

Introduction
Status epilepticus (SE) is a continuous seizure or 2 or more discrete seizures between which there is incomplete recovery of consciousness lasting more than 30 minutes(1, 2). SE commonly occurs in infants and toddlers, with more than 50% of cases of SE occurring below the age of 2 years (3, 4). SE is associated with high mortality, brain injury and the subsequent development of epilepsy in an age dependent fashion(5). Because SE may have deleterious effects on the brain, it is considered a neurological emergency that needs to be treated as quickly as possible to avoid the detrimental effects (6). The longer SE continues the less likely it will stop (7). This is the case also in infants and children; Eriksson et al have shown that SE not treated within 30 minutes of onset is more difficult to stop with antiepileptic medication (8).
The reasons for the diminished efficacy of antiepileptic medications to stop SE over time are not elucidated. Experimental studies in adult rats (9) suggest that the development of refractoriness as SE progress is perhaps related to the emergence of pharmacoresistance to GABAA agents such as benzodiazepines and barbiturates (10). Goodkin et al have also shown that the development of resistance to diazepam (DZP) treatment may also exist in young rats as well. They demonstrated that a low dose of DZP (2mg/kg) was sufficient to stop SE at 5 minutes after onset, but ineffective when given after 15 minutes of SE (11). For this reason it has been proposed that any seizure lasting greater than 5 minutes be considered and treated as SE(12). However, it is also possible that initial higher doses of medications may be more effective than those routinely recommended for the treatment of SE (2), when the SE is prolonged. Indeed, it is common for children with seizures to reach the emergency room only after the seizure has been ongoing for a prolonged period of time(13).
In this study, we evaluated the effectiveness of DZP and PTB in stopping 1-hour long SE induced either by kainic acid (KA) or lithium-pilocarpine (Li-Pilo) in developing male rats at postnatal day (P)9, 15 and 21 which may correspond to a newborn(14–17), toddler(18) and young child respectively(19, 20). KA is a glutamate receptor agonist and pilocarpine is a muscarinic acetylcholine receptor agonist; both agents induce a partial onset SE with secondary generalization. Lithium potentiates the epileptogenic action of pilocarpine(21). Two different drugs were used: DZP and pentobarbital (PTB). DZP is often used clinically to stop seizures that have not stopped on their own after 5 minutes(22). PTB is commonly used to treat refractory SE(23).
Methods
Animals
The experimental procedures were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine and performed according to the Revised Guide for the Care and Use of Laboratory Animals [NIH GUIDE, 25(28), 1996]. Sprague–Dawley lactating dams with a litter of male rats were obtained from Taconic Farms (Germantown, NY). Rats were kept on regular light/dark cycle with free access to food and water. Animals were housed in Plexiglas cages with 10 pups per mother per cage. The rats for each group were chosen randomly from each litter to avoid litter effect. The data collected in this study are limited to male rats. In the ages we used, there are sex dependent differences in the brain(24) that may influence the efficacy of the drugs in treating SE in female rats(25).
Models of seizure
To induce KA SE, P9, P15 and P21 rats were injected i.p. with KA (Ocean Produce International, Shelburne, NS, Canada) diluted in PBS. The doses were: 1.5mg/kg, 3.5 mg/kg and 10mg/kg, respectively. To induce Li-Pilo SE, the same age groups were injected i.p. with pilocarpine (Sigma Chemical Co. St. Louis, MO, USA), diluted in saline. The doses were 60 mg/kg, 60mg/kg and 30 mg/kg, respectively. These rats were pretreated with lithium (3 mmol/kg) 18–22 hours earlier (26). The doses of Li-Pilo used have been shown to induce behavioral and electrographic SE in all age groups (27).
Monitoring studies
P15 and P21 rats were monitored during SE both behaviorally and electrographically. Based on pilot studies showing that DZP and PTB cannot stop behavioral SE in P9 rats, these rats were monitored only behaviorally.
One bipolar twisted electrode (MS303; Plastics One, Roanoke, VA, U.S.A.) was inserted in the left CA1 region of the dorsal hippocampus of P13 and P19 rats anesthetized with a mixture of ketamine (70 mg/kg Ketaset; Fort Dodge Animal Health, Fort Dodge, IA, U.S.A.) and xylazine (10 mg/kg Anased; Lloyd Laboratories, Shenandoah, IA, U.S.A.). The following coordinates were used: anterioposterior, 3.0 mm from the bregma; lateral, 2.0 mm from the midline; depth, 3.0 mm (P13) and 3.5mm (P19) from the external surface of the skull, with the tooth bar set at –3.5 mm. The electrode was fixed to the skull by using two jeweler screws and dental acrylic cement. After surgery, rats were allowed to recover with their dams for 48 h.
On the day of the testing, each rat pup was placed in an individual cage. The recording electrode was connected by swivel wiring to a monitoring apparatus. EEG was recorded and stored in a computer using the Harmonie computer program (Stellate Systems, Montreal, PQ, Canada) and was analyzed off-line. To determine whether DZP and PTB can stop both behavioral and electrographic seizures, the rats underwent continuous video-EEG monitoring starting from 20 minutes prior to administration of KA or pilocarpine and for 60 minutes following treatment. The rats were then returned to the dam and re-monitored for 30 minutes every 6 hours over the next 24 hours and again at 48 hours to confirm that SE had not recurred. Electrode placement was verified histologically.
