalyl chloride method20 and the use of peptide coupling reagents such as EDCI/DMAP (1-ethyl-3-(3’-dimethylaminopropyl) carbodiimide /4-dimethyl-aminopyridine).21 The synthesis results showed that the oxalyl chloride method was the most suitable for this class of anilines, from which all the desired products were successfully isolated in varying yields.
Ring A: Modification of the Phenyl Group
The InhA inhibition activities of all the pyrrolidine carboxamide compounds with ring A modifications are summarized in Table 1. Compound s1, without any substituent on the phenyl ring, has an IC50 of ∼10 μM and serves as the standard for potency comparison. In the phenyl monosubstituted series, compounds generally lose activity when the hydrogen at the para-position of the aniline is replaced by an X (halo), OEt, OPh, OCOCH3, or NHCOCH3. A substituent at this para-position may clash with the InhA side chain residues located at the bottom of the hydrophobic binding pocket. Electron-withdrawing halo substituents at the 4-position weakened the potency of the inhibitors with an increasingly adverse effect on the potency in the order I > Br > Cl (s9, s5, s7). A 4-Cl replacement, in particular, resulted in at least a 10-fold reduction in potency. Like a 2-Br substituent, the introduction of a 2-CO2Me (s2) also led to a 3-fold decrease in potency. Comparison of the potencies of the three monobromo-substituted compounds (2-Br, s3; 3-Br, s4; 4-Br, s5) clearly indicates that the meta-position is the best position for substitution, giving rise to a 10-fold improvement in potency (IC50 0.89 μM), and a 3-CF3 or 3-Cl replacement (s6, s11) also displayed a 3−6 fold improvement over the reference compound (s1). Unlike a 3-Cl or 3-Br, the activity of s8 (3-I) was much lower (26% inhibition at 15 μM), whereas the inhibitory activity remained unchanged in the presence of a 3-NO2 (s12). These results suggest that there is a size limit requirement for the electron-withdrawing group at the 3-position in order to achieve the optimal activity. In agreement, other meta-substitutions, such as a 3-Me or 3-NHCOCH3 (s10, s13), weaken the activity. The results of the in situ screening of the microtiter library reveal that only two compounds, s14 and s15 (3-isopropoxy and 3-isopropyl), inhibit more than 50% of InhA activity when tested at 15 μM. Compound s15 was synthesized and exhibited an IC50 of 5.55 μM. No activity was detected for compounds with other meta-substituents, including –OH, –OCH3, -COCH3, -HNSO2CH3, and -CONH2 (g51, g52, g55, g57, g60 Figure S1).
Among the dual substitutions, the presence of symmetric 3- and 5-chloro substituents improves potency. Compound d11 with 3,5-dicloro substituents is the most potent compound identified in this series (IC50 = 0.39 μM). The greatly improved potency is unlikely to result from hydrogen bond interactions between the 3,5-halo atoms and InhA active site residues because the 3,5-difluro replacement (d10) with stronger hydrogen bond acceptors gave a 4-fold reduction in activity. In the absence of a substituent adjacent to the meta-chloro (d3, d15), the compounds had an efficacy similar to that of s6 (3-Cl). However, the presence of a 2-Me (d8) adjacent to the 3-Cl greatly diminished the potency. A para-substituent even for the most potent compound consistently reduced inhibitory activity (g53, g54 Figure S1). Consistent with our earlier findings, the presence of a substituent found to be unfavorable in the monosubstituted series, such as an ortho- (d1, d2, d5, d9) or para-substituent (d4, d16, d17, d18) decreases the potency of the disubstituted inhibitors and even reverses the improvement introduced by a beneficial substituent, such as a 5-Cl (d2). Compared with the precursor with the corresponding single substituent at the 3 position, the introduction of an additional CH3 or CF3 group at the meta-position has no effect on inhibitory activity (d7, d12, d13, d14).
Furthermore, aniline analogues g47-49 (Figure S1) were investigated to test the consequences of methylene group insertion between the sidechain and the pyrrolamide carboxamide core using one of the best compounds, s4, as the framework. The absence of detectable activity for any of the resulting compounds at a concentration of 15 μM indicates the unfavorable nature of chain extension.
Analysis of the InhA Inhibitor Binding Site
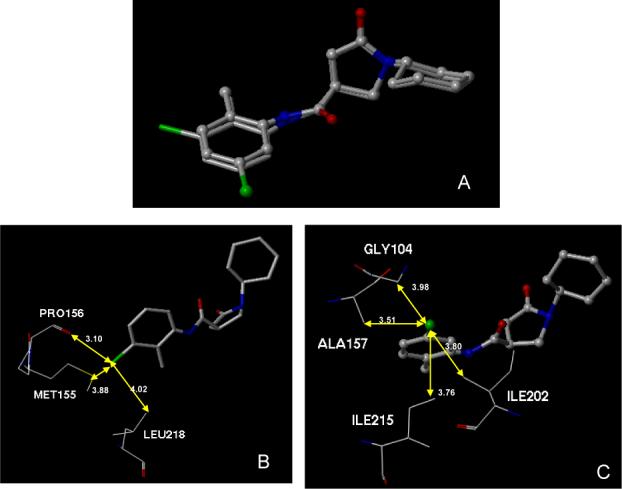
The crystal structures of InhA-inhibitor complexes incubated with NAD+ were solved for three InhA inhibitors exhibiting various degrees of inhibitory activity. All three of the inhibitors were found to bind in essentially the same manner within the enzyme active site (Figure 4A). Comparison of these structures with the published InhA·NAD+·triclosan structures reveals the same hydrogen-bonding pattern with the substrate NAD+ and side-chain of Tyr158 (Figure 4C), which is one of the catalytic residues of the enzyme. Such a hydrogen bond network is also consistent with our modeling prediction and is conserved in all known ENR-inhibitor complexes,22-26 which further confirms the important role of the ring B moiety (Figure 2) in the interaction between the inhibitor and InhA. Compared to native InhA, with the exception of Tyr 158, the positions of all other active site residues remained unchanged upon binding of the inhibitors (Figure 4B). To accommodate the inhibitor within the active site, the Tyr 158 side-chain rotated −90° to optimize the hydroxyl orientation for hydrogen bond formation with the inhibitor (Figure 4B).
Figure 4.

Inhibitor d11 bound to the active sites of the M. tuberculosis InhA. A, Purple molecular surface shows the active site cleft in which compound d11 (in capped stick model) binds. B, conformation comparison of catalytic residue Tyr 158 before (in green) and after (in purple) the binding of d11 to InhA, showing the rotation of Tyr158 in the d11-InhA complex. C, Details of InhA-d11 interactions. Key residues within 4.5 sphere of d11 in the binding pocket are shown. The oxygen on the carbonyl group of the five member ring lactam makes hydrogen-bonding interactions with the 2’-hydroxyl moiety of the nicotinamide ribose and the hydroxyl group of Tyr158 (blue line).
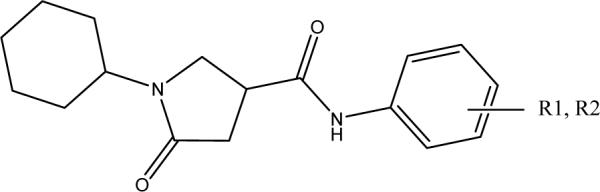
In all three of the complexes, the carbonyl group oxygen of the five member ring lactam is hydrogen-bonded to the 2’-hydroxyl moiety of the nicotinamide ribose and the hydroxyl group of Tyr158. The cyclohexyl ring had van der Waals interactions with the side chains of Gly96, Phe97 and the nicotinamide ribose. The lactam ring of the inhibitor also interacted with the NAD+ nicotinamide ring and the side-chains of Met161 and Met199. The only structural difference among these inhibitors lies in the substituted phenyl (ring A) of the inhibitor. Except for the meta-substituent, the major scaffold of the unsubstituted lead compound s1 overlaid well with that of d11 (Figure 5A), which suggested that the 25-fold increase in potency for the latter might stem from additional interactions between the halogen atom(s) and the side-chains of active site residues. The binding modes of ring A of s4 and d11 are nearly identical (Figure 5B). The 3-halo atom (Br, Cla) of ring A was surrounded by Ala157, Gly104, Tyr158, and Met103 within 4.1 Å suitable for van der Waal's contacts. Given the relative size of Br and Cl, a 3-Br is located closer to the key residue Tyr158 than a Cla, which might contribute to the higher potency of 3-Br (IC50 0.89 μM) over the corresponding singly-substituted 3-Cl (IC50 1.35 μM). For the dichloro-substituted compound (d11, IC50 0.39 μM), additional hydrophobic contacts were observed between the other 3-Cl (Clb) atom and the side-chains of Pro156, Met155 and Leu218. These interactions may contribute to the increased potency of this compound over the singly substituted 3-Br compound.
Figure 5.
Overlay of pyrrolidine carboxamide InhA inhibitors and triclosan in pairs showing the similar binding mode at the active site of InhA. A, Superimposition of d11 (3,5-dichloro substitution) with s1 (unsubstituted phenyl ring). B, Overlay of d11 and s4 (3-bromo substituted phenyl ring). C, Superimposition of d11 with dual triclosan molecules.
The unusually broad substrate specificity of M. tuberculosis InhA, which allows it to process long to very long chain (C16 and greater) fatty acyl substrates, requires a considerably larger substrate-binding cavity than the ENRs from other organisms. In a previous study, a unique doubly-bound InhA·triclosan complex, with two triclosan molecules bound within the active site, showed a remarkably similar binding mode to that of a long chain fatty acyl substrate analog.14 Such a ternary triclosan complex has not been observed with any other ENR. The large size of the binding pocket can apparently accommodate a second triclosan molecule. Thus, compounds incorporating the features of both triclosan molecules may be good candidates for the development of inhibitors selectively targeting M. tuberculosis. As seen from Figure 5C, the ring B and ring C section of d11 superimposed well with the first triclosan, and ring A was partially superimposable with the second triclosan molecule.