Treatment protocols
Depending on the rats’ age, various doses of DZP (Sigma, St. Louis MO) and PTB (Abbott laboratories North Chicago, IL) were used (Tables 1 and 2 respectively). The drugs were diluted in saline and injected i.p. Treatment was initiated 1 hour after the onset of SE, corresponding to the time the rats began experiencing successive clonic or tonic-clonic seizures (including scratching and salivation) without recovery to normal baseline. On the EEG, the behavioral seizures were accompanied by electrographic seizure patterns (Figure 1, middle tracings). SE usually occurred around 15–20 minutes after KA or pilocarpine administration.
Table 1.
Effects of DZP on SE, latency to seizure cessation, and mortality as a function of age.
| Age | SE | DZP dose mg/kg | n | SE Stopped | Dose trend for SE stopped (p) | Min. to SE stop | Dose trend for Min. to SE stop (p) | Mortality | Dose Trend for mortality (p) |
|---|---|---|---|---|---|---|---|---|---|
| P9 | KA | 30 | 6 | 0 (0%) | - | NA | - | 2 (33%) | 0.24 |
| 40 | 5 | 0 (0%) | NA | 4 (80%) | |||||
| Li- Pilo | 30 | 6 | 0 (0%) | - | NA | - | 6 (100%) | - | |
| 40 | 6 | 0 (0%) | NA | 6 (100%) | |||||
| P15 | KA | 20 | 8 | 5 (63%) | 0.016 * | 1.1 ± 0.1 | 0.32 | 0 (0%) | 0.000054 * |
| 45 | 5 | 4 (80%) | 1.3 ± 0.1 | 4 (80%) | |||||
| 60 | 6 | 6(100%) | 1.2 ± 0.1 | 6 (100%) | |||||
| Li-Pilo | 20 | 10 | 6 (60%) | 0.05 * | 1.0 ± 0.1 | 0.19 | 1 (10%) | 0.000068 * | |
| 45 | 5 | 5 (100%) | 1.1 ± 0.1 | 5 (100%) | |||||
| 60 | 6 | 6 (100%) | 1.1 ± 0.1 | 6 (100%) | |||||
| P21 | KA | 20 | 6 | 0 (0%) | 0.00044 * | * | 0.56 | 0 (0%) | 0.00075 * |
| 45 | 5 | 4 (80%) | 0.9 ± 0.1 | 1 (20%) | |||||
| 60 | 5 | 3 (60%) | 0.8 ± 0.1 | 5 (100%) | |||||
| Li- Pilo | 20 | 6 | 3 (50%) | 0.86 | 1.0 ± 0.1 | 0.93 | 0 (0%) | - | |
| 45 | 6 | 3 (50%) | 0.8 ± 0.1 | 0 (0%) | |||||
| 60 | 5 | 5 (100%) | 0.9 ± 0.2 | 0 (0%) |
NA – not applicable
Table 2.
Effects of PTB on SE, latency to seizure cessation, and mortality as a function of age.
| Age | SE | PTB dose mg/kg | n | SE Stopped | Dose trend for SE stopped(p) | Min. to SE stop | Dose trend for Min. to SE stop(p) | Mortality | Dose Trend for mortality(p) |
|---|---|---|---|---|---|---|---|---|---|
| P9 | KA | 40 | 10 | 0 (0%) | - | NA | - | 0 (0%) | 0.000023 * |
| 45 | 8 | 0 (0%) | NA | 8 (100%) | |||||
| Li-Pilo | 40 | 6 | 0 (0%) | - | NA | - | 6 (100%) | - | |
| 45 | 9 | 0 (0%) | NA | 9 (100%) | |||||
| P15 | KA | 30 | 5 | 0 (0%) | 0.0000031 * | NA | 0.11 | 0 (0%) | 0.34 |
| 40 | 6 | 1 (17%) | 3.5 | 1 (17%) | |||||
| 50 | 13 | 13 (100%) | 2.5 ± 0.2 | 3 (23%) | |||||
| Li-Pilo | 30 | 4 | 0 (0%) | 0.00017 * | NA | 0.47 | 0 (0%) | 0.00017 * | |
| 40 | 6 | 3 (50%) | 5.9 ± 0.2 | 6 (100%) | |||||
| 50 | 11 | 11 (100%) | 5.0 ± 0.6 | 11 (100%) | |||||
| P21 | KA | 30 | 6 | 0 (0%) | 0.000022 * | NA | 0.47 | 0 (0%) | - |
| 40 | 11 | 6 (55%) | 13.0 ± 0.6 | 0 (0%) | |||||
| 50 | 11 | 11 (100%) | 12.5± 0.3 | 0 (0%) | |||||
| Li-Pilo | 30 | 6 | 1 (17%) | 0.0033 * | NA | 0.04 * | 1 (17%) | 0.013 * | |
| 40 | 5 | 4 (80%) | 7.5 ± 0.3 | 3 (60%) | |||||
| 50 | 11 | 10 (91%) | 6.9 ± 0.3 | 9 (82%) |
NA – not applicable
Figure 1. EEG recordings from P21 rats after Li-Pilo induced SE, treated with (A)DZP 45mg/kg, (B) PTB 40mg/kg.
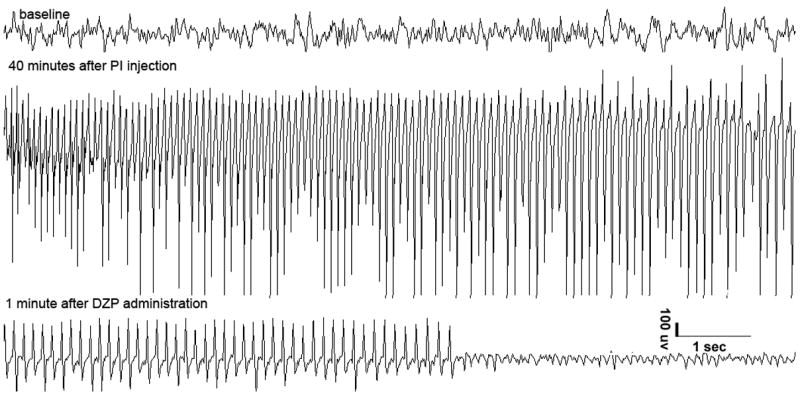
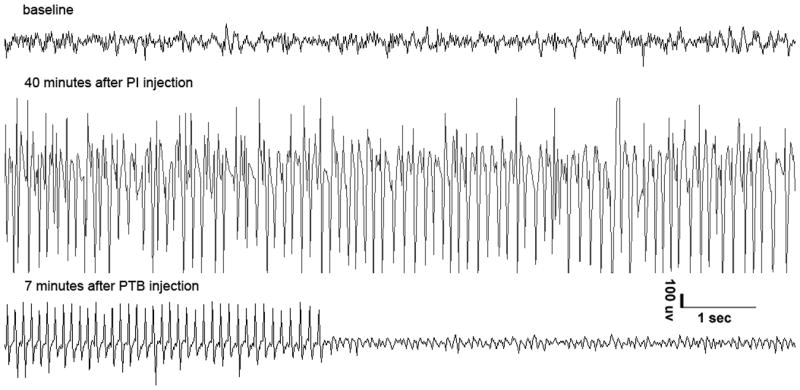
*please note that DZP stopped the electrographic SE within 1 minute, while PTB was effective 7 minutes after PTB injection.
The treatment of SE was considered effective when associated with complete cessation of behavioral seizures and with complete cessation of EEG seizure discharges for the duration of the observation period (Figure 1, bottom tracings). Acute mortality was assessed during the 2-hour period following the administration of the treatment.
Two age-matched controls were used: 1) To determine how long SE will continue if left untreated, KA or Li-Pilo rats were injected with saline. 2) To control for the effects of treatments, saline injected rats (not exposed to KA or Li-Pilo) were given doses of DZP or PTB that were effective and identified post hoc in this study.
Following the completion of the single treatment protocols, a group of P15 rats with KA induced SE were treated sequentially with lower doses of DZP and PTB. The doses of DZP and PTB were based on the collected data with a single drug treatment (see results). These rats first received a low dose of DZP (20mg/kg) and, if SE did not stop within 5 minutes, they received a low dose of PTB (20mg/kg); this dose was purposefully chosen to be lower than the doses used in single treatments to test for possible synergistic effects and to avoid a possible addititive respiratory compromise from the combined treatment.
Statistical analysis
The dose response trend for seizure stopping and mortality was assessed by computing the exact p-value for the Cochran-Armitage test for trend. The dose-response response trend for minutes until the seizure stopped was evaluated by fitting a linear regression model with dose as the predictor variable. To compare any differences between the two SE models in terms of percentage of SE cessation and mortality for each treatment, a Mantel-Haenszel test was performed stratified by dose. Age dependent effects of stopping SE and mortality for each treatment (PTB/DZP), after adjusting for dose were evaluated using a logistic regression model. Data from both the KA and Li-Pilo models were combined for this analysis. Comparison of mortality between experimental groups and control groups were evaluated by calculating p-values based on the Fisher’s exact test. The reported values are mean ± standard error (X ± SE) Significance was determined based on p ≤ 0.05.
Results
Efficacy following single drug treatment
Table 1 depicts the effects of DZP and Table 2 the effects of PTB in the 3 age groups on their efficacy to stop and latency to SE cessation as a function of dose.
At P9, neither DZP nor PTB were effective at stopping SE at all doses tested in both seizure models. The highest used DZP and PTB doses were associated with a high mortality and thus higher doses could not be used.
At P15 and P21 both DZP and PTB were effective in stopping behavioral and electrographic SE simultaneously (figure 1) and Tables 1 and 2 in a dose dependent fashion. There were no partial responses. The effects of each treatment on KA or Li-Pilo induced SE in these two age groups are specifically discussed below.