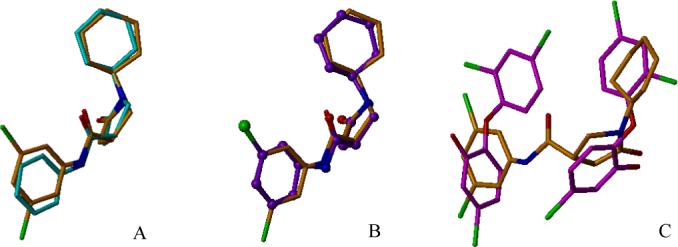
Given the slight structural difference between d3 and d8 and more than 20 fold potency difference between them (IC50 d3, 0.97 μM; d8, 23.12 μM), we also crystallized and solved the structures of the two inhibitor-InhA complexes to elucidate the role of the chloro atom. As seen in Figure 6A, other than the chloro atom position, the rest of the structures of the two compounds can be overlapped at the active site of InhA. Therefore, the observed potency difference must result from the different interactions between the chloro atoms and the various amino acid residues surrounding them. For compound d8, only three atoms are found to be within 4.1 Å sphere of the 3-Cl. The only favorable but rather weak hydrophobic interaction with the sidechain of I218 is 4.02 Å away (Figure 6B). The unfavorable electron repulsion caused by the close interaction between 3-Cl and the respective carbonyl oxygen of P156 (3.10 Å) and sulfur atom on Met155 (3.88 Å) might explain the modest potency observed in this compound. In contrast, unlike d8, the 5-chloro in compound d3 avoids such an unfavorable interaction. In addition, the d3-InhA complex revealed multiple favorable hydrophobic interactions formed between the 5-Cl and the InhA residues in the vicinity. Three aliphatic amino acid sidechains from Ala157, I215, I202 are within 4.0Å and may provide hydrophobic contacts with the inhibitor (Figure 6C).
Figure 6.
Overlay of compound d3 and d8 at the active site of InhA. 6A, Superimposition of d3 (2-Me, 5-Cl disubstitution) with d8 (2-Me, 3-Cl disubstitution). 6B, InhA residues within 4.1 Å of chloro atom in d8. 6C, InhA residues within 4.1 Å of chloro atom in d3.
In conclusion, among the mono substituents, electron withdrawing groups of a small to moderate size at the meta-position of the phenyl ring (ring A) gave the best InhA inhibitory activity; potency was generally reduced or lost in the presence of ortho-substituents or, especially, para-substituents. For disubstituted compounds, the inhibitory potency was usually improved by ring A 3,5-disubstitution. Thus, the 3,5 dichloro-substituted compound displayed the best activity of the series. However, inhibitory effect was generally weakened when the two substituents were adjacent to each other.
Ring A Modification: Phenyl Group Replacement
The effect on activity of replacing the phenyl ring with other ring structures was also examined. The commercially available compounds utilized for this purpose are listed in Figure 7. Both primary and secondary amines with aromatic or aliphatic ring structures were tested (r1-r13). Except for r7 (IC50 = 5.1 μM), none of the compounds inhibited InhA when tested at a 20 μM concentration. The activity was lost when the aromatic phenyl ring was replaced by a cyclopentyl or cyclohexyl ring (r8, r12). No inhibitory activity was observed when the phenyl ring was opened and replaced with an isobutyl group (r13). Compound r7 inhibited 65% of InhA activity, suggesting that potency improvements might be achieved by extending the aromatic ring structures and thus increasing the potential for hydrophobic interactions between the inhibitor and the enzyme.
Figure 7.

Structures of pyrrolidine carboxamides with phenyl ring surrogate.
To further explore the alternatives to the phenyl ring, a new library was generated using 45 diverse amines. Considering the hydrophobic nature of the phenyl group binding site in the InhA-inhibitor complex, all these entries have at least two ring structures in order to increase hydrophobic contacts (Figure S1). The library was rapidly generated via microtiter synthesis and was evaluated in the follow up in situ screening without product isolation. Out of the 45 members of the library, 16 compounds exhibited at least 20% InhA inhibitory activity when tested at a concentration of 15 μM, and four compounds inhibited 90% of InhA activity. Compounds showing at least 50% InhA inhibition were synthesized and purified by chromatography for IC50 determinations. The initial screening results and follow-up IC50 values are summarized in Table 2. Relative to the lead compound s1(IC50 10.66 μM), InhA inhibition was greatly increased by the introduction of appropriate polyaromatic moieties. Three compounds with novel multiple ring structures had submicromolar IC50 values (p20, p21, p24). The 3-phenyl substituent in compound p24 gave the same potency as the 3,5-dichloro substitution of compound d11 (IC50 0.39 μM), a 25-fold increase in activity over s1. The activity was maintained with the insertion of a CH2 between the two phenyl rings in compound p21. However, the potency was reduced nearly 10-fold upon insertion of another oxygen atom between the two phenyl rings (p9). In contrast, inhibitory activity was completely abolished when the phenoxyl replacement shifted from the meta- to the para-position (s19, Table 1). Also, the potency was much lower when a carbonyl group rather than a methylene carbon was inserted between the phenyls, as in compound p19. Enhanced InhA inhibition is also observed upon the introduction of three fused aromatic ring structures (p20, p31). However, the fusion site between the aromatic amine and the pyrrolidine carboxylic acid seems to be a key factor in the improved potency of p31 (90% inhibition at 15 μM, IC50 1.39 μM), as a much lower activity was observed at a 15 μM concentration when the amine group was slightly shifted, as in compound p30 (38% inhibition at 15 μM). Compounds p31 and p33 had similar potencies, with IC50 1.39 and 2.57 μM, respectively. Unlike the weak activity observed with the simple 2-, 3- or 2,4-disubstituted aniline analogues, compounds p31 and p33 are much better inhibitors than s1 (IC50 10 μM, Table 1). It is possible that a significant induced conformational change of the enzyme occurs to accommodate the bulky substituents of these compounds, giving rise to additional interactions that stabilize the complex.
Table 2.
Pyrrolindine carboxiamide microtiter library screening results
| ID | Structure | Inhibition % 15μM | IC50 |
|---|---|---|---|
| p24 |  |
97 | 0.39 ± 0.01 |
| p21 | 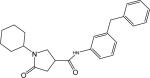 |
96 | 0.41 ± 0.01 |
| p20 | 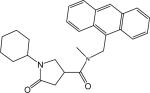 |
96 | 0.75 ± 0.04 |
| p31 | 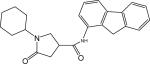 |
90 | 1.39 ± 0.02 |
| p9 | 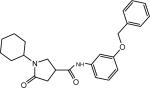 |
89 | 3.39 ± 0.10 |
| p37 | 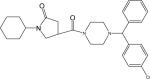 |
83 | 4.47 ± 0.28 |
| p33 | 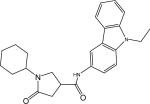 |
78 | 2.57 ± 0.17 |
| p27 | 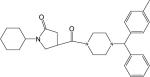 |
77 | 5.18 ± 0.34 |
| p28 | 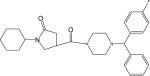 |
72 | 6.41 ± 0.12 |
| p19 | 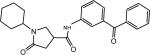 |
66 | ND |
| p36 | 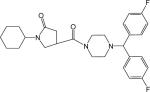 |
50 | 5.51 ± 0.22 |
| p30 | 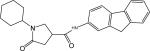 |
38 | ND |
| p26 | 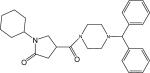 |
34 | ND |
| p25 | 32 | ND | |
| p4 | 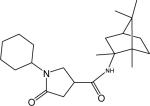 |
26 | ND |
| p35 | 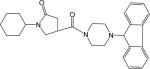 |
20 | ND |
ND: not determined
Several compounds that include the piperazine ring in their scaffold also exhibited strong InhA inhibition (p27, p28, p36 and p37). Generally, better inhibition activity was observed with additional substituents at the para-position of the phenyl ring attached to the piperazine. Compound p37, with IC50 = 4.47 μM, is the best of these compounds. Based on the structure of the InhA inhibitor 2 (Figure 1), the 1-(9H-fluoren-9-yl) piperazine function was included in the library. However, the resulting pyrrolidine carboxamide p35 only displayed modest inhibition (20% at 15 μM). In contrast, 2 inhibited 97% of InhA activity under the same conditions, which suggested a much less optimized binding mode for p35 (pyrrolidine carboxylic acid) than for 2 (indole 5-carboxylic acid). The library screening results focused on ring A modification illustrate that the introduction of appropriate hydrophobic moieties as phenyl ring replacements can give compounds of improved potency. The enhanced potency of InhA inhibitors with bulky aromatic groups is consistent with the large size of the InhA binding pocket and the flexibility of the enzyme to accommodate novel ligands at its active site.
Derivatization of Ring C
Replacement of the Cyclohexyl with Substituted Phenyl Rings
To further probe the SAR for the cyclohexyl terminus (ring C), several compounds were tested in which the cyclohexyl was replaced by unsubstituted or substituted phenyl rings while maintaining optimal substituents on ring A (R1 = 3-Cl, Br) (Table 3). Comparison of the activities of these phenyl replacements with the cyclohexyl counterparts clearly demonstrated that the substitution was unfavorable. A 3- or 15-fold potency decrease, respectively, was noted when the cyclohexyl was replaced by the unsubstituted phenyl in 3a (3-Cl) and 3i (3-Br). The same trend was also observed for 3d, for which the IC50 increased to above 100 μM. A substituent at the para-position of the phenyl ring appeared to be intolerable (3b, 3c, 3f-h), as the inhibitory activity of these compounds was completely abolished (IC50 > 100 μM) even with a substituent as small as a methyl (3b). For the other series (R1 = 3-Br), except for 3j, for which the potency dropped 2-fold from 13.6 to 30 μM, no inhibitory effect was detected for 3k and 3l in which a 4-Me or 3,4-dimethyl group was present on the phenyl ring, respectively. In conclusion, InhA inhibition activities were greatly decreased upon replacement of the cyclohexyl by a phenyl group. The chair conformation of the cyclohexyl appears to be more favorable than the flat phenyl ring for interactions within the active site.
Table 3.