Efficacy of DZP (20mg/kg, 45mg/kg, and 60mg/kg, Table 1)
a) KA-induced SE
At P15, the doses stopped KA induced SE in 63%, 80%, and 100% of rats respectively. There was a statistically significant increase in efficacy with increase in dose (p=0.016). The average latency to seizure cessation was 1.2 ± 0.05 minutes without a significant influence with increasing dose.
At P21, the same doses stopped SE in 0%, 80%, and 60% of rats respectively with a statistically significant (p=0.00044) dose response trend; however, this was mainly due to differences observed between the low dose and medium dose groups with a ceiling effect. When SE was stopped, the average latency to seizure cessation was 0.83 ±0.06 minutes without a significant influence with increasing dose.
b) Li-Pilo induced SE
At P15, the doses stopped SE in 60%, 100%, and 100% of rats respectively. Once again, the dose response trend was statistically significant (p=0.05) with a ceiling effect. The average latency to seizure cessation was 1.06 ± 0.06 minutes, irrespective of dose.
At P21, the doses stopped SE in 50%, 50%, and 100% of rats respectively with a statistically significant dose response trend. Average latency to seizure cessation was 1.65 ± 0.76 minutes without a statistically significant influence with increasing dose.
Efficacy of PTB (30mg/kg, 40mg/kg, and 50mg/kg, Table 2)
a) KA-induced SE
At P15, the doses stopped SE in 0%, 17% and 100% of rats respectively with a statistically significant (p=0.0000031) increase with dose. Average latency to seizure cessation was 2.57 ± .16 minutes without a significant influence with increasing dose.
At P21, the doses stopped SE in 0%, 55%, and 100% of rats respectively with a statistically significant (p=0.000022) increase with dose as well. Average latency to seizure cessation was 12.69 ± 0.30 minutes regardless of the dose.
b) Li-Pilo induced SE
At P15, the doses stopped SE in 0%, 50%, and 100% of rats respectively with a statistically significant (p=0.00017) dose response trend. Average latency to seizure cessation was 5.23 ± 0.47 minutes without any significant increase with increasing dose.
At P21, these doses stopped SE in 0%, 80%, and 91% of rats respectively with a statistically significant (p=0.0033) increase with dose with a ceiling effect. Average latency to seizure cessation was 7.08 ± 0.25 minutes with a statistically significant influence with increasing dose (p=0.04).
To determine whether the effectiveness of the treatment was model dependent, the data were evaluated using a Mantel-Haenszel test, stratified by dose. There was no significant difference in seizure cessation following the treatments between the two SE models.
To determine the role of age on the effectiveness of DZP and PTB to stop SE we used a logistic regression model after adjusting for dose. The data from both the KA and Li-Pilo models were combined. Following treatment with DZP, there was a significantly higher probability of DZP stopping seizures at P15 over P21 (p=0.0068). Following treatment with PTB, there was no overall significant difference in efficacy between P15 and P21.
Mortality following treatments
Although the treatments stopped SE, they were associated with significant mortality often in a dose dependent fashion (Tables 1 and 2). Mortality was high even though SE was stopped and did not recur prior to death. Mortality rates were the lowest in the P21 rats exposed to Li-Pilo induced SE and treated with DZ and to KA induced SE treated with PTB.
To determine whether the treatments may have been more detrimental than SE per se, we compared the mortality among rats exposed to SE and treated with DZP or PTB, to rats exposed to SE but not treated (Table 3). We also compared the mortality among rats exposed to SE and treated with DZP or PTB, to rats that received DZP or PTB in the absence of SE (Table 4). For the latter analysis, the doses of DZP 20mg/kg and PTB 50mg/kg were given to P15 and P21 rats. At P9, we used lower doses (DZP 30mg/kg and PTB 40mg/kg) as these doses were associated with lower mortality than the higher dose. Most groups had similar mortality regardless of whether or not they were exposed to KA or Li-Pilo SE. Indeed, the treatment of SE increased mortality in several groups although it decreased mortality in others, suggesting that these treatments should be administered with caution. Rats that survived the first 2 hours following treatment were monitored intermittently as described in the methods section. Behavioral observation and EEG recording during this period showed no evidence of seizure recurrence in the animals that had seizure cessation following treatment. However, it should be noted that during this 48-hour observation period several of the rats were cannibalized by the dam.
Table 3.
Comparison of mortality between rats exposed to SE with or without treatment.
| Age | Model of SE/Drug | Dose of drug | Mortality with treatment | Mortality without treatment | p |
|---|---|---|---|---|---|
| P9 | KA/DZP | 30 | 2/6=33% | 0/9=0% | 0.14 |
| 40 | 4/5=80% | 0.005 * | |||
| Li-Pilo/DZP | 30 | 6/6=100% | 0/5=0% | 0.0022 * | |
| 40 | 6/6=100% | 0.0022 * | |||
| KA/PTB | 40 | 0/10 = 0% | 0/9=0% | 1.0 | |
| 45 | 8/8=100% | 0.000041 * | |||
| Li-Pilo/PTB | 40 | 6/6=100% | 0/5=0% | 0.0022 * | |
| 45 | 9/9=100% | 0.000050 * | |||
| P15 | KA/DZP | 20 | 0/8=0% | 2/12=17% | 0.49 |
| 45 | 4/5=80% | 0.028 * | |||
| 60 | 6/6=100% | 0.0015 * | |||
| Li-Pilo/DZP | 20 | 1/10=10% | 10/12=83% | 0.0019 * | |
| 45 | 5/5=100% | 1.0 | |||
| 60 | 6/6=100% | 0.52 | |||
| KA/PTB | 30 | 0/5=0% | 2/12=17% | 1.0 | |
| 40 | 1/6=17% | 1.0 | |||
| 50 | 3/13=23% | 1.0 | |||
| Li-Pilo/PTB | 30 | 0/4=0% | 10/12=83% | 0.0082 * | |
| 40 | 6/6=100% | 0.53 | |||
| 50 | 11/11=100% | 0.48 | |||
| P21 | KA/DZP | 20 | 0/6=0% | 1/7=14% | 1.0 |
| 45 | 1/5=20% | 0.15 | |||
| 60 | 5/5=100% | 0.02 * | |||
| Li-Pilo/DZP | 20 | 0/6=0% | 4/6=67% | 0.06 | |
| 45 | 0/6=0% | 0.06 | |||
| 60 | 0/5=0% | 0.06 | |||
| KA/PTB | 30 | 0/6=0% | 1/7=14% | 1.0 | |
| 40 | 0/6=0% | 1.0 | |||
| 50 | 0/11=0% | 0.39 | |||
| Li-Pilo/PTB | 30 | 1/6=17% | 4/6=67% | 0.24 | |
| 40 | 3/5=60% | 1.0 | |||
| 50 | 9/11= 82% | 0.58 |
At P9: The treatment with DZP or PTB increased mortality in 6/8 groups; only rats with KA induced SE that were treated with DZP 30mg/kg or PTB 40mg/kg had no change in mortality.
At P15: In 8/12 groups, treatment did not have any significant change on mortality rates compared to controls. The exceptions were: rats with KA induced SE that were treated with DZP 45mg/kg or 60 mg/kg had increased mortality, while rats with KA induced SE that were treated with DZP 20mg/kg and rats with Li-Pilo induced SE that were treated with PTB 30mg/kg had lower mortality.
At P21: In rats with KA induced SE, the treatment did not significantly change mortality in any group except in the rats treated with 60mg/kg of DZP. In rats with Li-Pilo induced SE that were treated with DZP, differences in mortality between treated and untreated rats were nearly significant within each individual dose and when the 3 doses were combined, a significant decrease in mortality was observed.
Table 4.
Comparison of mortality between rats exposed to SE and treated with rats not exposed to SE not treated with either DZP or PTB.
| Age | Model of SE | Drug | Dose mg/kg | Mortality of DZP or PTB with SE | Mortality DZP or PTB without SE | p |
|---|---|---|---|---|---|---|
| P9 | KA | DZP | 30 | 33% | 0% | 0.5 |
| PTB | 40 | 0% | 10% | 1.0 | ||
| Li-Pilo | DZP | 30 | 100% | 0% | 0.01 * | |
| PTB | 40 | 100% | 10% | 0.00087 * | ||
| P15 | KA | DZP | 20 | 0% | 0% | - |
| PTB | 50 | 23% | 23% | - | ||
| Li-Pilo | DZP | 20 | 10% | 0% | 0.45 | |
| PTB | 50 | 100% | 23% | 0.0001 * | ||
| P21 | KA | DZP | 20 | 0% | 0% | - |
| PTB | 50 | 0% | 11% | 0.45 | ||
| Li-Pilo | DZP | 20 | 0% | 0% | - | |
| PTB | 50 | 82% | 11% | 0.006 * |
At P9: In the KA model the mortality following the treatment was not different from rats without SE that were treated with DZP or PTB. In the Li-Pilo model, both treated groups had a significantly higher mortality than the groups without SE. At P15 and P21: In the KA model there was no difference in mortality between the two groups. In the Li-Pilo model rats that received PTB following SE had a significantly higher mortality than the rats that were given PTB but not exposed to Li-Pilo. There was no difference in the rats receiving DZP.
Sequential Treatment
We administered PTB 20mg/kg in a group of P15 rats with KA induced SE that was not stopped with DZP 20mg/kg (n=10, 41% of total) within 5 min after its administration. In all 10 rats, PTB stopped SE and only 3 rats died.
Discussion
In this study, we addressed two primary outcomes related to the treatment of 1 hour long SE in developing rats in two models of SE (KA and Li-Pilo): a) drug efficacy and b) latency to seizure cessation. We also determined the acute mortality at 2 hours in rats that SE was stopped as well as overall mortality rates within this period. Using DZP or PTB, we have shown that it is possible to stop SE after 1 hour in P21 and P15 rats but not in P9 rats in both models.