InhA inhibition activities of pyrrolidine carboxamide compounds (phenyl)
 | |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) |
| 3a | 3-Cl | H | 3.94 ± 0.34 |
| 3b | 3-Cl | 4-Me | >100 |
| 3c | 3-Cl | 4-OPr | >100 |
| 3d | 3-Cl, 2-Me | H | >100 |
| 3e | 3-Cl, 2-Me | 3-NO2 | >100 |
| 3f | 3-Cl, 2-Me | 4-Br | >100 |
| 3g | 3-Cl, 2-Me | 4-Me | >100 |
| 3h | 3-Cl, 2-Me | 4-OiPr | >100 |
| 3i | 3-Br | H | 13.55 ± 0.85 |
| 3j | 3-Br | 4-F | 29.23 ± 2.17 |
| 3k | 3-Br | 4-Me | >100 |
| 3l | 3-Br | 3,4 diMe | >100 |
Replacement of the Cyclohexyl by Other Saturated Rings
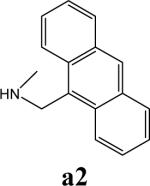
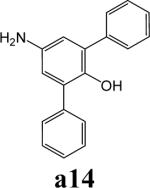
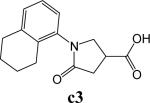
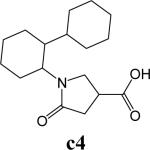
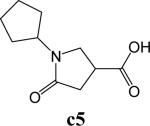
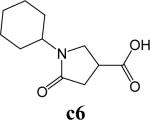
To further explore alternatives to the cyclohexyl ring, another library was prepared by microtiter synthesis. A series of pyrrolidine carboxylic acid analogues was first synthesized in which monocyclic saturated rings of 5−8 carbons and multiple rings (c1, c2, c4) replaced ring C (Figure 8a). Compound c3 with a modified phenyl group was also prepared. To select the amine part of the final amide product, 13 amines were chosen (a1-a13, Figure 8b) on the basis of the structures of the most potent InhA inhibitors identified in the previous microtiter libraries. The amides derived from these 13 amines inhibited at least 55% InhA activity during the initial in situ screening at a concentration of 15 μM (not shown). Compound a14 was also included to investigate the effect of an additional phenyl group at the meta-position (Figure 8b). In a15 (Figure 8b), an additional – NH2 is present on the phenyl ring of compound 21, which presents the amine moiety of the potent inhibitor identified earlier (p21, Table 2). This structure was included to test the possibility of greater H-bond formation at the InhA active site.
Figure 8.


A. Structures of pyrrolidine carboxylic acids synthesized for the modification of pyrrolindine carboxamide Ring C section. B. Structures of amines applied for the ring C exploration of pyrrolidine carboxamide.
The carboxylic acids were synthesized by fusion of itaconic acid with the corresponding amine at high temperature and processed following the standard protocol as described in a previous report.27 The formation of the major desirable product was confirmed by LC-MS. The target compound library (15 × 8, 120 compounds) was generated in 96 well plates and was assayed by in situ screening after serial dilution, as described in the experimental section. Those compounds showing at least 90% inhibition in the screening test were synthesized and purified by chromatography for IC50 determinations.
The screening results demonstrated that all the most potent amide inhibitors derived from the amines a1, a2 and a14. Table 4 summarizes the activities of the products at two different concentrations (1.5 and 15 μM). The activities of the compounds were reduced or abolished when the cyclohexyl group was replaced by a phenyl ring or a bicyclohexyl structures (c3, c4). The activities of the bicyclic replacement (p62, 0.46 μM; p63, 0.85 μM) (Table 5) are comparable to those of the corresponding cyclohexyl compounds (p24, 0.39 μM; p20, 0.75 μM) (Table 2). The adamantyl substituent greatly reduces the activities, possibly as the result of steric interactions of this bulky group within the active site. Among the monocyclic compounds of different sizes, the cyclopentyl compound is the least effective, whereas the cyclohexyl and cycloheptyl displayed the best and comparable activities among each of the three amine classes (a1, a2 and a14). A slight decrease in activity was observed with the cyclooctyl replacement. All together, the SAR exploration of ring C demonstrated that hydrophobic interactions between the enzyme binding pocket and ring C play a key role in inhibitory activity. A saturated ring structure of proper dimensions is required to optimize InhA inhibition activity. At this position, the 6 or 7 member ring is the most suitable, whereas a too large (adamantly ring) or too small (pentyl ring) group invariably led to a decrease in activity.
Table 4.
Summary of InhA inhibition activities of pyrrolindine carboxamide Ring A analogues
| Structure | Effectiveness |  |
 |
 |
|---|---|---|---|---|
 |
Inhibition % | >90 | >90 | 80 |
| (15μM) | ||||
| Inhibition % | 75 | 70 | ND | |
| (1.5μM) | ||||
| IC50 (μM) | 0.46±0.01 | 0.85±0.05 | ||
 |
Inhibition % | 70 | 38 | ND |
| (15μM) | ||||
| Inhibition % | ND | ND | ND | |
| (1.5μM) | ||||
| IC50 (μM) | ||||
 |
Inhibition % | 80 | 36 | 3 |
| (15μM) | ||||
| Inhibition % | ND | ND | ND | |
| (1.5μM) | ||||
| IC50 (μM) | ||||
 |
Inhibition % | 5 | 0 | 3 |
| (15μM) | ||||
| Inhibition % | ND | ND | ND | |
| (1.5μM) | ||||
| IC50 (μM) | ||||
 |
Inhibition % | 65 | 51 | 50 |
| (15μM) | ||||
| Inhibition % | ND | ND | ND | |
| (1.5μM) | ||||
| IC50 (μM) | ||||
 |
Inhibition % | >90 | >90 | >90 |
| (15μM) | ||||
| Inhibition % | ND | 64 | 90 | |
| (1.5μM) | ||||
| IC50 (μM) | 0.39±0.01 | 0.75±0.04 | 0.14±0.01 | |
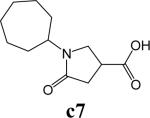 |
Inhibition % | 90 | >90 | >90 |
| (15μM) | ||||
| Inhibition % | 30 | 72 | 80 | |
| (1.5μM) | 0.32±0.02 | 0.27±0.03 | ||
| IC50 (μM) | ||||
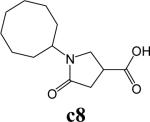 |
Inhibition % | 90 | >90 | 70 |
| (15μM) | ||||
| Inhibition % | 25 | 65 | ND | |
| (1.5μM) | ||||
| IC50 (μM) | 0.62±0.05 | 1.29±0.10 |
ND: not determined
Table 5.
InhA inhibition activities of pyrrolindine carboxamide compounds identified after Ring C modification
| Structure | ID | MW | IC50 (μM) |
|---|---|---|---|
| p62 | 374.48 | 0.845 ± 0.05 | |
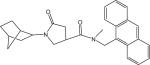 |
p63 | 426.55 | 0.46 ± 0.01 |
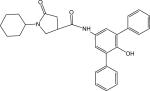 |
p64 | 454.26 | 0.14 ± 0.01 |
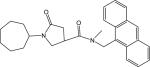 |
p65 | 428.57 | 0.62 ± 0.05 |
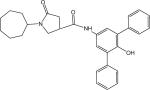 |
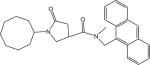
p66 | 468.59 | 0.36 ± 0.03 |
 |
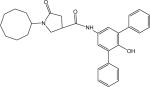
p67 | 442.59 | 0.32 ± 0.02 |
 |
p68 | 482.26 | 1.29 ± 0.10 |
Compound p64 with IC50 = 140 nM (Table 5) is the best InhA inhibitor discovered so far. Its enhanced potency presumably reflects increased hydrophobic interactions of the additional phenyl ring with the active site residues. Although the presence of a para-hydroxyl group on the phenyl ring suggests it could increase potency by functioning as an H-bond donor, the complete loss of activity in the amide product from amine g54 (Figure S1) precludes this possibility. Surprisingly, the amide products generated from the a15 (Figure 8b) amine were also inactive. Since the only difference between amine a15 and g21 (Figure S1) is the extra – NH2, it is possible that the different charged form of the former caused a subtle change in binding within the active site.
Resolution of Chiral Pyrrolidine Carboxamide Derivatives by Chiral HPLC
The presence of one chiral center in the pyrrolidine carboxamide (Figure 9) prompted us to investigate the biological activity of the individual enantiomers. The three most potent InhA inhibitors were purified by chiral HPLC. For compound d11, the enantiomer peaks appeared at 11.86 and 13.77 min, respectively (Figure 9b). Fractions containing the individual enantiomers were collected. After the purification, the optical purity was determined by a follow-up HPLC. Based on the peak area, the enantiomeric excess (ee%) of the two enantiomers of d11 was determined to be 96.8% (d11a, retention time, 11.94 min) and 98.5% (d11b, retention time, 13.82 min). For compound p24, the ee% of the two enantiomers was 77.8% (p24a, retention time, 10.40 min) and p24b 97.1% (retention time, 11.45 min) (Fig. 9a), and for compound p64 the ee% of the two enantiomers was 98.5% (p64a, retention time, 8.47 min) and 96.8% (p64b retention time, 9.16 min) (Figure 9c).
Figure 9.
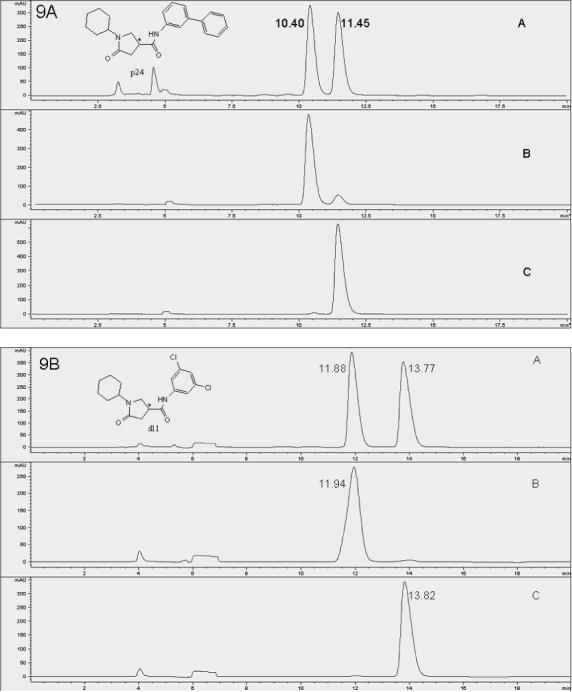
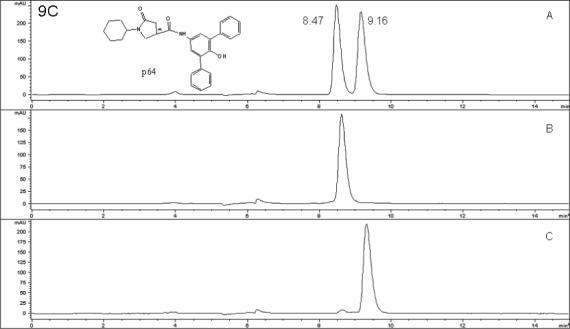
Racemate and enantiomers chromatograms of compounds p24 (9A), d11 (9B) and p64 (9C). Cyclobond I 2000 stationary phase; eluent, ACN/MeOH/HOAC/TEA (95/5/0.3/0.2 v/v/v/v); flow rate of 0.8 mL/min; temperature 23 °C; wavelength 260 nm (polar organic mode). 9A: A, p24 racemate, B, p24a, C, p24b, 9B: A, d11 racemate, B, d11a, C, d11b, 9C: A, p64 racemate, B, p64a, C, p64b. *: Chiral center.