The results show that the drugs, when effective, stop both the behavioral and also the EEG seizures. To completely stop SE after one hour, higher doses than those used to stop shorter seizures are required(28–30). For example, in adult rats DZP 20mg/kg is effective in stopping SE in all rats if treatment is initiated after 10 minutes of seizures. However after 45 minutes of seizures this same dose is ineffective(29). Goodkin et al have shown that, in developing rats, a low dose of DZP (2mg/kg) was sufficient to stop SE at 5 minutes after onset, but was ineffective past 15 minutes of SE(11). Other studies have treated prolonged seizures with lower doses of DZP (5–10mg/kg) but the stated desired outcome was attenuation of seizures to promote long term studies of epileptogenesis, rather than complete cessation(31–33).
In this study, the action of DZP appears to be very quick, suggesting that subsequent treatments can be rapidly initiated if the seizures do not stop within 2 minutes. On the other hand, PTB took significantly longer to work, and more time was needed to determine if the therapy stopped the seizures. One possible factor that may explain this difference is the fact that after systemic administration, DZP is redistributed in a manner similar to lipid soluble agents while PTB belongs to the group of barbiturates that are less lipid soluble and equilibrate much more slowly, since its uptake is limited by permeability(34). These data suggest that after a benzodiazepine such as DZP is unsuccessful, one should consider the next treatment fairly quickly. Indeed, the combined treatment with DZP and PTB had the benefit of increasing efficacy without the mortality associated with increasing the dose of DZP. This was shown in P15 rats with KA induced SE as the dose of PTB (20mg/kg, which was lower than an ineffective dose of 30 mg/kg) was effective in stopping SE 100% of the rats after DZ was tried and failed to stop SE. These results imply that DZP administration may prime the effectiveness of PTB and that lower doses can be used when both drugs are administered sequentially.
The efficacy of DZP and PTB was similar in the two seizure models (KA and Li-Pilo) and suggests that it may not be necessary to test every putative treatment in many SE models. From the clinical standpoint, the data suggest that it may not be crucial to determine the cause of SE before choosing the treatment. However, mortality following treatment was significantly different in the two seizure models. In general, untreated Li-Pilo induced SE is associated with higher mortality than KA induced SE. On the other hand, at P21, in the Li-Pilo model, DZP administration was associated with a lower mortality than in the KA model or in the control (untreated) rats. This observation is supported by studies commonly using DZP to increase survival following Li-Pilo induced SE as discussed above.
Controlling SE was not necessarily associated with improvements in mortality
For example, even when PTB is effective in stopping SE (i.e. P15 and P21), there is no effect on mortality. Our study did not identify the cause of the high mortality following treatment. It is possible that death is secondary to apnea from the central sedative effects of the GABAergic drugs. It is also possible that hypotension is a factor in the increased mortality, especially in the group receiving both Li-Pilo and PTB, which are both known to cause hypotension(34). In human studies as well, although PTB is very effective, at the dose needed to stop SE, it causes significant mortality and morbidity(35). In future studies it may be possible to improve mortality rates by performing the experiments in an intensive care environment, such as by using the “pup in a cup” preparation(36, 37). These data suggest that, in a clinical setting, the administration of these high doses should be in a controlled environment where appropriate support can be provided.
Although DZP and PTB were effective in controlling the seizures in P15 and P21 rats they were unable to stop the seizures in P9 rats. Both DZP and PTB are drugs that act on GABAA receptors(34), although PTB also acts by direct inhibition of excitatory AMPA-type glutamate receptors(34). In the developing brain, the functionality of the GABAA receptor changes with age(38), and this may be responsible for differences in seizure control following administration of GABAA agonists in the youngest age group (39) (P9, an age equivalent to human neonates(20)). Although it is possible that increasing the doses of DZP or PTB may have stopped the seizures, the already high mortality precluded their use. In human neonates high doses of PTB cause many adverse complications including hypotension and pulmonary edema(40). Furthermore, antiepileptic drugs may cause significant morbidity and mortality in the developing brain especially when used at very high doses(41). It is interesting that in the current study the mortality of untreated SE at P9 was, lower than that in the treated groups suggesting that the adverse effects of the drugs overwhelmed the beneficial effects of stopping the SE using high doses. Indeed, there is a real concern regarding the risk benefit ratio of using high doses of GABAergic drugs such as DZP and PTB in the treatment of SE in human neonates(40).
The data suggest that when treating SE, the cause may not play a predictive role in the success of the medication, while age may be a significant factor in choosing a treatment. While it is desirable to treat SE early on, this may not always be possible because there is often a lag in recognition of SE or in getting the patient to medical attention. Our data, together with the data of Goodkin (11) indicate that it may be necessary to immediately use higher doses to treat the prolonged seizures than currently used doses. Our data also suggest that after a benzodiazepine such as DZP is unsuccessful, one should consider the next treatment fairly quickly, since if DZP is going to be effective, it will stop the SE within a few minutes. After several minutes have passed, it may be appropriate to introduce a new medication such as PTB before attempting higher doses of DZP. The combined treatment may have the benefit of increasing efficacy with lower morbidity. This is in contrast to the common clinical practice of pushing one drug to toxicity before adding another drug(42). Nevertheless, because of the significant morbidity associated with administration of high doses of DZP or PTB to stop SE, these treatments should be offered in an intensive care environment.