IC50 Values of the Enantiomers
The IC50 values, determined according to the standard protocol as described in the experimental section, are summarized in Table 6. The IC50 values for p24a and p24b were determined to be 2.1 and 0.255 μM, respectively. The IC50 of p24b is approximately half of the IC50 of racemic p24 (0.39 μM). Furthermore, given the ee of p24a (77.8%), its IC50 can be converted to 0.23 μM, which is close to p24b. The comparable values of the two IC50 values indicates that only one of the two enantiomers (p24b) can bind to the enzyme and is biologically active, whereas the other enantiomer (p24a) can do neither, as it did not attenuate the activity of the active enantiomer. The IC50 measurements for the enantiomers of p64 gave the same result, with p64b displaying an IC50 of 62 nM. Due to the high ee value and the insolubility of compound p64 at high concentrations, the inhibition activity of p64a was measured at a concentration of 2.4 μM, at which 70% of the enzyme activity was inhibited. Based on the purity of p64a and the IC50 curve of p64b, the inhibition percentage was converted to 72.7%. Thus, the small activity observed for p64a was solely due to the presence of traces of p64b in the sample. Comparable results were obtained for d11, for which the second enantiomer to elute from the HPLC was the active component. The IC50 determination of compound d11 gave an IC50 of 8.16 μM for d11a and 0.20 μM for d11b, which is half the IC50 of the d11 racemate (0.39 μM). The converted IC50 value for d11a is 0.13 μM, also close to the IC50 of d11b. In agreement with the crystallographic result from cocrystalization of d11 with InhA, only one configuration of d11(R) binds within the active site of InhA. The bound configuration in the crystal structure was used to assign the absolute configurations to the enantiomers.
Table 6.
Summary of InhA inhibitor purification by chiral HPLC and activities of the enantiomers
| Rt min | ee | IC50 nM (converted) | |
|---|---|---|---|
| p24 | 390 | ||
| p24a | 10.40 | 77.8% | 2100 (230) |
| p24b | 11.45 | 97.1% | 255 |
| d11 | 390 | ||
| d11a | 11.94 | 96.8% | 8160 (130) |
| d11b | 13.82 | 98.5% | 200 |
| p64 | 140 | ||
| p64a | 8.47 | 98.5% | 70% (72.7%)* |
| p64b | 9.16 | 96.8% | 62 |
ee: Enantiomeric excess
Percentage of control activity at 2.4μM
Antibacterial Activity
Selected compounds with the best enzyme inhibitory activities were submitted for evaluation as inhibitors of the growth of M. tuberculosis in culture. Determination of the minimum inhibitory concentrations (MIC) against M. tuberculosis strain H37Rv showed modest antibacterial activity.28 The majority of compounds exhibited MIC above 125 μM. Compounds d12 and p67 displayed the best activity, with MIC values of 62.5 μM, and compounds p9, p20, p63, and p65 had MIC values of 125 μM. These results suggest that that these inhibitors do not yet have optimal membrane permeability or are actively pumped out of the bacterial cells by efflux pumps.
Conclusion
We have identified pyrrolidine carboxamides as a novel class of InhA inhibitors through high-throughput screening and microarray parallel synthesis methods. Based on the preliminary SAR studies, a focused microtiter pyrrolidine carboxamide library was synthesized by diversifying the core scaffold of the pyrrolidine carboxamide through an amide-forming reaction. More potent pyrrolidine carboxamide inhibitors with a novel scaffold were discovered through subsequent in situ screening without purification. The application of a structure-based iterative library approach allowed highly focused and rapid optimization of the initial leads to low nanomolar InhA inhibitors, with the best inhibitor showing an IC50 of 62 nM. A 160 fold gain in potency was thus realized through library optimization. The resolution of racemic mixtures and the biological evaluation of the pure enantiomer clearly indicate that only one stereoisomer is responsible for the inhibition. These newly identified compounds serve as promising candidates for the development of a second generation InhA inhibitors with improved bioavailability properties.
Experimental Section
Materials
Reagents and solvents were used as obtained from commercial suppliers without further purification. Column chromatography was carried out on Merck silica gel 60 (230−400 mesh). 1H-NMR spectra were recorded on a Varian Gemini 400 (400 MHz) spectrometer. Mass spectra were recorded using an LC/MS system consisting of a Waters 1100 HPLC instrument and a Waters ZQ mass detector (ESI positive). Coenzyme A hydrate trilithium salt (93−97%), NADH and ethyl chloroformate were from Sigma Chemical Co. (St. Louis, MO). Triethylamine was from Aldrich (Milwaukee, WI). trans-2-Octenoic acid was from TCI Chemicals (Portland, OR). Hits identified in the high throughput screening were purchased from Chembridge and ChemDIV Inc. (San Diego, CA). All other buffer salts (reagent grade or better), solvents (HPLC grade or better), and chemicals were purchased from Fisher Scientific Co. (Pittsburgh, PA).
General Methods
Column chromatography was carried out on Merck silica gel 60 (230−400 mesh). 1H-NMR spectra were recorded on a Varian Gemini 400 (400 MHz) spectrometer. Chemical shifts are given in ppm with tetramethylsilane (organic solvents) and coupling constants (J) are given in Hertz (Hz). Mass spectra were recorded using an LC/MS system consisting of a Waters 1100 HPLC instrument and a Waters ZQ mass detector (ESI positive). Purity was determined with a Hewlett Packard HPLC (1090C) system. Normal phase: Alltech Silica, 4.6 × 250 mm column (5 □m); HPLC1 90:10 v/v; HPLC2 85:15 v/v HPLC3 75:25 v/v hexane: isopropanol over 20 min, flow rate 1.0 mL/min; wavelength 254 nm. Reverse phase: Varian, Dynaman microsorb 100−5 C18, 250 × 4.6 mm column; HPLC4: 60:40 v/v CH3CN:H2O over 2 min, followed by 60:40−95:5 v/v CH3CN:H2O over 8 min, then hold at 95/5 v/v CH3CN:H2O over 10 min; flow rate 1.0 mL/min; wavelength 230/254 nm. HPLC5: 60:40 v/v MeOH:H2O over 2 min, followed by 60:40−5:10 v/v MeOH: H2O over 8 min, then hold at 95/5 v/v CH3CN: H2O over 10 min; flow rate 1.0 mL/min; wavelength 230/254 nm
Overexpression and Purification of InhA in E. coli.
The expression plasmid of inhA from M. tuberculosis H37Rv was kindly provided by Dr. John Blanchard. It was a pET15b vector (Novagen) containing an 810 bp inhA gene insert between the NcoI and BamHI restriction sites at the 5' and 3' ends, respectively. The plasmid was transformed into expression E. coli BL21-Gold (DE3) (Stratagene) host cells. The E. coli BL21-Gold (DE3): inhA strain was grown in 4 L of LB broth with ampicillin (100 μg/mL) at 37 °C and induced at OD600 = 0.8−1.0 with 0.5 mM IPTG and 250 rpm shaking. The culture was then transferred to a cold room and cultured overnight at 18 °C. Approximately 20 g of cells were harvested by centrifugation and frozen at −70 °C.
Standard protein purification procedures were performed at 4 °C and fractions were analyzed by SDS-PAGE. The frozen cells were thawed, resuspended in buffer A (20 mM Tris, pH 7.5; 5% glycerol; 1 mM EDTA), and disrupted by sonication. The supernatant was obtained after centrifugation (40 min, 30 000 rpm) and subjected to protein purification. The sample was loaded on a Source 15Q high resolution anion exchange column. Gradient elution with a flow rate of 3 ml/min was set up as follows: 5 column volumes (CV) of 60 mM NaCl Buffer A, 10 CV of 60−240 mM NaCl Buffer A, 5 CV of 240−600 mM NaCl Buffer A (CV = 10 mL).
The protein was further purified with the following optimized procedure. The ammonium sulfate (AS) concentration in the pooled elute from the last step was first adjusted to 1 M with solid ammonium sulfate. The sample was then loaded onto a 5 × 5 ml Hitrap butyl Sepharose column (Amersham Biosciences). Gradient elution was set up as follows with a flow rate of 4 ml/min: 1 CV (0−25 ml) of 1 M to 0.4 M AS, 5 CV (25−150 ml) of 0.4 M AS, 5 CV (150−275 ml) of 0.4 M to 0 M AS, 9 CV (275−500 ml) of 0 M AS. Finally, the sample was desalted with a Hitrap desalting column and the buffer was changed to 30 mM PIPES, 10% glycerol pH 6.8. Glycerol was added and InhA samples containing 50% glycerol were kept at −20 °C. InhA was stable for more than 6 months under such conditions. The extinction coefficient of the protein was determined to be ε280 = 37.3 ± 0.2 mM−1cm−1 by amino acid analysis,29 the concentration of the protein was 1 mM, and the yield was 146 mg / 20 g cell pellet.
Preparation of 2-trans-Octenoyl-CoA (OCoA)
2-Trans-octenoyl-CoA (OCoA) was prepared from trans-2-octenoic acid by the mixed anhydride method.29,30 A sample of 122.58 mg (862 μmol) of the acid was dissolved in 25 mL of anhydrous diethyl ether and stirred in an ice water bath. To this was added 180 μL (1316 μmol) of triethylamine. Ethyl chloroformate (123 μL, 1290 μmol) was first added to 2 mL of prechilled anhydrous diethyl ether and the solution was then added dropwise with stirring over a period of 10 min to the acid and triethylamine mixture. The reaction solution was stirred for another 30 min in the ice water bath. The mixed anhydride was filtered and was then washed 3 times with an equal volume of diethyl ether-saturated water. The diethyl ether layer (light yellow) was collected.
Coenzyme A (CoA) (160 mg, 203.5 μmol) was dissolved in 10 mL of a mixture of 50 mM Na2CO3 (pH 8), ethanol, and ethyl acetate (1:1:1 v/v/v). The resulting solution was aliquoted into 5 reaction flasks and the mixed anhydride was then added dropwise to the CoA solution and stirred at room temperature. The reaction was monitored by the 5, 5’ dithiobis (2-nitrobenzoic acid) (DTNB) test until the yellowness on the TLC plate disappeared completely, indicating complete consumption of the free thiol. After concentration of the reaction mixture in vacuo, the white residue was dissolved in distilled water and the solution was filtered before HPLC purification. The sample was purified using an Alltima C18 150 mm × 10 mm (5 μm) preparative column (Alltech). Preparative HPLC was carried out using 20 mM ammonium acetate as buffer A and 100% acetonitrile as buffer B with a flow rate of 4 mL/min. The gradient elution program was as follows: 0 to 3 min with buffer A; 3 to 5 min running a 0 to 35% gradient of buffer B; 5 to 25 min, 35% to 55% buffer B; 25−26 min, 55% to 95% buffer B; 26 to 29 min, 95% buffer B; 2 min 95% to 0% buffer B. Elution was monitored at 260 and 285 nm and fractions containing OCoA were pooled and lyophilized. The retention time for OCoA was 18 to 27 min. The lyophilized flaky, white powder was redissolved in H2O and relyophilized twice to remove the ammonium acetate. The expected product (63.9 mg) was obtained in a yield of 35.2%.: LC-MS ([M + H] +) calcd for [C29H48N7O17P3S]+ 892.1, found 892.1.