Acknowledgments
Supported by: Dr. Hasson is an NSADA trainee supported by K12 NS048856. Supported by RO1 NS20253 from NINDS and the Heffer Family Foundation. S.L.M. is a recipient of a Martin A. and Emily L. Fischer Fellowship in Neurology and Pediatrics.
Footnotes
This submission is a basic science research article in the field of developmental epilepsy.
The recommendations from the Declaration of Helsinki and the internationally accepted principles in the care and use of experimental animals have been adhered to
Institutional approval was obtained
All coauthors have seen and agree with the attached manuscript.
None of the coauthors have any financial disclosures to disclose
This submission (aside from abstracts) is not under review at any other publication.
Group authorship: N/A
Credits and Permissions: N/A
Financial interests: None to report
Conflicts of interest: None to report
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guidelines for epidemiologic studies on epilepsy. Commission on Epidemiology and Prognosis, International League Against Epilepsy. Epilepsia. 1993;34(4):592–6. doi: 10.1111/j.1528-1157.1993.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 2.Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. Jama. 1993;270(7):854–9. [PubMed] [Google Scholar]
- 3.DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46(4):1029–35. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- 4.DeLorenzo RJ, Towne AR, Pellock JM, Ko D. Status epilepticus in children, adults, and the elderly. Epilepsia. 1992;33(Suppl 4):S15–25. doi: 10.1111/j.1528-1157.1992.tb06223.x. [DOI] [PubMed] [Google Scholar]
- 5.Hasson H, Haut S, Moshé SL, Veliskova J, Shinnar S. Temporal lobe epileptogenesis and epilepsy in the developing brain: bridging the gap between the laboratory and the clinic. In: Arzimanoglou A, Aldenkamp A, Cross H, Lassonde M, Moshé SL, Schmitz B, editors. Progress in Epileptic, Cognitive Dysfunction in Children with Temporal Lobe Epilepsy: John Libbey Eurotext. 2005. pp. 183–202. [Google Scholar]
- 6.Riviello JJ, Jr, Ashwal S, Hirtz D, Glauser T, Ballaban-Gil K, Kelley K, et al. Practice parameter: diagnostic assessment of the child with status epilepticus (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2006;67(9):1542–50. doi: 10.1212/01.wnl.0000243197.05519.3d. [DOI] [PubMed] [Google Scholar]
- 7.Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology. 1993;43(3 Pt 1):483–8. doi: 10.1212/wnl.43.3_part_1.483. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson K, Metsaranta P, Huhtala H, Auvinen A, Kuusela AL, Koivikko M. Treatment delay and the risk of prolonged status epilepticus. Neurology. 2005;65(8):1316–8. doi: 10.1212/01.wnl.0000180959.31355.92. [DOI] [PubMed] [Google Scholar]
- 9.Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol. 1988;101(2):267–75. doi: 10.1016/0014-4886(88)90010-6. [DOI] [PubMed] [Google Scholar]
- 10.Jones DM, Esmaeil N, Maren S, Macdonald RL. Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res. 2002;50(3):301–12. doi: 10.1016/s0920-1211(02)00085-2. [DOI] [PubMed] [Google Scholar]
- 11.Goodkin HP, Liu X, Holmes GL. Diazepam terminates brief but not prolonged seizures in young, naive rats. Epilepsia. 2003;44(8):1109–12. doi: 10.1046/j.1528-1157.2003.62402.x. [DOI] [PubMed] [Google Scholar]
- 12.Lowenstein DH, Bleck T, Macdonald RL. It’s time to revise the definition of status epilepticus. Epilepsia. 1999;40(1):120–2. doi: 10.1111/j.1528-1157.1999.tb02000.x. [DOI] [PubMed] [Google Scholar]
- 13.Shinnar S. Who is at risk for prolonged seizures? J Child Neurol. 2007;22(5 Suppl):14S–20S. doi: 10.1177/0883073807303065. [DOI] [PubMed] [Google Scholar]
- 14.Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25(10):518–24. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 16.Gottlieb A, Keydar I, Epstein HT. Rodent brain growth stages: an analytical review. Biol Neonate. 1977;32(3–4):166–76. doi: 10.1159/000241012. [DOI] [PubMed] [Google Scholar]
- 17.Nehlig A. Cerebral energy metabolism, glucose transport and blood flow: changes with maturation and adaptation to hypoglycaemia. Diabetes Metab. 1997;23(1):18–29. [PubMed] [Google Scholar]
- 18.Velisek L, Moshe SL. Effects of brief seizures during development. Prog Brain Res. 2002;135:355–64. doi: 10.1016/S0079-6123(02)35032-5. [DOI] [PubMed] [Google Scholar]
- 19.Ojeda SR, Urbanski HF. Puberty in the rat. In: Knobil E, Neil J, editors. The Physiology of Reproduction. 2. New York: Raven Press, Ltd; 1994. pp. 363–409. [Google Scholar]
- 20.Ojeda SR, Andrews WW, Advis JP, White SS. Recent advances in the endocrinology of puberty. Endocr Rev. 1980;1(3):228–57. doi: 10.1210/edrv-1-3-228. [DOI] [PubMed] [Google Scholar]
- 21.Clifford DB, Olney JW, Maniotis A, Collins RC, Zorumski CF. The functional anatomy and pathology of lithium-pilocarpine and high-dose pilocarpine seizures. Neuroscience. 1987;23(3):953–68. doi: 10.1016/0306-4522(87)90171-0. [DOI] [PubMed] [Google Scholar]
- 22.Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001;345(9):631–7. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- 23.Prasad AN, Seshia SS. Status epilepticus in pediatric practice: neonate to adolescent. Adv Neurol. 2006;97:229–43. [PubMed] [Google Scholar]
- 24.Velísková J, Moshé SL. Sexual dimorphism and developmental regulation of substantia nigra function. Ann Neurol. 2001;50:596–601. doi: 10.1002/ana.1248. [DOI] [PubMed] [Google Scholar]
- 25.Diler AS, Uzum G, Akgun Dar K, Aksu U, Atukeren P, Ziylan YZ. Sex differences in modulating blood brain barrier permeability by NO in pentylenetetrazol-induced epileptic seizures. Life Sci. 2007;80(14):1274–81. doi: 10.1016/j.lfs.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 26.Sankar R, Shin DH, Liu H, Mazarati A, Pereira de Vasconcelos A, Wasterlain CG. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18(20):8382–93. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirsch E, Baram TZ, Snead OCd. Ontogenic study of lithium-pilocarpine-induced status epilepticus in rats. Brain Res. 1992;583(1–2):120–6. doi: 10.1016/s0006-8993(10)80015-0. [DOI] [PubMed] [Google Scholar]
- 28.Morrisett RA, Jope RS, Snead OC., 3rd Effects of drugs on the initiation and maintenance of status epilepticus induced by administration of pilocarpine to lithium-pretreated rats. Exp Neurol. 1987;97(1):193–200. doi: 10.1016/0014-4886(87)90293-7. [DOI] [PubMed] [Google Scholar]
- 29.Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17(19):7532–40. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338(14):970–6. doi: 10.1056/NEJM199804023381407. [DOI] [PubMed] [Google Scholar]
- 31.Suchomelova L, Baldwin RA, Kubova H, Thompson KW, Sankar R, Wasterlain CG. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr Res. 2006;59(2):237–43. doi: 10.1203/01.pdr.0000196333.16608.30. [DOI] [PubMed] [Google Scholar]
- 32.Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Research. 1998;814(1–2):179–85. doi: 10.1016/s0006-8993(98)01080-4. [DOI] [PubMed] [Google Scholar]
- 33.Pitkanen A, Kharatishvili I, Narkilahti S, Lukasiuk K, Nissinen J. Administration of diazepam during status epilepticus reduces development and severity of epilepsy in rat. Epilepsy Res. 2005;63(1):27–42. doi: 10.1016/j.eplepsyres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Brunton L, Lazo J, Parker K. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11. McGraw-Hill; 2005. [Google Scholar]
- 35.Yaffe K, Lowenstein DH. Prognostic factors of pentobarbital therapy for refractory generalized status epilepticus. Neurology. 1993;43(5):895–900. doi: 10.1212/wnl.43.5.895. [DOI] [PubMed] [Google Scholar]
- 36.West JR. Use of pup in a cup model to study brain development. J Nutr. 1993;123(2 Suppl):382–5. doi: 10.1093/jn/123.suppl_2.382. [DOI] [PubMed] [Google Scholar]
- 37.Beierle EA, Chen MK, Hartwich JE, Iyengar M, Dai W, Li N, et al. Artificial rearing of mouse pups: development of a mouse pup in a cup model. Pediatr Res. 2004;56(2):250–5. doi: 10.1203/01.PDR.0000132753.81333.39. [DOI] [PubMed] [Google Scholar]
- 38.Veliskova J, Claudio OI, Galanopoulou AS, Lado FA, Ravizza T, Velisek L, et al. Seizures in the developing brain. Epilepsia. 2004;45(Suppl 8):6–12. doi: 10.1111/j.0013-9580.2004.458002.x. [DOI] [PubMed] [Google Scholar]
- 39.Galanopoulou AS, Kyrozis A, Claudio OI, Stanton PK, Moshe SL. Sex-specific KCC2 expression and GABA(A) receptor function in rat substantia nigra. Exp Neurol. 2003;183(2):628–37. doi: 10.1016/s0014-4886(03)00213-9. [DOI] [PubMed] [Google Scholar]
- 40.Holmes GL, Riviello JJ., Jr Midazolam and pentobarbital for refractory status epilepticus. Pediatr Neurol. 1999;20(4):259–64. doi: 10.1016/s0887-8994(98)00155-6. [DOI] [PubMed] [Google Scholar]
- 41.Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A. 2002;99(23):15089–94. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wheless J, Clarke DF. Recurrent Seizures. In: Maria BL, editor. Current Management In Child Neurology. 3. B.C. Decker; 2005. p. 687. [Google Scholar]