High Throughput Screening of InhA Inhibitors
An endpoint assay was developed for the high-throughput screening of InhA inhibitors. A chemical diversity library of 30,000 compounds was obtained from the Bay Area Screening Center (BASC). The assays were performed at 23 °C in 200 μl of 30 mM piperazine-1,4-bis(2-ethanesulfonic acid) (PIPES) buffer (pH 6.8) containing 0.1% BSA. The samples in the chemical library plates were stored at a concentration of 30 mM in DMSO. They were first diluted then transferred to daughter plates at a final concentration of 600 μM in 20% DMSO (v/v). The screening was performed in UV-transparent 96-well plates. The assay components were added to the wells by a liquid handler in three steps: step 1, to wells A2 through H11: 10 μl of sample (600 μM in 20% DMSO; final concentration in the assay was 30 μM, 1% of DMSO), for wells A1 and A2 (blank, 0% activity), C1 to E1(controls without InhA), A12 through H12 (100% activity), 10 μL 20% DMSO was added for wells F1 to H1(control wells with internal standard inhibitor) 10 μL 1700 μM palmitoyl CoA in 20% DMSO was added (final concentration 86 μM); step 2, 40 μl of assay buffer PIPES pH 6.8, 50 mM 0.1% BSA) was added to all the wells; step 3, 100 μl of mixture of 400μM freshly prepared NADH (final concentration 100 μM) and 160 nM InhA (final concentration 40 nM) in assay buffer was added to all the wells except blank wells (100 μL assay buffer) and C1 to E1 wells (100 μL 400 μM NADH). The plate was then shaken for 1 min and finally the reaction was initiated by the addition of 50 μl of 250 μM substrate OCoA to all the wells except the blanks. The plate was incubated for 60 min at 23 °C and terminated by the addition of 100 μL of 100% ethanol. The absorbance OD readings were taken on a Molecular Device SPECTRAmax PLUS 384 microreader at 340 nm. For those samples showing at least 50% inhibition, a follow up verification step was conducted in triplicate in a kinetic mode in which the rate of the absorbance change at 340 nm was monitored for 30 min. Triton X-100 (0.01% v/v) was also included as the most convenient means to exclude false positive results caused by promiscuous inhibitors. Data points collected every 10 s during the linear range of OD change (10 min) were used to evaluate the inhibitory activity of the samples. The inhibition percentage was calculated by comparing the slopes (OD change/time) for the samples with the slopes for the enzyme alone. The internal standard for every plate was palmitoyl-CoA, which gave about 90% inhibition at 86 μM.
Separation of Chiral Pyrrolidine Carboxamide Derivatives by HPLC Using a Cyclodextrin Chiral Stationary Phase
Cyclobond I 2000 (250 mm × 4.6 mm) was obtained from Advanced Separation Technologies (Whippany, NJ). HPLC grade methanol (MeOH) and acetonitrile (ACN) were from Fisher (Fairlawn, NJ). Triethylamine (TEA), and acetic acid (HOAC) were ACS certified grade from Fisher. Chromatographic separations were conducted at room temperature (∼23 °C) using an HP 1090 HPLC system with UV detection at 285 and 260 nm. The racemic compounds were first dissolved in DMSO at a final concentration of 10 mM and were then diluted with the mobile phase solvent. In organic polar mode, the enantiomers were separated using ACN/MeOH/HOAC/TEA (95/5/0.3/0.2 v/v/v/v) as the mobile phase with a flow rate of 0.8 mL/min.
IC50 Determination
IC50 values were determined in 96-well plates with serial 2-fold dilutions with DMSO of each inhibitor. The final concentration of DMSO in the final assay was 1%. The IC50 values were determined using at least 8 concentrations, with each concentration assayed in triplicate under saturating substrate conditions. The concentrations of other components in the assay were as follows: NADH 250 μM (stock 1 mM, 50 μL, Km 7.6 μM); InhA 20 nM (stock 80 nM, 50 μL). OCoA 500 μM (stock 2 mM, 50 μL, Km 467 μM) was prepared freshly by dissolving the flaky solid substrate immediately before the assay; the remaining material was kept at −80° C and was used up within two days. The reaction, carried out as in the standard assay, was monitored over 10 min at room temperature. IC50 values were calculated from plots of enzyme activity versus the log of the inhibitor concentration using the GRAFIT-IC50-4 parameter fit software (Grafit 4.021, Erithacus).
Minimum Inhibitory Concentration (MIC) Determination
Compounds were submitted to the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) for in vivo determination of the minimum inhibitory concentration (MIC) against M. tuberculosis strain H37Rv using Alamar Blue Assay.28 The MIC is defined as the lowest concentration of compound required to give 90% inhibition of bacterial growth.
Molecular Modeling
Molecular modeling was carried out using SYBYL, version 7.0 (Tripos, Inc. St. Louis, MO.), running on a Silicon Graphics O2 (R10000) UNIX workstation. Figure 4, 5, 6 were prepared using SYBYL. Flexible docking was performed with FlexX, version 1.7.6, within SYBYL.
Enzyme Structure
The coordinates for the crystal structure of the 2-InhA complex possessing the highest crystallographic resolution (2.7 Å)14 were downloaded from the Protein Data Bank (entry 1p44) and used for initial docking studies.
Crystallization and Data Collection
InhA (0.1 mg/ml =3 μM) was mixed at a 1:1 ratio with NADH and 1% Me2SO in a 10 ml volume. The mixture was then concentrated to 0.1 ml and crystallized by hanging drop vapor diffusion. The protein crystallized in 100 mM HEPES, pH 7.0−8.0, 6−8% MPD, 50 mM Na citrate, pH 6.5, according to previously published conditions.31 Crystals were soaked with inhibitors (10 mM in 100% Me2SO) for 30−60 min at a final concentration of 1−2 mM. Diffraction data were collected, from single crystals, under cryoconditions at Beamline 8.3.1 at the Advanced Light Source (ALS) at the Lawrence Berkeley National Laboratory (Berkeley, CA).
Structure Determination and Refinement
Data were indexed and scaled by using ELVES32 and DENZO/SCALEPACK.33 The molecular replacement solutions were found by using a single subunit of InhA derived from the Protein Data Bank file 1P4514 as a search model. Molecular replacement and subsequent refinement calculations were performed by using CNS.34 The models were built with Quanta (Accelrys). Topology and parameter files for all ligands were obtained from Hetero-compound Information Centre – Uppsala (Hic-Up).35 The final statistics are listed in Table 7. The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org under the following PDB ID codes: s1 (2H7I), s4 (2H7L), d11 (2H7M), d3 (2H7N), and d8 (2H7P).
Table 7.
Statistics for data collection and refinement
| s1 | s4 | d3 | d8 | d11 | |
|---|---|---|---|---|---|
| Crystal data | |||||
| Space group | P6222 | P6222 | P6222 | P6222 | P6222 |
| Cell dimensions, Å | |||||
| a | 97.54 | 97.551 | 97.004 | 97.476 | 97.794 |
| c | 140.522 | 140.346 | 140.456 | 141.330 | 140.661 |
| alpha/gamma | 90 (120) | 90 (120) | 90 (120) | 90 (120) | 90 (120) |
| Data collection statistics | |||||
| Wavelength, Å | 1.11 | 1.11 | 1.11 | 1.11 | 1.11 |
| Number of reflections | 50,760 | 41,798 | 31,434 | 33,993 | 51,058 |
| Redundancy (last shell) | 3.7 (2.2) | 3.4 (2.4) | 4.3 (4) | 4.1 (3.3) | 3.6 (2.0) |
| Completeness (last shell), % | 92 (90.45) | 99.8 (100) | 97.4 (96.3) | 97.9 (96.4) | 99.7 (99.3) |
| Rsym | 0.064 | 0.11 | 0.089 | 0.129 | 0.085 |
| I/σ (last shell) | 10.4 (2) | 6.8 (2.5) | 9.6 (2.6) | 5.8 (2.3) | 8.9 (2.8) |
| Refinement statistics | |||||
| Resolution, Å | 54−1.62 | 54−1.73 | 41−1.9 | 41−1.86 | 54−1.62 |
| Reflections in working set | 43,330 | 38,644 | 28,335 | 30,746 | 47,211 |
| Reflections in test set (7.4%) | 3,475 | 3,146 | 2,335 | 2,538 | 3,782 |
| Rcrystal/Rfree, % | 19.8 (21.6) | 20.8 (23.2) | 17.8 (20.3) | 19.0 (20.6) | 19.1 (20.4) |
| RMSD bonds, Å | 0.006 | 0.006 | 0.006 | 0.009 | 0.006 |
| RMSD angels, ° | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 |
| B factors, | |||||
| Average | 27.5 | 25.1 | 25.2 | 27.7 | 20.6 |
| NAD | 21.2 | 18.9 | 17.6 | 20 | 13.8 |
| Inhibitor | 31.4 | 26.6 | 43.8 | 35.1 | 23.7 |
Testing of the HBTU, DIEA Effect on InhA
40 mM HBTU and 40 mM DIEA were prepared and then diluted with PIPES buffer to 4 mM. Serial dilution was carried out to make 400 μM, 40 μM, and 4 μM stock solutions for the enzyme assay. A 50 μL aliquot of stock solution was added to each well, followed by 400 μM NADH and 50 μL of 160 nM InhA. The reaction was initiated with 50 μL of 1 mM OCoA and the reaction rate was monitored for 15 min at room temperature.
General Procedure for Microtiter Library Synthesis and Follow Up in situ Screening
The synthesis of microtiter library compounds was performed in 96 well plates. A solution of pyrrolidine carboxylic acid (40 μL, 100 mM, 1 equiv) in DMF was added to each well followed by 22 μL HBTU (200 mM, 1.1 equiv) and 8 μL of DIEA (1 M, 2 equiv). Finally, the corresponding amine (40 μL, 100 mM, 1 equiv) in DMF was added and the reaction plate was shaken on the Molecular Device SPECTRAmax PLUS 384 microreader for 2 h at 45 °C. The reaction was monitored by TLC and the identity of the major product in each well was confirmed by LC-MS. Once the reaction was completed, the products were diluted, transferred to the assay plate, and tested following the standard assay procedure except that DMF (< 0.5% in the final assay) was used instead of DMSO in the control and the final testing concentration of each product was 15 μM (assuming a 100% yield in the reaction).
Synthesis of Pyrrolidine Carboxylic Acid as Exemplified by 1-Cyclohexyl-5-oxopyrrolidine-3-carboxylic Acid (Compound c6)27
A mixture of itaconic acid (655 mg, 5 mmol) and the cyclohexylamine (493.9 mg, 5 mmol) was heated at 200 °C in a flask attached to a reflux condenser for 30 min. After the molten mass had cooled, water was added and the mixture was chilled in an ice-bath and then dissolved in aqueous sodium hydroxide. After the product was filtered, the mixture was acidified with dilute hydrochloric acid. The precipitated product was recrystallized from MeOH and Et2O: 742mg of white crystals was obtained with a yield of 70%. LC-MS ([M + H] +) calcd for [C7H11NO3H]+ 212.1, found 212.1. 1H NMR (400 MHz CD3OD) δ 3.76−3.85 (m, 1H), 3.56−3.66 (m, 2H), 3.12−3.26 (m, 1H), 2.58−2.68 (m, 2H), 1.60−1.88 (m, 5H), and 1.08−1.50 (m, 5H).
Synthesis of 1-(9H-Fluoren-9-yl) Piperazine (Compound 35)36
9-Chlorofluorene (359 mg, 1.8 mmol) and piperazine (464 g, 3.6 mmol) were mixed and refluxed in 2-butanone (25 mL) for 16 h. The mixture was cooled and 20 mL of ether was added. After filtration, the ether layer was evaporated in vacuo. The residue was dissolved in ether, washed with water (15 mL × 3), and then extracted with a 2 N solution of methanesulfonic acid in water (10 mL × 3). This extract was basified with concentrated ammonium hydroxide and extracted with ether. After the extract was dried (Na2S04) and evaporated in vacuo, 297 mg (67%) of the desired product was obtained: LC-MS ([M + H] +) calcd for [C17H18N2H]+ 251.2, found 251.2. 1H NMR (400 MHz CD3OD) δ 7.65 (dd, 4H), 7.30−7.36 (m, 2H), 7.22−7.28 (m, 2H), 4.77 (s, 1H), 2.78−2.84 (m, 4H), and 2.56−2.61 (m, 4H).
General Procedure for Synthesis of the Amide, as Exemplified by the Synthesis of Compound p21
1-Cyclohexyl-N-(3-benzylphenyl)-5-oxopyrrolidine-3-carboxamide (Compound p21)
To a solution of amine (18.4 mg, 0.1 mmol) and acid (23 mg, 0.11 mmol) in 3 mL of dry DMF, HBTU (50 mg, 0.13 mmol) and DIEA (50 μL, 0.3 mmol) were added at 23 °C. The reaction mixture was stirred for 5 h and was then quenched by the addition of brine and extracted three times with EtOAc. The organic layers were combined and washed sequentially with 1 N HCl, saturated aqueous NaHCO3, and brine before being dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by preparative TLC with CH2Cl2/MeOH (40:1 v/v) to give the product (32.2 mg) in 85% yield: LC-MS ([M + H] +) calcd for [C24H28N2O2H]+ 377.22, found 377.22. 1H NMR (400 Mhz CDCl3) δ 8.47 (s, 1H), 7.36−7.42 (m, 2H), 7.10−7.28 (m, 5H), 6.88−6.96 (m, 1H), 3.82−3.96 (m, 1H, s, 2H), 3.59−3.64 (m, 1H), 3.44−3.52 (m, 1H), 3.08−3.20 (m, 1H), 2.68−2.78 (m, 1H), 2.52−2.64 (m, 1H), 1.58−1.82 (m, 5H), and 1.00−1.40 (m, 5H). HPLC1: 12.88 min, purity >97%. HPLC4: 10.55 min, purity >97%. Compounds p9, p20, p24, p27, p28, p31, p33, p36, p37, p50, p56 and p62-p68 were prepared following the same protocol.
N-(3-(Benzyloxy)phenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide (Compound p9)
Yield 61.3%. LC-MS ([M + H] +) calcd for [C24H28N2O3H]+ 393.21, found 393.21. 1H NMR (400 Mhz CD3OD) δ 7.724−7.46 (m, 6H), 7.16−7.23 (m, 1H), 7.06−7.12 (m, 1H), 6.72−6.78 (m, 1H), 5.05 (s, 2H), 3.81−3.86 (m, 1H), 3.61−3.72 (m, 1H), 3.56−3.64 (m, 1H), 3.24−3.36 (m, 1H), 2.62−2.74 (m, 2H), 1.60−1.88 (m, 5H), and 1.00−1.58 (m, 5H). HPLC1: 9.52 min, purity >95%. HPLC4: 12.48 min, purity >97%.
N-((Anthracen-9-yl)methyl)-1-cyclohexyl-N-methyl-5-oxopyrrolidine-3-carboxamide (Compound p20)
Yield 77.6%. LC-MS ([M + H] +) calcd for [C27H30N2O2H]+ 415.23, found 415.23. 1H NMR (400 Mhz CD3OD) δ 8.5 (s, 1H), 8.28−8.32 (d, 2H), 8.00−8.08 (d, 2H), 7.44−7.56(m, 4H), 5.60−5.74 (q, 2H), 3.82−3.92 (m, 1H), 3.56−3.66 (m, 2H), 3.26−3.32 (m, 1H), 2.58−2.76 (m, 2H, s,3H), 1.60−1.88 (m, 5H), and 1.00−1.58 (m, 5H). HPLC3: 10.79 min, purity >99%. HPLC4: 10.79 min, purity >95%.
1-Cyclohexyl-N-(3-phenylphenyl)-5-oxopyrrolidine-3-carboxamide (Compound p24)
Yield 59.2%. LC-MS ([M + H] +) calcd for [C23H26N2O2H]+ 363.21, found 363.21. 1H NMR (400 Mhz CDCl3) δ 7.78−7.88 (m,2H), 7.56−7.60 (m, 2H), 7.48−7.54 (m, 1H), 7.32−7.46 (m, 4H), 3.90−4.02 (m, 1H), 3.69−3.78 (m, 1H), 3.54−3.63 (m, 1H), 3.16−3.28 (m, 1H), 2.80−2.888 (m, 1H), 2.67−2.77 (m, 1H), 1.62−1.86 (m, 5H), and 1.04−1.45(m, 5H). HPLC3: 10.40 min, purity >98%. HPLC4: 9.19 min, purity >99%.
1-Cyclohexyl-4-(1-(phenyl(p-tolyl)methyl)piperazine-4-carbonyl)pyrrolidin-2-one (Compound p27)
yield 76.9%. LC-MS ([M + H] +) calcd for [C29H37N3O2H]+ 460.29, found 460.29. 1H NMR (400 Mhz CD3OD) δ 7.40−7.45 (m, 2H), 7.24−7.32 (m, 4H), 7.12−7.20 (m, 1H), 7.06−7.12 (m, 2H), 4.22 (s,1H), 3.76−3.86 (m,1H), 3.48−3.62 (m, 6H), 3.28−3.36 (m, 1H), 2.52−2.62 (m, 2H), 2.28−2.42 (m, 4H), 2.26 (s,3H), 1.76−1.84 (m, 5H), and 1.08−1.52 (m, 5H). HPLC2: 11.79 min, purity >95%. HPLC4: 10.32 min, purity >98%.
1-Cyclohexyl-4-(1-((4-fluorophenyl)(phenyl)methyl)piperazine-4-carbonyl)pyrrolidin-2-one (Compound p28)
yield 69.5%. LC-MS ([M + H] +) calcd for [C28H34FN3O2H]+ 464.26, found 464.26. 1H NMR (400 Mhz CD3OD) δ 7.36−7.46 (m, 4H), 7.22−7.29 (m, 2H), 7.13−7.19 (m, 1H), 6.95−7.02 (m, 2H), 4.29 (s,1H), 3.76−3.86 (m,1H), 3.50−3.62 (m, 6H), 3.28−3.32 (m, 1H), 2.54−2.64 (m, 2H), 2.30−2.42 (m, 4H), 1.60−1.84 (m, 5H), and 1.08−1.52 (m, 5H). HPLC2: 14.35 min, purity >95%. HPLC4: 14.11min, purity >96%.
1-Cyclohexyl-N-(9H-fluoren-1-yl)-5-oxopyrrolidine-3-carboxamide (Compound p31)
Yield 60%. LC-MS ([M + H] +) calcd for [C24H26N2O2H]+ 375.21, found 375.21. 1H NMR (400 Mhz CD3OD) δ 7.76−8.04 (d,1H), 7.64−7.70 (d, 1H), 7.50−7.57 (d, 1H), 7.44−7.50 (d, 1H), 7.25−7.42 (m, 3H), 3.80−3.92 (s, 2H, m, 1H), 3.64−3.8 (m, 2H), 3.42−3.52 (m, 1H), 2.70−2.78 (m, 2H), 1.60−1.88 (m, 5H), and 1.00−1.58(m, 5H). HPLC2: 11.29 min, purity >97%. HPLC4: 10.35 min, purity >97%..
1-Cyclohexyl-N-(9-ethyl-9H-carbazol-3-yl)-5-oxopyrrolidine-3-carboxamide (Compound p33)
Yield 80%. LC-MS ([M + H] +) calcd for [C25H29N3O2H]+ 404.23, found 404.23. 1H NMR (400 Mhz CD3OD) δ 8.28−8.32 (d,1H), 7.96−8.00 (m,1H), 7.46−7.50 (dd, 1H), 7.34−7.50 (m, 3H), 7.08−7.16 (m, 1H), 4.27−4.36 (q, 2H), 3.74−3.86 (m, 1H), 3.54−3.66 (m, 1H), 3.24−3.36 (m, 1H), 2.60−2.72 (m, 2H), 1.56−1.80 (m, 5H), and 1.00−1.44(m, 5H). HPLC2: 12.49 min, purity >97%. HPLC4: 13.00 min, purity >96%.
4-(1-(Bis(4-fluorophenyl)methyl)piperazine-4-carbonyl)-1-cyclohexylpyrrolidin-2-one (Compound p36)
yield 77.5%. LC-MS ([M + H]+) calcd for [C28H33F2N3O2H]+ 482.25, found 482.25. 1H NMR (400 Mhz CD3OD) δ 7.40−7.50 (dd, 4H), 6.92−7.06 (dd, 4H), 4.33 (s,1H), 3.76−3.88 (m,1H), 3.50−3.65 (m, 6H), 3.28−3.36 (m, 1H), 2.52−2.66 (m, 2H), 2.29−2.44 (m, 4H), 1.60−1.84 (m, 5H), and 1.08−1.52 (m, 5H). HPLC2: 15.82 min, purity >99%. HPLC4: 13.45 min, purity >95%.
1-Cyclohexyl-4-(1-(phenyl(4-chlorophenyl)methyl)piperazine-4-carbonyl)pyrrolidin-2-one (Compound p37)
Yield 54%. LC-MS ([M + H] +) calcd for [C25H29N3O2H]+ 480.23, found 480.23. 1H NMR (400 Mhz CDCl3) δ 7.33−7.39 (m,4H), 7.19−7.32 (m, 5H), 4.22 (s,1H), 3.88−3.98 (m,1H), 3.64−3.76 (m, 1H), 3.52−3.64 (m, 1H), 3.44−3.50 (m, 4H), 3.24−3.34 (m, 1H), 2.52−2.70 (m, 2H), 2.30−2.44 (m,4H),1.62−1.82 (m, 5H), and 1.04−1.44(m, 5H). HPLC2: 13.67 min, purity >96%. HPLC5: 15.12 min, purity >96%.
N-(3-Bromo-4-methylphenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide (Compound p50)
yield 99%. LC-MS ([M + H] +) calcd for [C19H26N2O2H]+ 315.20, found 315.20. 1H NMR (400 Mhz CD3OD) δ 7.16−7.20 (s, 2H), 6.72−6.76 (s, 1H), 3.80−3.90 (m, 1H), 3.56−3.70 (m, 2H), 3.25−3.35 (m, 1H), 2.64−2.69 (m, 2H), 2.26 (s, 6H), 1.60−1.86 (m, 5H), and 1.00−1.56 (m, 5H). HPLC1: 8.25 min, purity >97%. HPLC4: 13.04 min, purity >97%.
1-Cyclohexyl-N-(3-isopropoxyphenyl)-5-oxopyrrolidine-3-carboxamide (Compound p56)
Yield 69.7%. LC-MS ([M + H] +) calcd for [C25H29N3O2H]+ 345.21, found 345.21. 1H NMR (400 Mhz CDCl3) δ 8.05 (s,1H), 7.29−7.35 (m,1H), 7.15−7.22 (m, 1H), 6.95−7.00 (m, 1H), 6.63−6.68 (m, 1H), 4.50−4.58 (m, 1H), 3.88−3.99 (m,1H), 3.65−3.72 (m, 1H), 3.52−3.60 (m, 1H), 3.15−3.28 (m, 1H), 2.76−2.84 (m, 1H), 2.62−2.71(m,1H), 1.62−1.82 (m, 5H), and 1.04−1.44 (m, 5H). HPLC1: 14.75 min, purity >95%. HPLC4: 13.24 min, purity >98%.
1-(Bicyclo[2.2.1]heptan-2-yl)-5-oxo-N-(3-phenylphenyl)pyrrolidine-3-carboxamide (Compound p62)
Yield 35%. LC-MS ([M + H] +) calcd for [C24H26N2O2H]+ 375.201, found 375.20. 1H NMR (400 Mhz CDCl3) δ 8.18−8.28 (m,1H), 7.81 (s, 1H), 7.22−7.62 (m, 8H), 3.98−4.12 (m, 1H), 3.56−3.84 (m, 2H), 3.12−3.32 (m, 1H), 2.62−2.88 (m, 1H,s,3H), 1.40−1.82 (m, 5H), and 1.00−1.40 (m, 5H). HPLC3: 5.87 min, purity >99%. HPLC4: 10.63 min, purity >97%.
N-((Anthracen-9-yl)methyl)-1-(bicyclo[2.2.1]heptan-2-yl)-N-methyl-5-oxopyrrolidine-3-carboxamide (Compound p63)
Yield 95 %. LC-MS ([M + H] +) calcd for [C28H30N2O2H]+ 427.23, found 427.23. 1H NMR (400 Mhz CDCl3) δ 8.48 (s, 1H), 8.18−8.28 (m,2H), 8.02−8.08 (m, 2H), 7.44−7.60 (m, 4H), 5.64−5.72 (m, 2H), 4.04−4.16 (m, 1H), 3.82−3.94 (m, 1H), 3.32−3.56 (m, 2H), 2.52−2.80 (m, 1H, s,3H), and 1.08−1.82 (m, 10H). HPLC3: 10.58 min, purity >97%. HPLC4: 11.96 min, purity >96%.
1-Cyclohexyl-N-(3,5-diphenyl-4-hydroxyl)phenyl-5-oxopyrrolidine-3-carboxamide (Compound p64)
Yield 22%. LC-MS ([M + H] +) calcd for [C29H30N2O3H]+ 455.23, found 455.23. 1H NMR (400 Mhz CDCl3) δ 8.06 (s, 1H), 7.48−7.56 (m, 4H), 7.42−7.48 (m, 6H), 7.32−7.40 (m, 2H), 3.84−3.92 (m, 1H), 3.62−3.70 (m, 1H), 3.46−3.56 (m, 1H), 3.08−3.20 (m, 1H), 2.72−2.82 (m, 1H), 2.56−2.2.66 (m, 1H), 1.56−1.88 (m, 5H), 1.20−1.40 (m, 4H), and 1.00−1.32 (m, 1H). HPLC1: 5.19 min, purity >99%. HPLC4: 16.52 min, purity >98%.
N-((Anthracen-9-yl)methyl)-1-cycloheptyl-N-methyl-5-oxopyrrolidine-3-carboxamide (Compound p65)
Yield 62.9%. LC-MS ([M + H] +) calcd for [C28H32N2O2H]+ 429.25, found 429.25. 1H NMR (400 Mhz CDCl3) δ 8.48 (s, 1H), 8.20−8.28 (m, 2H), 8.02−8.08 (m, 2H), 7.44−7.60 (m, 4H), 5.64−5.72 (m, 2H), 4.12−4.24 (m, 1H), 3.82−3.88 (m, 1H), 3.32−3.54 (m, 2H), 2.42−2.80 (m, 1H, s,3H), and 1.20−1.98 (m, 12H). HPLC2: 7.72 min, purity >96%. HPLC4: 11.46 min, purity >98%.
1-Cycloheptyl-N-(3,5-diphenyl-4-hydroxyl)phenyl-5-oxopyrrolidine-3-carboxamide (Compound p66)
22.9% yield. LC-MS ([M + H] +) calcd for [C30H32N2O3H]+ 469.24, found 469.24. 1H NMR (400 Mhz CD3OD) δ 7.72 (s.1H), 7.52−7.58 (m, 4H), 7.42−7.48 (m, 6H), 7.34−7.42 (m, 2H), 4.04−4.16 (m, 1H), 3.68−3.76 (m, 1H), 3.52−3.60 (m, 1H), 3.08−3.20 (m, 1H), 2.60−2.80 (m, 2H), and 1.40−1.84 (m, 13H). HPLC3: 10.23 min, purity >99%. HPLC4: 12.43 min, purity >98%.
N-((Anthracen-9-yl)methyl)-1-cyclooctyl-N-methyl-5-oxopyrrolidine-3-carboxamide (Compound p67)
yield 95%. LC-MS ([M + H] +) calcd for [C29H34N2O2H]+ 443.26, found 443.26. 1H NMR (400 Mhz CDCl3) δ 8.48 (s, 1H), 8.20−8.28 (m, 2H), 8.02−8.08 (m, 2H), 7.44−7.60 (m, 4H), 5.64−5.72 (m, 2H), 4.18−4.28 (m, 1H), 3.82−3.88 (m, 1H), 3.32−3.54 (m, 2H), 2.42−2.80 (m, 1H, s,3H), and 1.40−1.84 (m, 14H). HPLC2: 9.59 min, purity >99%. HPLC4: 13.08 min, purity >96%.
1-Cyclooctyl-N-(3,5-diphenyl-4-hydroxyl)phenyl-5-oxopyrrolidine-3-carboxamide (Compound p68)
Yield 18%. LC-MS ([M + H] +) calcd for [C31H34N2O3H]+ 483.26, found 483.26. 1H NMR (400 Mhz CD3OD) δ 7.36−7.58 (m, 12H), 4.08−4.28 (m, 1H), 3.48−3.80 (m, 2H), 3.08−3.22(m, 1H), 2.60−2.82 (m, 2H), and 1.40−1.84 (m, 15H). HPLC2: 13.46 min, purity >97%. HPLC4: 14.72 min, purity >97%.
General Procedure for the Preparation of Pyrrolidine Carboxamide Using Oxalyl Chloride
To a solution of cyclohexyl-5-oxopyrrolindine carboxylic acid (0.25 mmol) in dry DMF (2 mL) cooled to 0 °C in a NaCl/ice bath was slowly added oxalyl chloride (0.5 mmol). After stirring for 45 min, the reaction mixture was evaporated in vacuo and the residue was dissolved in 3 mL of dry CH2Cl2, followed by dropwise addition of the corresponding amine (0.75 mmol) dissolved in dry CH2Cl2 (5 mL) with stirring at 0 °C for an additional 5 h. After evaporation of the reaction mixture, the amide product was purified by prep-TLC with a 70:30 (v/v) mixture of n-hexane and ethyl acetate as the eluent. The identity of the final product was confirmed by LC-MS and proton NMR.
N-(3-Trifluoromethylphenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide (s11)
Yield 47%. LC-MS ([M + H] +) calcd for [C18H21F3N2O2H]+ 355.16, found 355.16. 1H NMR (400 Mhz CDCl3) δ 9.00 (s,1H), 7.88 (s,1H), 7.72−7.81 (m, 1H), 7.32−7.48 (m, 2H), 3.86−3.96 (m, 1H), 3.68−3.80 (m, 1H), 3.52−3.64 (m, 1H), 3.23−3.34 (m, 1H), 2.60−2.84 (m, 2H), 1.61−1.88 (m, 5H), and 1.00−1.48 (m, 5H). HPLC2: 10.29 min, purity >96%. HPLC4: 9.70 min, purity >96%.
1-Cyclohexyl-N-(3,5-difluorophenyl)-5-oxopyrrolidine-3-carboxamide (d10)
Yield 41%. LC-MS ([M + H] +) calcd for [C17H20F2N2O2H]+ 322.15, found 322.15. 1H NMR (400 Mhz CDCl3) δ 8.50 (s,1H), 7.16−7.24 (m, 2H), 6.52−6.61 (m, 1H), 3.88−4.00 (m, 1H), 3.68−3.78 (m, 1H), 3.16−3.28 (m, 1H), 3.23−3.34 (m, 1H), 2.62−2.82 (m, 2H), 1.61−1.88 (m, 5H), and 1.00−1.48 (m, 5H). HPLC2: 7.72 min, purity >96%. HPLC1: 8.33 min, purity >99%.
N-(3-Bromo-5-(trifluoromethyl)phenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide (d12)
Yield 48%. LC-MS ([M + H] +) calcd for [C18H20BrF3N2O2H]+ 433.07, found 433.07. 1H NMR (400 Mhz CDCl3) δ 9.58 (s,1H), 8.05 (s,1H), 7.85 (s,1H), 7.48 (s,1H), 3.84−3.96 (m, 1H), 3.72−3.80 (m, 1H), 3.56−3.64 (m, 1H), 3.26−3.38 (m, 1H), 2.62−2.82 (m, 2H), 1.60−1.86 (m, 5H), and 1.00−1.48 (m, 5H). HPLC2: 6.66 min, purity >97%. HPLC4: 14.63min, purity >97%.
1-Cyclohexyl-N-(3-(trifluoromethyl)-5-methoxyphenyl)-5-oxopyrrolidine-3-carboxamide (d13)
Yield 48.2%. LC-MS ([M + H] +) calcd for [C19H23F2N3O3H]+ 385.17, found 385.17. 1H NMR (400 Mhz CDCl3) δ 9.20 (s,1H), 7.56 (s, 2H), 7.36 (s,1H), 6.86 (s,1H), 3.86−3.96 (m, 1H), 3.68−3.76 (m, 1H), 3.56−3.64 (m, 1H), 3.24−3.36 (m, 1H), 2.62−2.84 (m, 2H), 1.60−1.84 (m, 5H), and 1.00−1.48 (m, 5H). HPLC2: 10.30 min, purity >97%. HPLC4: 13.47min, purity >96%.
N-(3,5-bis(trifluoromethyl)phenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide (d14)
Yield 46.5%. LC-MS ([M + H] +) calcd for [C19H20F6N2O2H]+ 423.14, found 423.14. 1H NMR (400 Mhz CDCl3) δ 9.62 (s,1H), 8.14 (s, 2H), 7.60 (s,1H), 7.48 (s,1H), 3.88−3.96 (m, 1H), 3.76−3.84 (m, 1H), 3.58−3.66 (m, 1H), 3.30−3.40 (m, 1H), 2.64−2.82 (m, 2H), 1.60−1.86 (m, 5H), and 1.00−1.50 (m, 5H). HPLC2: 6.38 min, purity >99%. HPLC4: 15.26min, purity >99%.
Supplementary Material
Acknowledgment
This research was supported by National Institutes of Health grant GM56531. We thank Dr. John Blanchard for the generous gift of expression plasmid of inhA from M. tuberculosis H37Rv. Antimycobacterial data were provided by the Tuberculosis Antimicrobial Acquisition and Coordinating Facility through a research and development contract with the U.S. National Institute of Allergy and Infectious Disease. We thank Bob Reynolds for facilitating these assays. We also thank Sarah Griner, Joseph O'Connell III, and the staff of Beam line 8.3.1 at the Advanced Light Source Berkeley National Laboratory for assistance. This research was supported by National Institutes of Health grant P01 GM56531.
Abbreviations
- ENR
enoyl acyl carrier protein reductase
- TB
tuberculosis
- MDR-TB
multidrug-resistant tuberculosis
- FASI
type I fatty acid synthase FASII, type II fatty acid synthase
- INH
isonicotinic acid hydrazide
- AFI
aggregate-forming inhibitors
- ecENR
enoyl acyl carrier protein reductase from E. coli
- pfENR
enoyl acyl carrier protein reductase from Plasmodium falciparum
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DMF
N,N-dimethylformamide
- DIEA
N,N-diisopropylethylamine
- EDCI
1-ethyl-3-(3’-dimethylaminopropyl) carbodiimide
- DMAP
4-dimethyl-aminopyridine
- MIC
minimum inhibitory concentrations
- OCoA
2-trans-octenoyl-CoA
- CoA
Coenzyme A
- DTNB
5, 5’ dithiobis 2-nitrobenzoic acid
- BASC
Bay Area Screening Center
- PIPES
piperazine-1,4-bis(2-ethanesulfonic acid
- TEA
triethylamine
- MeOH
methanol
- HOAC
acetic acid
- ACN
acetonitrile
- TAACF
Tuberculosis Antimicrobial Acquisition and Coordinating Facility
Footnotes
Supporting Information Available. Tables S1A and S1B of amines used in constructing the libraries are presented in the supplementary material.
References
- 1. Tuberculosis, NIAID Fact Sheet http://www.niaid.nih.gov/factsheets/tb.htm.
- 2. Global Alliance for TB Drug Development ( www.tballiance.org)
- 3. WHO fact sheet http://www.who.int/mediacentre/factsheets/who104/en/print.html.
- 4.Campbell JW, Cronan JE., Jr. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 2001;55:305–332. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- 5.Heath RJ, Rock CO. Fatty acid biosynthesis as a target for novel antibacterials. Curr. Opin. Investig. Drugs. 2004;5:146–153. [PMC free article] [PubMed] [Google Scholar]
- 6.White SW, Zheng J, Zhang YM, Rock CO. The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YM, Lu YJ, Rock CO. The reductase steps of the type II fatty acid synthase as antimicrobial targets. Lipids. 2004;39:1055–1060. doi: 10.1007/s11745-004-1330-3. [DOI] [PubMed] [Google Scholar]
- 8.Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 11.Escalante P, Ramaswamy S, Sanabria H, Soini H, Pan X, Valiente-Castillo O, Musser JM. Genotypic characterization of drug-resistant Mycobacterium tuberculosis isolates from Peru. Tuber. Lung Dis. 1998;79:111–118. doi: 10.1054/tuld.1998.0013. [DOI] [PubMed] [Google Scholar]
- 12.Roujeinikova A, Sedelnikova S, de Boer GJ, Stuitje AR, Slabas AR, Rafferty JB, Rice DW. Inhibitor binding studies on enoyl reductase reveal conformational changes related to substrate recognition. J. Biol. Chem. 1999;274:30811–30817. doi: 10.1074/jbc.274.43.30811. [DOI] [PubMed] [Google Scholar]
- 13.Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 14.Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Jr., Sacchettini JC, Fidock DA. Targeting tuberculosis and malaria through inhibition of enoyl reductase: compound activity and structural data. J. Biol. Chem. 2003;278:20851–20859. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- 15.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 16.Ryan AJ, Gray NM, Lowe PN, Chung CW. Effect of detergent on “promiscuous” inhibitors. J. Med. Chem. 2003;46:3448–3451. doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- 17.Brik A, Lin YC, Elder J, Wong CH. A quick diversity-oriented amide-forming reaction to optimize P-subsite residues of HIV protease inhibitors. Chem. Biol. 2002;9:891–896. doi: 10.1016/s1074-5521(02)00184-9. [DOI] [PubMed] [Google Scholar]
- 18.Lee LV, Mitchell ML, Huang SJ, Fokin VV, Sharpless KB, Wong CH. A potent and highly selective inhibitor of human alpha-1,3-fucosyltransferase via click chemistry. J. Am. Chem. Soc. 2003;125:9588–9589. doi: 10.1021/ja0302836. [DOI] [PubMed] [Google Scholar]
- 19.Wu CY, Chang CF, Chen JS, Wong CH, Lin CH. Rapid diversity-oriented synthesis in microtiter plates for in situ screening: discovery of potent and selective alpha-fucosidase inhibitors. Angew. Chem. Int. Ed. Engl. 2003;42:4661–4664. doi: 10.1002/anie.200351823. [DOI] [PubMed] [Google Scholar]
- 20.He X, Lin ZY, Zhu LY, Fu HJ. Synthesis and vasodilative activities of benzamide derivatives. Acta Pharmaceutica Sinica. 1998;33:666–674. [PubMed] [Google Scholar]
- 21.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J. Med. Chem. 2005;48:3621–3629. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward WH, Holdgate GA, Rowsell S, McLean EG, Pauptit RA, Clayton E, Nichols WW, Colls JG, Minshull CA, Jude DA, Mistry A, Timms D, Camble R, Hales NJ, Britton CJ, Taylor IW. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–12525. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 23.Qiu X, Janson CA, Court RI, Smyth MG, Payne DJ, Abdel-Meguid SS. Molecular basis for triclosan activity involves a flipping loop in the active site. Protein Sci. 1999;8:2529–2532. doi: 10.1110/ps.8.11.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roujeinikova A, Levy CW, Rowsell S, Sedelnikova S, Baker PJ, Minshull CA, Mistry A, Colls JG, Camble R, Stuitje AR, Slabas AR, Rafferty JB, Pauptit RA, Viner R, Rice DW. Crystallographic analysis of triclosan bound to enoyl reductase. J. Mol. Biol. 1999;294:527–535. doi: 10.1006/jmbi.1999.3240. [DOI] [PubMed] [Google Scholar]
- 25.Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. Molecular basis of triclosan activity. Nature. 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 26.Stewart MJ, Parikh S, Xiao G, Tonge PJ, Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 1999;290:859–865. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- 27.Paytash PL, Sparrow E, Gathe JC. The reaction of itaconic acid with primary amines. J. Am. Chem. Soc. 1950;72:1415–1416. [Google Scholar]
- 28.Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parikh S, Moynihan DP, Xiao G, Tonge PJ. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 1999;38:13623–13634. doi: 10.1021/bi990529c. [DOI] [PubMed] [Google Scholar]
- 30.Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs WR, Jr., Blanchard JS. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry. 1995;34:8235–8241. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- 31.Dessen A, Quemard A, Blanchard JS, Jacobs WR, Jr., Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 32.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Kleywegt GJ, Jones TA. Databases in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1998;54:1119–1131. doi: 10.1107/s0907444998007100. [DOI] [PubMed] [Google Scholar]
- 36.Bogeso KP. Neuroleptic activity and dopamine-uptake inhibition in 1-piperazino-3-phenylindans. J. Med. Chem. 1983;26:935–947. doi: 10.1021/jm00361a002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.